重组载体的构建解析
重组载体构建的方法和步骤

重组载体构建的方法和步骤全文共四篇示例,供读者参考第一篇示例:重组载体构建是基因工程领域中非常重要的一项技术,它可以用来将特定的基因插入到目标细胞中,实现基因的转移和表达。
在科学研究、医学诊断和治疗等领域中都有广泛的应用。
下面我们来详细介绍一下重组载体构建的方法和步骤。
一、选择载体首先我们需要选择一个适合的载体作为基础,常见的载体有质粒、病毒、原核生物等。
在选择载体时需要考虑载体的大小和特性,以及目标基因的大小和需要表达的水平。
同时还需要考虑载体的复制原点、抗生素抗性基因等相关元件。
二、线性化载体接下来我们需要将选择的载体进行线性化处理,以便将目标基因插入到载体中。
线性化可以通过受控的限制酶酶切处理来实现,将载体的环状DNA骨架切割成线性DNA片段。
三、插入目标基因将目标基因与线性化的载体进行连接。
目前常用的方法包括:内切酶切割连接法、PCR扩增连接法、接头连接法等。
这些方法可以有效地将目标基因插入到载体中,并确保插入的正确性和稳定性。
四、转化目标宿主将构建好的重组载体导入到目标宿主细胞中,使其稳定地存在和复制。
转化的方法多样,包括热激转化、电穿孔转化、化学法转化等。
转化效率和载体稳定性是评价转化效果的主要因素。
五、筛选重组子对转化后的细胞进行筛选,筛选出含有目标基因的重组子。
常用的筛选方法包括抗生素筛选、荧光筛选、酵素检测等。
筛选过程中需要注意筛选压力和筛选条件的优化,以提高筛选效率。
六、鉴定重组子对筛选出的重组子进行鉴定,确保其构建正确。
常用的鉴定方法包括PCR扩增、酶切鉴定、序列分析等。
通过这些方法可以验证重组子的结构和功能是否正确,确保后续实验的准确性和可靠性。
七、表达目标基因对鉴定合格的重组子进行表达。
通过选用适当的启动子和调控元件,可以实现目标基因的高效表达。
表达的方法有多种选择,包括转染法、感染法、转基因法等。
表达的效果可以通过荧光显微镜观察、酶活性测定、Western blot等方法进行检测和验证。
载体的构建

基因工程所用的克隆载体常称之为分子克隆载体(molecular cloning vector),它是一类可供外源DNA插入并携带重组DNA分子进入适当宿主细胞的DNA分子。
如果打一个十分浅显的比喻,分子克隆载体犹如航天技术中的运载火箭,外源DNA分子就如火箭上搭载的卫星,宿主细胞则如外部深邃的空间。
分子克隆载体的主要功能就是将外源基因携带入宿主细胞,并在宿主细胞中进行DNA扩增和使外源基因得以高效表达。
在这一点上又与火箭不一样,因为火箭在运载卫星的过程中分级脱落和烧毁。
尽管克隆载体是由DNA分子所构成,但与宿主细胞的染色体DNA分子相比,它们一般都比较小。
比如常用的质粒载体多数都在10 kb以下,而常用的噬菌体载体,一般都在20 kb 左右。
我们知道,大肠杆菌的染色体DNA是4200kb。
正是由于克隆载体的分子量比较小,在操作过程中不会造成载体DNA分子的断裂,而大分子的DNA容易在操作中发生断裂。
克隆载体的基本结构和功能在短短的20年间,用于不同生物和不同目的的分子克隆载体至少也有好几千种,那么它们应该有些什么样的基本结构呢?经过仔细分析发现,所有的分子克隆载体都具备了以下3个最基本的结构:1)至少有一个复制起点,因而至少可在一种生物体中自主复制;2)至少应有一个克隆位点,以供外源DNA插入;3)至少应有一个遗传标记基因,以指示载体或重组DNA 分子是否进入宿主细胞。
既然说每一个载体至少应有一个复制起点,一个克隆位点和一个标记基因,那么一个载体可否有多个复制起点,多个克隆位点和多个标记基因呢?答案是肯定的。
不过,我们还是先看看载体中的三个基本结构克隆载体的复制起点由于DNA的复制是始于复制起点的,因此只要一个DNA分子有了复制起点,那么这个DNA分子就可以自主复制。
如果一个分子克隆载体有了复制起点,那么该载体就可以在某种生物的细胞中自主复制,因此这个载体就可以多拷贝地存在于某种细胞内。
多拷贝DNA有两个好处:一是可以用于大量制备克隆载体DNA分子,以利于外源基因的克隆,这样可大大减少工作量;二是如果载体中插入了外源基因,那么外源基因的拷贝数也就大量增加了,这就有利于大量地表达外源基因,从而获得大量的基因表达产物,这也正是基因工程的目的之一。
重组表达质粒的构建——原核表达载体选择
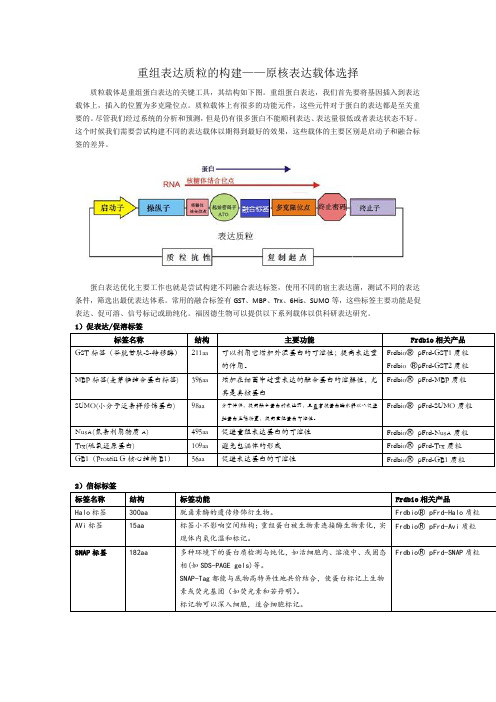
重组表达质粒的构建——原核表达载体选择质粒载体是重组蛋白表达的关键工具,其结构如下图。
重组蛋白表达,我们首先要将基因插入到表达载体上,插入的位置为多克隆位点。
质粒载体上有很多的功能元件,这些元件对于蛋白的表达都是至关重要的。
尽管我们经过系统的分析和预测,但是仍有很多蛋白不能顺利表达、表达量很低或者表达状态不好。
这个时候我们需要尝试构建不同的表达载体以期得到最好的效果,这些载体的主要区别是启动子和融合标签的差异。
蛋白表达优化主要工作也就是尝试构建不同融合表达标签,使用不同的宿主表达菌,测试不同的表达条件,筛选出最优表达体系。
常用的融合标签有GST、MBP、Trx、6His、SUMO等,这些标签主要功能是促表达、促可溶、信号标记或助纯化。
福因德生物可以提供以下系列载体以供科研表达研究。
1)促表达/促溶标签2)信标标签3)纯化标签我们选择表达载体的时候不但要考虑蛋白怎么表达成功,更要考虑蛋白怎么纯化出来,纯化的问题主要是考虑纯化标签和酶切位点的选择,下表我们列举了常见的纯化标签和酶切位点。
4)酶切位点以上为原核表达常用的标签和酶切位点,其性质也都作了简要的介绍,各专业网站或专业书籍已对此做详尽解释,科研工作者可根据具体实验设计方案,组合设计以上标签和酶切位点的使用。
特别值得注意的是,选用和设计蛋白酶切位点的时候首要考虑的是序列内部有没有蛋白酶位点,同时要考虑酶切的效率和蛋白酶试剂成本。
一般商业化载体,在标签蛋白与载体多克隆位点之间都设计有酶切位点。
标签可设计在N-端也可在C-端,设计在N-端的优势是,可通过标签高效翻译起始位点带动插入蛋白的表达,可溶性标签的高效表达更可促进蛋白的可溶性表达;同时,大部分的蛋白内切酶的切割位点在C-端,所以标签设计在N-端可将标签切割完全。
在设计标签序列与酶切位点的时候还要考虑N-端稳定性原则,也就是所谓宿主细胞的N-端规则(N-end rule),这个要避免;同时,还应该检查是否引入了可与别的蛋白相互作用的序列或者蛋白酶切位点。
汉恒生物实例分享-一步法构建同源重组载体

一步法构建同源重组载体王海艳(中国农业大学)同源重组载体构建需要分别克隆目的片段两端的同源臂(长度~1kb),并与Marker基因进行连接。
传统的构建方法是两段同源臂分别克隆构建到目的载体,费时费力。
在本例中,本人已经成功应用汉恒生物科技(上海)有限公司的HB-Infusion TM无缝克隆试剂盒,将同源臂及Marker基因一步成功构建到载体上,现将实验过程及结果分享如下:1. 目的片段引物的设计用NEB builder(https:///watch?v=8_-t5xtJ3y8),或snapgene,genome compiler等软件,将三个目的片段的基因序列、线性化载体的基因序列按照软件使用说明依次填入,将自动生成所需要的引物序列(如下表)。
将引物提交华大基因进行合成。
引物序列列表2. 目的片段的扩增反应体系DNA(248ng/uL)1uLForward Primer (10uM)2uLReverse Primer(10uM)2uLExTaq premix25uLDDW 20uLTotal50uL(注:此步反应中的酶最好用高保真酶,以免产物出现错配)PCR反应程序95℃4min95℃30s65℃30s72℃1~2.5min72℃10min将扩增到的PCR产物,用1%琼脂糖凝胶电泳后,进行胶回收并用Nanodrop 测量核酸浓度。
图1. 三个目的片段凝胶电泳结果3. 载体的线性化酶切:Plasmid2ug34cyclesSacI2uLXbaI2uL10×M buffer5uLDDW39uLTotal50uL37℃酶切4h后,用1%凝胶电泳进行分析,并用Axygen凝胶回收试剂盒进行胶回收。
图2. 载体酶切及胶回收实验结果A:质粒;B:质粒酶切产物;C:目的片段胶回收产物。
4. 冰上融化并且充分混匀HB-infusion TM Master mix,配制反应体系将各个片段按照说明书的要求计算用量并配制反应体系,如下表所示:片段碱基对数(bp)浓度(ng/uL)OD260/280所需mol数所需质量(ng)所需体积(uL)3'-flank1000129.5 1.840.2130.00 1.00 5’-flank1000141.4 1.890.2130.00 0.92 Marker fragment2518184.9 1.880.2327.34 1.77 Lineared vector287595.2 1.850.1186.88 1.96 HB-infusion MIX10 DDW 4.34 Total205. 将上述反应体系置于50℃孵育60min 。
重组载体的构建原理及目的

重组载体的构建原理及目的
重组载体是一种用于携带和传递DNA片段的分子工具。
其构建原理是将目标DNA片段插入到载体DNA分子中,形成一个新的重组载体。
重组载体的目的是用于基因工程、基因疗法以及基因表达等领域的研究。
构建重组载体需要具备以下几个步骤:
1. 选择载体:根据需要选择合适的载体,例如质粒、病毒、细胞质等。
2. 切割载体:将载体DNA分子用限制性内切酶或其他方法切割成线性或圆形片段。
3. 插入DNA片段:将目标DNA片段用限制性内切酶切割后,与载体DNA片段连接成重组载体。
4. 转化宿主细胞:将重组载体转化到宿主细胞中,让其复制并表达目标基因。
重组载体的构建可以实现外源基因的转染和表达,从而实现基因修饰、基因敲除、基因定位等目的。
同时,重组载体也可以用于基因疗法,将修饰后的基因载体导入到人体细胞,实现治疗作用。
因此,重组载体的构建是现代生命科学领域的重要研究内容之一。
- 1 -。
实例分享-一步法构建同源重组载体

汉恒生物科技(上海)有限公司 400-092-0065一步法构建同源重组载体同源重组载体构建需要分别克隆目的片段两端的同源臂(长度~1kb ),并与Marker 基因进行连接。
传统的构建方法是两段同源臂分别克隆构建到目的载体,费时费力。
在本例中,本人已经成功应用汉恒生物科技(上海)有限公司的HB-Infusion TM 无缝克隆试剂盒,将同源臂及Marker 基因一步成功构建到载体上,现将实验过程及结果分享如下:1. 目的片段引物的设计用NEB builder (https:///watch?v=8_-t5xtJ3y8),或snapgene ,genome compiler 等软件,将三个目的片段的基因序列、线性化载体的基因序列按照软件使用说明依次填入,将自动生成所需要的引物序列(如下表)。
将引物提交华大基因进行合成。
引物序列列表2. 目的片段的扩增反应体系汉恒生物科技(上海)有限公司 400-092-0065DNA(248ng/μL) 1 μL Forward Primer (10μM)2 μLReverse Primer(10μM) 2 μL ExTaq premix 25 μL DDW 20 μL Total50 μL(注:此步反应中的酶最好用高保真酶,以免产物出现错配)PCR 反应程序95℃ 4 min95℃ 30 s 65℃ 30 s 72℃ 1~2.5 min 72℃10 min将扩增到的PCR 产物,用1%琼脂糖凝胶电泳后,进行胶回收并用Nanodrop 测量核酸浓度。
图1. 三个目的片段凝胶电泳结果34cycles汉恒生物科技(上海)有限公司 400-092-00653. 载体的线性化酶切:Plasmid 2 μg SacI 2 μL XbaI2 μL 10×M bμffer 5 μL DDW 39 μL Total50 μL37℃酶切4h 后,用1%凝胶电泳进行分析,并用Axygen 凝胶回收试剂盒进行胶回收。
目的基因重组表达载体的构建及在原核上的表达实验报告

目的基因重组表达载体的构建及在原核上的表达实验报告目的基因重组表达载体的构建及在原核上的表达实验报告引言•介绍目的基因重组表达载体的意义和作用•阐述本实验的研究目的和重要性材料和方法1.实验所需材料清单2.载体构建方法的详细步骤3.基因重组方法的详细步骤4.原核表达实验的操作细节5.实验数据的收集和处理方法结果与讨论载体构建•描述目的基因的克隆过程和结果•分析目的基因在载体中的定位和正确性基因重组•阐述基因重组的具体过程和结果•讨论基因重组是否成功,并进行分析和验证原核表达实验•详细描述原核表达实验的步骤和操作细节•提供实验数据的结果和分析•讨论目的基因在原核中的表达情况并与预期进行比较结论•总结实验的目的、方法和结果•分析实验结果的意义和影响•提出未来进一步研究的建议致谢•感谢实验室的支持和帮助•感谢实验过程中的合作伙伴的贡献•感谢本研究所使用的仪器和设备的供应商参考文献•引用实验中使用到的相关文献和资料注意:以上内容仅供参考,具体报告的内容可以根据实验设计和实验数据进行调整和补充。
目的基因重组表达载体的构建及在原核上的表达实验报告引言•采用目的基因重组表达载体的方法可以将特定基因导入到细胞中,并在表达载体的指导下进行高效表达,从而实现研究目的。
•本实验旨在构建一种能够在原核中高效表达目的基因的表达载体,并在原核表达系统中进行实验验证。
材料和方法1.实验所需材料清单:•目的基因序列•原核表达载体•适当的限制酶•购买的DNA分析和纯化试剂盒等2.载体构建方法的详细步骤:•参考文献方法进行载体构建•分别提取目的基因和载体的DNA•利用限制酶切将目的基因插入载体中•进行连接酶切和连接反应•进行DNA纯化和质检3.基因重组方法的详细步骤:•将重组产物转化到感受态细胞中•进行细胞筛选和鉴定•提取目的基因重组载体并进行测序验证4.原核表达实验的操作细节:•利用适当的方法将目的基因重组载体导入感受态细胞中•在适当培养基中培养并加入相关诱导剂•收集样本并进行目的基因的表达检测5.实验数据的收集和处理方法:•记录实验过程中的观察结果•进行数据统计和分析•制作图表和图示以展示实验结果结果与讨论载体构建•成功构建出目的基因重组载体,经测序验证其准确性和完整性。
MAGE-A3重组腺病毒载体的构建及表达的开题报告

CRT/MAGE-A3重组腺病毒载体的构建及表达的开
题报告
一、研究背景和意义:
癌症是世界公认的重大疾病之一,在其中恶性黑色素瘤的死亡率也非常高。
通过疫苗调节免疫系统来预防或治疗癌症的方法已经成为重要的研究方向,包括恶性黑色素瘤。
而MAGE-A3和CRT是在恶性黑色素瘤中表达的抗原,且它们的表达在癌细胞和正常细胞中有明显的差异。
因而它们被认为是恶性黑色素瘤的潜在免疫治疗靶点。
二、研究内容和方案
本研究通过将MAGE-A3和CRT基因插入到腺病毒载体中,构建CRT/MAGE-A3重组腺病毒载体,并在细胞水平表达融合蛋白。
具体步骤如下:
1. 通过RT-PCR方法从恶性黑色素瘤组织中提取MAGE-A3和CRT 基因。
2. 将MAGE-A3和CRT基因克隆到腺病毒载体中,形成CRT/MAGE-A3重组载体。
3. 利用PCR进行扩增,通过酶切和测序进行CRT/MAGE-A3重组载体的鉴定。
4. 将CRT/MAGE-A3重组载体转染到恶性黑色素瘤细胞株中,通过Western Blotting方法检测融合蛋白CRT/MAGE-A3的表达和纯度。
三、研究预期结果
1. 成功构建CRT/MAGE-A3重组腺病毒载体。
2. 观察到CRT/MAGE-A3的表达并在细胞中得到验证。
3. 为下一步开展体内实验打下坚实基础,为疫苗调节免疫系统来预防或治疗癌症奠定理论基础。
四、研究意义
本研究有望为恶性黑色素瘤的免疫治疗提供新的方法和思路,也有望开展潜在的抗癌疫苗研究。
同时还有望在癌症免疫治疗的新技术方面提供一定的探索,为癌症的预防和治疗提供新的思路和方法。
DNA重组-载体构建常见问题及解决方法

载体构建中常见问题及解决方法重组质粒构建是常用的分子生物学手段,是研究相关分子及蛋白功能的基础,是生化分子实验中最基本的方法,但是其中还是有些基本的技巧需要掌握。
下面我总结一下自己的经验,以期能为虫友们提供借鉴,希望多少给大家一下借鉴。
所涉及内容如下:1)克隆基因的酶切位点问题2)目的片段的扩增3) 载体酶切的问题4) 连接片段浓度比的问题5)菌液的提取6)酶切鉴定7)表达鉴定一、克隆基因的酶切位点问题1、克隆位点选择的问题。
首先要对目标基因进行酶切位点扫描分析,列出其所含酶切位点清单。
然后对照质粒多克隆位点,所选择的克隆位点必须是目标基因所不含的酶切位点。
双酶切时尽量选择酶切温度相同,buffer一致,双酶切效率高的两个内切酶,二者最好是您实验室常用的酶。
2、保护碱基数目的问题。
在设计PCR引物时,引入酶切位点后,常常要加入保护碱基,这是大家所熟知的。
但是保护碱基数量多少,可能被新手所忽视。
这种忽视可能会大大影响后续的实验进展。
参考常用的酶切位点保护碱基,选择酶切时间短且酶切效率高的碱基数。
注释:1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率3. 加保护碱基时最好选用切割率高时加的相应碱基二目的片段的扩增目的片段扩增失败的原因和可采取措施:1)无条带:引物设计错误,找人帮忙看看引物设计是否合理;退火温度不合适,设置梯度,选择最合适的退火温度;2)条带不单一:引物不特异,适当增长或引物序列长度;3)出现引物二聚体,适当提高退火温度或者重新设计引物。
三载体酶切的问题酶切效率直接影响后续实验的成功率,酶切过程的注意事项:1)酶切质粒浓度和纯度要好2)酶切温度和时间,如果两个酶的最适温度不同,建议单酶切,回收后在用另一个酶切,时间最好过夜切四连接片段浓度比的问题1)连接比例的确定:上一步酶切产物回收后,用琼脂糖凝胶电泳比较载体和目的片段的亮度,确定体积比,一般载体:片段=1:3~1:5;2)连接时间和温度:现在很多连接酶都比较高效了,室温2个小时就可以了,如果后续实验检测阳性克隆较少,可采用16℃过夜连接提高连接成功率;3)未避免后续菌落PCR的假阳性情况,建议连接转化时做对照组:酶切后的载体做自连接对照,如果对照组与实验组长出的克隆数相差不大则很可能存在载体自连或者没完全切开的情况,建议先不要做后续验证,重新从酶切开始,增加酶量及酶切时间、适当减少所切载体的量、改变连接比等;如果对照组没有克隆形成或鲜有克隆形成,则可继续进行后续验证。
载体构建简介

载体构建SOP载体构建(vectorconstruction),为把DNA分子运送到受体细胞中去,必须寻找一种能进入细胞、在装载了外来的DNA片段后仍能照样复制的运载体。
理想的运载体是质粒(plasmid),在基因工程中,常用人工构建的质粒作为载体。
载体构建即是构建含外源DNA的质粒。
载体构建是分子生物学研究常用的手段之一。
主要包括已有载体多克隆位点MCS的改造和已有载体启动子、增强子、筛选标记等功能元件的改造。
载体构建完成后可利用PCR原理进行测序验证。
原核重组表达常用载体构建策略有多种选择,(1)常用的酶切连接是较为广泛使用的克隆技术,主要优点是技术稳定,缺点是周期长、步骤多,任何一个环节产生的误差都会影响克隆构建的失败,如用酶切连接的策略进行载体批量构建,不同载体和不同外源基因尽可能选用相同的上下游酶切位点,需特别留意的是基因内部不能有与上述相同的酶切位点;(2)同源重组是目前流行的克隆技术。
同源重组(Homologous Recombination)是指发生在非姐妹染色单体(sister chromatid)之间或同一染色体上含有同源序列的DNA分子之间或分子之内的重新组合。
该技术可以任意载体任意基因片段快速实现多片段长片段定点定向克隆,最大的优点在于不依赖于酶切位点进行克隆,操作时间也非常短,将目的片段和线性载体按照一定比例混合后,在重组酶的作用下即可发生重组,本实验室常用37℃的温度,30min反应即可完成。
一、目的载体进行双酶切(一)实验准备1.实验材料:质检合格的目的载体。
2.试剂与耗材:buffer,ddH2O,琼脂糖(Spain,9012-36-6)、TBE缓冲液、PCR 管(国产,81245)、1.5ml离心管(国产)、枪头(国产)、Axygen AxyPrep PCR cleanup Kit(Axygen,AP-PCR-250)、限制性内切酶A、限制性内切酶B。
3.仪器与设备:PCR仪(杭州博日,TC-96/G/H(b)A)、mini离心机(其林贝尔,LX600)、电泳槽(北京君意,JY-SPCT)、凝胶成像仪(Bio-rad,GelDoc/ChemiDocuniversal hood II)、分光光度计(天根,OSE-260-01)。
重组腺相关病毒(AAV)载体构建技术原理

重组腺相关病毒(AAV)载体构建技术原理腺相关病毒(adeno-associated virus, AAV)是微⼩病毒科(parvoviridae)家族的成员之⼀,是⼀类⽆法⾃主复制、⽆被膜的⼆⼗⾯体微⼩病毒,其直径约20-26nm,含有4.7kb左右的线状单链DNA作为基因组。
研究中采⽤的重组腺相关病毒载体(recombination adeno-associated virus, rAAV)是在⾮致病的野⽣型AAV基础上改造⽽成的基因载体,由于其种类多样、免疫原性极低、安全性⾼、宿主细胞范围⼴(对分裂细胞和⾮分裂细胞均具有感染能⼒)、扩散能⼒强、体内表达基因时间长等,rAAV被视为最有前途的基因研究和基因治疗载体之⼀。
AAV基因组结构悬浮框:包装纯化及滴度检测--侵染细胞过程-- rAAV载体优势—rAAV⾎清型选择—应⽤案例rAAV包装纯化及滴度检测AAV包装过程中,包装质粒负责编码⽬的基因以及两个末端反向重复序列(ITR,对于病毒的复制和包装具有决定性作⽤),辅助质粒包含AAV包装所需的cap(编码病毒⾐壳蛋⽩)和rep(参与病毒的复制)基因,以及腺病毒Helper质粒。
三种质粒共同转染293T细胞后,AAV病毒开始复制和包装。
和元⽣物腺相关病毒的⽣产和质控采取了国际学术界公认的标准流程,采⽤三质粒系统在293T细胞中进⾏包装。
所获得的病毒颗粒经过超速离⼼纯化,并通过qPCR对病毒基因拷贝进⾏滴定。
另外,我们也可以根据使⽤者要求,提供蛋⽩染⾊检测⾐壳蛋⽩的完整性以及内毒素含量等。
⼀般情况下rAAV的滴度在1012~1013VG/ml,完全可以满⾜各类整体实验的使⽤要求。
AAV包装流程图滴度检测⽅法:定量PCR检测病毒基因组中外源DNA拷贝数。
滴度检测原理:腺相关病毒的基因组为单链DNA,外源DNA拷贝数代表病毒基因组拷贝数。
重组载体的构建

PCR 反应系统的组成 引物、寡核苷酸、Taq DNA聚合酶、模板DNA、缓冲液。
材料与试剂
PCR仪、台式离心机、灭菌的微量离心管、凝胶电泳系统、电泳仪、电泳 槽、紫外透射仪、一次性塑料手套等
TaKaRa Taq (5U/ ul)
10XPCR Buffer (Mg2+ Plus) dNTP Mixture (each 10mM) 模板 (10ng,质粒模板或DNA或cDNA模板) 引物(Primer 1 and 2,10uM) Primer 1: CGCGGGCAGTCTATCTAATCCAGTGGACTCAAG Primer 2: TGCAGCCTAGATCCATGGCATCAATGCAGAAGC pBluescript KS 质粒(3.0kb)
C.纯化步骤
1) 将取出拉杆的注射器筒(3ml)装在纯化柱上,加 入上述DNA与树脂混合液,然后装上拉杆,慢慢将 混合液推进纯化柱内; 2) 卸下注射器筒,拔出拉杆,再将注射器筒装上纯化 柱,加2ml 80%的Isopropanol, 然后装上拉杆,慢 慢推出液体以清洗柱子; 3) 再次卸下注射器筒,将纯化柱装在1.5ml离心管上, 10000g离心2 min,以干燥树脂; 4) 再将纯化柱装在一个新的1.5ml离心管上,加50μl 水或TE缓冲液,1分钟后,10000g离心20秒,溶出 DNA片段; 5) 移去纯化柱,将纯化出的DNA溶液在-20℃下保存备 用。
D.说明
若 向 1ml 树 脂 中 加 入 500bp PCR 产 物 50ng 至
16μg,其回收率可在95%以上。同样500bp,若从
低溶点琼脂糖凝胶中回收其回收率在70~90%之间。
学生PCR实验结果(4l)
重组载体构建的方法和步骤

重组载体构建的方法和步骤全文共四篇示例,供读者参考第一篇示例:重组载体是基因工程领域中的一个非常重要的技术手段,通过对DNA序列的重组,可以构建出具有特定功能的载体,进而实现对目标基因的操控和表达。
在生物学研究、新药开发、农业生产等领域都有着广泛的应用。
下面将介绍一下重组载体构建的方法和步骤。
一、选择载体和目标基因在进行重组载体构建之前,首先要选择适合的载体和目标基因。
一般来说,选择的载体应该具有良好的表达性能和较高的复制稳定性,常用的载体有质粒、病毒等。
而目标基因则应该是我们需要研究或者操控的基因,例如编码重要酶类、蛋白质、抗性蛋白等。
二、获取目标基因目标基因的获取可以通过多种方式,包括PCR扩增、基因合成、基因克隆等。
其中PCR扩增是最为常用的方法,通过设计引物,可以在DNA模板上扩增出目标基因的片段,然后进行纯化和检测,确保其纯度和完整性。
三、线性化载体将选择的载体进行线性化处理,通常是通过限制性内切酶将载体切割成线性片段,以便于后续的连接和转化。
线性化后的载体需要经过纯化和鉴定,确保其完整性和纯度。
四、连接目标基因和载体将目标基因和线性化载体连接起来,一般通过DNA连接酶催化反应实现。
连接时需要考虑目标基因和载体的互补性,以确保连接的正确性和稳定性。
连接后的重组载体需要经过转化操作,将其导入到宿主细胞中。
五、转化宿主细胞将构建好的重组载体导入到宿主细胞中,通常通过化学法、电击法、病毒介导等方式实现。
转化后的宿主细胞需要进行筛选、鉴定和培养,确保其稳定表达目标基因,并能够满足后续实验和研究的需要。
六、鉴定和验证对构建好的重组载体进行鉴定和验证是非常重要的一步,可以通过PCR、限制性酶切割、测序等方法进行验证。
同时也需要进行功能性和表达性的分析,确保重组载体的构建是成功的,能够稳定地表达目标基因。
七、应用和拓展构建好的重组载体可以应用于多个领域,如基因治疗、转基因作物、蛋白质表达等。
同时也可以通过改进和拓展构建方法,提高载体的稳定性和表达效率,开拓更广阔的应用领域。
重组载体构建实验报告

一、实验目的1. 学习重组载体的构建原理和方法;2. 掌握基因克隆、酶切、连接等基本操作;3. 鉴定重组载体的构建成功与否。
二、实验原理重组载体构建是指将目的基因插入到载体中,形成具有特定功能的重组DNA分子。
实验中,通过PCR扩增目的基因,将其与载体进行酶切连接,构建重组载体。
三、实验材料1. 基因组DNA(含有目的基因);2. 载体DNA;3. 限制性内切酶;4. DNA连接酶;5. DNA标记物;6. 转化试剂;7. 载体宿主菌(如大肠杆菌);8. PCR试剂;9. 电泳试剂;10. 实验器材。
四、实验步骤1. 目的基因的扩增:以基因组DNA为模板,设计特异性引物,利用PCR技术扩增目的基因。
2. 载体的酶切:分别对载体DNA和目的基因进行限制性内切酶酶切,获得具有相同黏性末端的DNA片段。
3. DNA连接:将酶切后的载体DNA和目的基因进行连接,形成重组载体。
4. 转化:将连接好的重组载体转化到载体宿主菌中。
5. 重组载体的筛选与鉴定:a. 抗生素筛选:在含有抗生素的培养基上接种转化菌,观察菌落生长情况;b. PCR鉴定:提取转化菌的DNA,进行PCR扩增,检测目的基因是否插入载体;c. 酶切鉴定:对PCR鉴定为阳性的转化菌进行酶切,观察酶切图谱与预期结果是否一致;d. 序列分析:对酶切鉴定为阳性的转化菌进行DNA测序,验证目的基因插入载体的正确性。
五、实验结果与分析1. PCR扩增:成功扩增出目的基因片段,大小与预期一致。
2. 酶切:载体DNA和目的基因分别进行酶切,获得具有相同黏性末端的DNA片段。
3. DNA连接:连接反应成功,形成重组载体。
4. 转化:转化菌在含有抗生素的培养基上生长良好。
5. 重组载体的筛选与鉴定:a. 抗生素筛选:转化菌在含有抗生素的培养基上生长,说明已成功转化;b. PCR鉴定:PCR扩增结果显示,转化菌中含有目的基因;c. 酶切鉴定:酶切图谱与预期结果一致,证明目的基因已插入载体;d. 序列分析:DNA测序结果与预期序列一致,验证目的基因插入载体的正确性。
同源重组构建载体步骤

同源重组构建载体步骤1. 嘿,同源重组构建载体就像搭乐高积木一样,第一步先把那基因“小砖块”找齐喽,这基因就像调皮的小精灵,得一个个揪出来。
2. 然后呢,你得准备好载体这个“大货车”,要把那些基因小精灵装进去呢,这载体就像个超级大货仓,就盼着基因们入住。
3. 接着就像厨师做菜似的,要把基因的两端切得整整齐齐,这切割工具就像是厨师的超级利刀,一下下去精准无比,那基因两端就像被裁剪过的布料边边。
4. 同源臂就像桥梁的两边支架,要稳稳当当的,这可是连接基因和载体的关键呢,要是没搭好,那就像断桥一样,啥都过不去。
5. 把切好的基因和载体放在一起的时候,就像是安排相亲,盼着它们看对眼,然后亲密结合,可别互相排斥啊,不然就像俩吵架的小冤家。
6. 这时候要用酶来催化它们的结合,那酶就像神奇的月老红线,轻轻一牵,基因和载体就开始有结合的趋势了,酶的力量大得像能推动两座大山靠拢。
7. 在合适的反应环境里,就像给它们创造一个温馨的小窝,温度啊、酸碱度啊这些条件都得恰到好处,不然就像把人扔到冰火两重天里,肯定结合不好。
8. 反应的时候就像一场无声的魔法大战,基因和载体在酶的魔法下慢慢融合,那场面就像两个变形金刚合体一样神奇。
9. 中间还得像个监工一样,时不时检查一下它们的结合进度,要是有哪里不对,就像发现建房子的工人把墙砌歪了一样,得赶紧纠正。
10. 同源重组的过程有时候就像挤牙膏,一点一点来,急不得,要是太心急就像用力过猛把牙膏挤爆了,整个反应就乱套了。
11. 那些没有成功结合的基因和载体就像被淘汰的选手,只能在旁边干瞪眼,而成功结合的就像冠军选手,站在舞台中央闪闪发光。
12. 经过一段时间的反应后,就像等庄稼成熟一样,要耐心等待载体构建成功,这个过程就像等待一个宝宝慢慢在肚子里长大。
13. 检测构建是否成功就像寻宝一样,要用各种工具和方法去找寻那成功构建的“宝藏”载体,这些检测方法就像寻宝的探测器。
14. 如果构建失败了,那就像煮熟的鸭子飞了一样,只能重新开始,所有的努力就像泡沫一样消失了,这时候可不能灰心,就当是游戏重来一次。
酵母菌表面展示操作步骤之基因重组和构建

酵母菌表面展示操作步骤之基因重组和构建酵母菌表面展示是一种常用的蛋白质表达和展示技术,可以用于各种研究和应用领域。
其中,基因重组和构建是实施酵母菌表面展示的关键步骤之一。
本文将详细介绍酵母菌表面展示操作中的基因重组和构建步骤。
一、基因重组和构建的原理基因重组和构建是指将目标蛋白质的基因序列插入到酵母菌表面展示载体上,使其能够被酵母菌表面展示。
这一步骤包括以下几个关键的操作过程:1. 寻找合适的表达载体:根据目标蛋白质的特性和需要展示的表面酵素活性,选择合适的表达载体。
常用的载体包括pYD1、pCTCON2等。
2. 提取目标基因:从目标蛋白质的源菌中提取基因序列,通过PCR扩增获得目标基因片段。
3. 消化酵母菌表面展示载体:使用限制性内切酶对表达载体进行切割,以产生适当的限制性内切片段。
4. 连接目标基因和载体:将目标基因片段与切割后的载体通过DNA连接酶进行连接。
连接时需确保目标基因与载体之间的方向一致,以便正确表达。
5. 转化酵母菌:将连接好的重组载体转化到酵母菌中,可使用化学转化或电穿孔法。
二、基因重组和构建的操作步骤下面将具体介绍酵母菌表面展示操作中的基因重组和构建步骤:1. 寻找合适的表达载体根据实验需求选择合适的表达载体。
常用的表达载体包括pYD1、pCTCON2等。
根据需要选择合适的酵母菌,如酿酒酵母(Saccharomyces cerevisiae)或表面展示酵母(Pichia pastoris)。
2. 提取目标基因从目标蛋白质的源菌中提取基因序列,可以通过细菌基因组DNA提取试剂盒等常用试剂盒进行提取。
提取后,使用PCR扩增获得目标基因片段。
3. 消化酵母菌表面展示载体将所选择的表达载体进行消化,使用适当的限制性内切酶切割载体。
消化后的载体含有适当的限制性内切片段,以便与目标基因连接。
4. 连接目标基因和载体将目标基因片段与切割后的载体进行连接,可采用DNA连接试剂盒等常用试剂盒进行连接。
重组克隆载体的构建实验结果分析

重组克隆载体的构建实验结果分析
在此次电泳中,我的 PCR产物并没有很成功地产生。
由于在实
验一中的结果并没有出现问题,因此我分析实验三的电泳结果之所以会失败,应该是在实验的过程中出现了操作失误或是其他不可控因素。
在实验的操作步骤中,主要部分为取一个0.2ml eppendorf 管,添加各种成分反应液。
由于各种反应液要添加的量都比较小,而且添加顺序比较固定,不能出现乱序添加的情况,这些都是可能造成实验失败的因素。
但经过回忆,我在添加各反应液的过程中使用的是设定好吸取容量的移液器,也没有出现容量错误、吸取排出不到位等现象。
由于是排队添加,顺序与周围同学进行添加的顺序也是一致的,因此我分析在添加反应液这一步没有出现失误。
根据实验步骤,在添加反应液过后应该稍作离心,再放入PCR仪中进行 PCR扩增。
我在添加反应液之后,因为想使各种反应液更好
融合,在离心之前将eppen dorf管在旋涡反应器上进行了时长约15秒的涡旋。
我猜想实验结果的失败很有可能与这次涡旋有关。
在涡旋的过程,振荡太过激烈,破坏了原有的基因组 DNA,因此在之后的PCR扩增中没有出现产物。
鉴于这一步骤的实验失败,为了顺利进行接下来的实验,我吸取了老师提供的PCR扩增产物以完成随后的实验。
载体构建技术

载体构建技术一.同源重组敲除的原理主要是应用DNA同源重组原理,用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。
载体构建分为以下步骤:1)载体的选择和引物设计以扩增目的基因;2)目的片段的PCR扩增;3)载体和目的片段的限制性酶切;4)连接转化;5)挑取克隆提质粒;6)单克隆的验证及送样测序。
二.实验材料为形象化说明同源重组敲除技术,以敲除金黄色葡萄球菌中agr为例。
表1实验中所需试剂表2 实验中所用引物三.实验方法3.1 agr基因上臂、下臂及庆大霉素抗性基因PCR扩增参照NCBI数据库中已公布的金黄色葡萄球菌的agr基因序列,分别按照表2中的引物,采用Prime STAR DNA 聚合酶PCR分别扩增出重叠PCR的上下两个片段,50 μL体积反应体系为:上下游引物(100 pmol/μL)各2 μL;模板DNA 0.5 μL;5*Prime buffer 10 μL;dNTP 4 μL;Prime STAR 0.5 μL;ddH2O 31 μL。
PCR扩增条件为: 预变性95℃,5 min;(95℃,30 s,55℃,30s,72℃,1 min),30个循环;72℃,5 min。
冷却至4°C,结束反应,琼脂糖凝胶电泳鉴定PCR产物。
以成功获得的上下两个片段为模板,∆agr-F1,∆agr-R2为上下游引物,利用重叠PCR技术获得∆agr片段,PCR体系及扩增条件类似上述过程(庆大霉素抗三.实验结果3.1 重组质粒pBT2-△agr的构建及验证以金黄色葡萄球菌基因组DNA为模板,利用重叠PCR技术扩增出∆agr基因。
琼脂糖电泳检测扩增的片段大小约为1 kb(图2);片段插入质粒载体pBT2转化大肠杆菌JM109感受态细胞,菌体复苏后浓缩至100 μL涂布于50 µg/mL的氨苄青霉素LB平板培养基上,挑取阳性转化子,重组质粒经酶切电泳鉴定(图3),命名为pBT2-△agr。