原核表达载体的构建
β-synuclein蛋白原核表达载体的构建与表达

的蛋 白的 表 达 。 结果
R —C T P R扩增 出 人 8snc i 因 , 其 亚 克 隆 至 p E -P 1 建 成 重 组 表 达 质 粒 , 在 . uln基 y e 将 G X6. 构 并 成 功 构 建 人 3sn e i 原 核 表 达 载 体 , 在 大 肠 杆 菌 中 表 达 了人 1 - u ln的 y e 并 3 -
中提 取 总 R A , R —C N 用 TP R方 法 获 得 人 1sn c i 因 , 隆 至 p U . 3 yul n基 - e 克 C m T载体 中 ,C P R筛选 阳性 克 隆 并 测 序 。将 酶
切后 的 目的 片段 克隆 至 原 核 表 达载 体 p E -P1中 , 化 大 肠 杆 菌 B 2 。 IT G X6 一 转 L 1 P G诱 导 后 , S SP G 电泳 分 析 目 经 D -A E
E cl .o BL 1 2 .M e b d T tlR a s ltd fo u n ftlb an t s e h u 1 e gh h ma .y u l i DNA to s oa NA w sioae r m h ma e a r i i u .T ef l 1 n t u n B s n cen c s .
【 中图分类号 】 72 R 4
【 文献标识码】A
【 文章编 号】10 4 4 (00 0 -45 3 0 5 8 7 2 1 )60 9 - 0
D i1 . 9 9 jis . 0 5~4 4 . 0 0 0 . 1 o :0 3 6 / .sn 1 0 8 72 1 .60 0
C ntu t no rk r oi E p es nVetro l y u li n o sr ci f o ay t x rsi co flS n c n a d o P c o j - e
新型rPA(K)原核表达载体的构建及初步表达

1 1 1 菌 株及质 粒 ..
含有 r A( 基 因的质粒 载体 p r K) P K) E A( 由笔 者所在 实验室李 敏等人 构建 ; 表达 载体 p T一 0 ( , 主 E 3 a +) 宿
菌大肠 杆菌 B 2 ( E ) L 1 D 3 均为笔 者所在 实验室保 存 .
1 12 主 要 试 剂 ..
于以上原因 , 同时为了降低研究成本 , 方便 rA( ) P K 的表达 、 测和纯化 等后续工作 , 检 笔者实验 利用 P R技术将 C
rA K) D A克隆至原核表达载体 p T 3a +) , 了 rA K 的新型原核表达载体 . P ( cN E 一0 ( 中 构建 P ()
1 材 料 与 方 法 11 材 . 料
定 ( L s 试剂 盒为上 海太 阳生 物制 品公 司产 品. E IA)
1 2 主 要 方 法 .
12 1 rA( c NA片段 的获 得 . . P K) D 12 1 1 引物设 计 . . .
根据 rA( 的 c N P K) D A序列【 3J 0 及引物设计原则设计 了一对 引物 , 于亚克隆 , 引物序 列 中加入 B l , 为便 在 g Ⅱ
是 以融合蛋 白的形式进行表达 的 , 即在表达产物之前存在非 目的片段 , 得临床使 用时将会 引起机体 的过敏反 使 应; 而且 如果对Байду номын сангаас其进行纯化 , 则必须使用 价格 昂贵的 L s ehms 或 H sBn 层 析柱 , 而提 高 了研究 成本 . y- p a e s i・id 从 鉴
限制性 内切酶 X o , t , h No I I
I, p , g , L 5 0 re, o —ag rt nMakr H K nI B l ID 10 0MakrL w R n eP e re 为宝 泰 克公 I o i
大肠杆菌的原核表达实验过程结果

大肠杆菌的原核表达实验过程结果
大肠杆菌的原核表达实验一般分为以下几个步骤:
1. 构建表达载体:将待表达的基因克隆到适当的表达载体上,如常用的pET、pGEX等载体中。
2. 转化大肠杆菌:将表达载体转化到大肠杆菌细胞中。
可以通过化学方法、电转化、冷冻复苏、热激转化等方法进行。
3. 诱导表达:在大肠杆菌细胞进行生长至适当时期后,添加适宜浓度的诱导剂,如IPTG等,诱导待表达基因蛋白的合成。
4. 细胞收获和破碎:诱导一定时间后,收获大肠杆菌细胞并进行破碎,以获得待表达蛋白。
5. 蛋白提取:对细胞破碎物进行离心、超滤等步骤,去除残余细胞构成和杂质,得到含有待表达蛋白的上清液。
6. 纯化和分析:将上清液进行分离、纯化、鉴定,以确定表达蛋白相应的分子量、酶活性等。
在以上步骤中,实验者需要进行质控和评估,确认实验步骤是否正确和表达蛋白是否达到预期,以确保实验结果的可靠性和准确性。
小鼠TIM-3原核表达载体的建立及多克隆抗体的制备与鉴定

MAD — n H O Y u u eQ g, A o- a, H
a 3 0 0. h n n 4 0 3 C ia
Mi , U C u —i G O Yh, H N Ye £ egf ,A GD n-i g. is n n X h nL , U a C E u , M n - Y N ogL n Dv i i a io
.
5 0 bp e t c lua 1 xr e l rDNA q e c s o tie rm e s le el fBA a e s u n e wa ban d fo t pe n c l o LB/c mie b h s c y RT- CR , n he p o ay t x rs in e tr P a d t rk ro i e p e so v co c
( 中科技大 学 同济 医学 院附属 同济 医院临床免 疫研 究室 , 汉 403) 华 武 300
中 国 图书 分 类 号 R 9 .3 323 文 献标 识 码 A 文 章编 号 10-8X(0 10.180 0044 2 1 )205-5
[ 摘
要 ] 目的: 克隆小 鼠 TM 3 I . 基因 , 构建原核表达载体 , 制备相应 的多克隆抗体并初 步鉴定 。方法 : 以小 鼠脾 细胞总
・18 5 Fra bibliotek中 国免疫 学杂 志 21 年 第 2 01 7卷
di1.99jin 10—8X.0 10 .1 o:036/. s.0044 2 1 . 04 s 2
小 鼠 T M. 核 表 达 载 体 的 建 立 及 多 克 隆 抗 体 的 制 备 与 鉴 定 ① I 3原
马德强 郝友华② 刘 敏 徐 春利 郭 艳 陈 悦③ 陆孟 吉④ 杨 东亮
丙型肝炎病毒F蛋白原核表达载体的构建、表达和纯化

一
35 一 8
基础研究
文 号: 5 3 6 0 ) —3 — 章编 0 3 6 ( 0 0 0 50 2 —2 294 8 4
表达 , 出现 了与 预 期 分 子量 相 符 的蛋 白条 带 , 蛋 白通 过 Wet nbo检 测 具 有 6 itg 白 的免 疫 学 活 性 。结 论 : 功 构建 该 s r lt e H sa 蛋 X 成
了丙型肝炎病毒 F蛋白的原核表达载体 p T 2 ( ) H V E 3 a+ 一 C F并表达和纯化出重组融合蛋 白, 为进一步研究 F蛋 白的生物学 功能
C og i h n qn gMe ia U ie i ) dc nvr t l sy
【 bt c】 be t e T nt capoayt xrsi ls i otnn C n ,n ui e eo i n s n rtni A s at O jci : oc s ut rkro c pes npam dcna ig VFg eadp ryh cmbn tui o i n r v o r ie o i H e ft r a f o p e
丙型肝 炎病毒 F蛋 白原核 表达载体 的构建 、 表达和纯化
高亚 丽 , 娇 , 刘 刘 湘 , 汤 华 , 陈 沂 , 雪 飞 , 蔡 唐 霓
( 重庆医科大学感染性疾病分子生物学教育部重点实验室 重庆 医科大学病毒性肝炎研究所 , 庆 重 40 1) 0 06
【 摘
要】 目的 : 构建丙型肝炎病毒( ptiCv u , C F蛋 白的原核表达载体 , Hea t i s H V) is r 表达 p T 2 ( )HC F融合蛋 白。方法 : E 3a + 一 V 根
原核表达载体pET28a-EGFP的构建与表达

利 用 热 击 法把 得 到 的 重组 质 粒 p T 8 —G P转 化 至 E clB 2 ( shr haclB 2 ) 受 态 细 胞 中, 大 肠 E 2 aE F .o L 1 Ecec i o L 1 感 i i i 当
埃 希 茵 L ( ui・et i培养 液 在 6 0n 下 的光 密 度值 D 6 =0 4时 , 过 添 加 异 丙基 硫 代 BD 半 乳 糖 苷 B L r B r n) a a 0 m D0 . 0 通 —一 (P G 作 为 诱 导 剂诱 导 E F IT ) G P表 达 。结 果 表 明 : 重组 质 粒 酶 切 鉴 定 及 测 序 结 果 正 确 。 在 自然 光 下 , 化 子 在 转
重组表达质粒的构建——原核表达载体选择
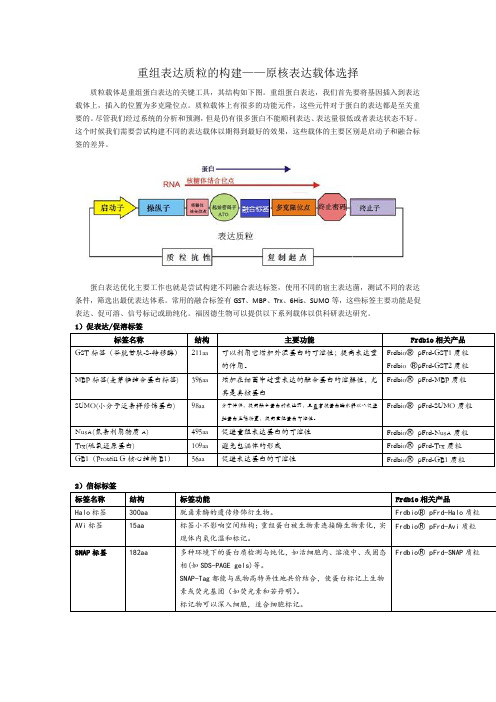
重组表达质粒的构建——原核表达载体选择质粒载体是重组蛋白表达的关键工具,其结构如下图。
重组蛋白表达,我们首先要将基因插入到表达载体上,插入的位置为多克隆位点。
质粒载体上有很多的功能元件,这些元件对于蛋白的表达都是至关重要的。
尽管我们经过系统的分析和预测,但是仍有很多蛋白不能顺利表达、表达量很低或者表达状态不好。
这个时候我们需要尝试构建不同的表达载体以期得到最好的效果,这些载体的主要区别是启动子和融合标签的差异。
蛋白表达优化主要工作也就是尝试构建不同融合表达标签,使用不同的宿主表达菌,测试不同的表达条件,筛选出最优表达体系。
常用的融合标签有GST、MBP、Trx、6His、SUMO等,这些标签主要功能是促表达、促可溶、信号标记或助纯化。
福因德生物可以提供以下系列载体以供科研表达研究。
1)促表达/促溶标签2)信标标签3)纯化标签我们选择表达载体的时候不但要考虑蛋白怎么表达成功,更要考虑蛋白怎么纯化出来,纯化的问题主要是考虑纯化标签和酶切位点的选择,下表我们列举了常见的纯化标签和酶切位点。
4)酶切位点以上为原核表达常用的标签和酶切位点,其性质也都作了简要的介绍,各专业网站或专业书籍已对此做详尽解释,科研工作者可根据具体实验设计方案,组合设计以上标签和酶切位点的使用。
特别值得注意的是,选用和设计蛋白酶切位点的时候首要考虑的是序列内部有没有蛋白酶位点,同时要考虑酶切的效率和蛋白酶试剂成本。
一般商业化载体,在标签蛋白与载体多克隆位点之间都设计有酶切位点。
标签可设计在N-端也可在C-端,设计在N-端的优势是,可通过标签高效翻译起始位点带动插入蛋白的表达,可溶性标签的高效表达更可促进蛋白的可溶性表达;同时,大部分的蛋白内切酶的切割位点在C-端,所以标签设计在N-端可将标签切割完全。
在设计标签序列与酶切位点的时候还要考虑N-端稳定性原则,也就是所谓宿主细胞的N-端规则(N-end rule),这个要避免;同时,还应该检查是否引入了可与别的蛋白相互作用的序列或者蛋白酶切位点。
pGEX-4T-3- pMD19-T-S11原核表达载体的构建、表达与纯化及多克隆抗体的制备

pGEX-4T-3- pMD19-T-S11原核表达载体的构建、表达与纯化及多克隆抗体的制备水生呼肠孤病毒是水域生态环境中广泛存在的一类病毒,其中草鱼呼肠孤病毒(Grass carp reovirus, GCRV)是在1991年被国际病毒分类委员会(ICTV)分到水生呼肠孤病毒属,是由中国分离鉴定的第一株鱼类病毒[1]。
GCRV是具有双层衣壳、无囊膜、立体对称的20面体球形颗粒,主要由蛋白质和核酸组成,还含有少量以糖蛋白的形式存在的糖类,不含脂类[2]。
草鱼自古以来就是中国池塘养殖的当家品种,而GCRV被认为是水生病毒中对草鱼毒力最强、致病死亡率最高的病毒之一,该病毒引起的出血病不仅直接危害鱼体,而且会激发细菌性“三病”的发生,使草鱼的成活率降低到40~50[3],甚至只有10%左右[4],给中国的草鱼养殖业造成了很大的危害和困扰。
草鱼病毒性出血病,主要侵染于草鱼心脏、肝脏、肾脏、肌肉、脾、鳃、肠道等组织,且化学药物很难达到治疗效果,为了给草鱼养殖增产和减小不必要的经济损失,找到有效防治GCRV 的方法显得尤为重要。
GCRV基因组含有11条双链RNA(dsRNA),其分段基因组RNA的3' 端不含poly(A)尾巴, 在5’和3’末端各含有特异的重复保守序列,为5’-GUUAUU和3’-UCAUC。
11个片段根据凝胶电泳迁移率可分为3组,即大片段(L1,L2,L3)、中等片断(M4,M5,M6)、较小片段(S7,S8,S9,S10,S11)。
GCRV共编码5 种非结构蛋白, 分别是NS16、NS26、NS31、NS38、NS80[5]。
病毒的非结构蛋白在病毒的复制过程中发挥重要作用。
其中NS26是由S11编码的,含有244个氨基酸残基,分子量为26.4KD, 是水生呼肠孤病毒特有的蛋白质, 没有同源蛋白,含ATP-依赖性的RNA解螺旋酶功能性位点[6]。
国际上对GCRV编码蛋白的功能(尤其是非结构蛋白的功能)方面的研究还不够系统和全面;但根据呼肠孤病毒家族中同源蛋白功能相似的原则,除非结构蛋白NS26外,GCRV各蛋白在病毒复制周期中的地位和作用已经初步明确[7]。
实验十五、、表达载体的构建

选择表达载体应考虑的因素
(4).所产生的融合蛋白是否需要切割加工以及如何切割 加工。对需要切割的融合蛋白,在目的基因和载体 序列间应有适当的切割识别序列。 (5).是否需要用强启动子提高表达水平。当需要提高表 达水平时,可选用强启动子。但在某些情况下,由 于被表达蛋白质对宿主有毒或其大量表达对宿主不 利,可选用诱导型载体,只有经诱导后,蛋白质才 能被表达。
实验十二、表达载体的构建及鉴定 实验十二、
基因工程中使用的表达系统类型
一、基因来源: 基因来源: 原核生物 真核生物 二、原核表达系统 原核表达系统: 1、原核表达系统: 遗传背景清楚,易改造,培养容易,费用 低,蛋白质表达水平高。但在原核中表达后所合 成的蛋白无自我修饰如糖基化等,或因折叠的方 式不正确或折叠效率的低下,最后产生的蛋白质 其生物活性很低。 2、真核表达系统
原核表达系统类型
大肠杆菌表达系统 大肠杆菌
遗传背景很清楚,基因操作的方法非常完善,很容易大 规模培养产生大量的目的蛋白质。 但大肠杆菌表达的蛋白质大多不分泌到细胞体外,如 要使蛋白分泌到细胞外,必需使目的基因带有信号肽。信 号肽(signal peptide)又称前导肽(1eader peptide),这是一种 位于分泌蛋白质或输出蛋白质N端上长约15~30个氨基酸的 序列,可被细胞的蛋白质加工机制所识别,使蛋白质通过 细胞膜被分泌出来。
原核表达载体 克隆载体: 克隆载体
通常都带一个松复制子、一个多克隆位点和一个筛选标 记,以便被克隆和筛选的DNA序列能够大量增殖。
表达载体: 表达载体:
除了上述因子外,还需要有强启动子、核糖体结合位 点(核糖体结合位点,又称SD序列,它是指mRNA分子中核糖 体结合的序列,通常位于翻译起始密码子前面3~12个碱基, 该序列与16S rRNA的互补)、转录起始信号、转录信号、翻 译起始密码子(ATG)和终止密码子等一系列调控序列。
原核表达载体构建步骤

原核表达载体构建步骤构建原核表达载体就像是在微观世界里盖房子,而且是给那些超级小的生物分子住的。
咱得先找好“建筑材料”,也就是目的基因啦。
这目的基因就像是一颗特别的种子,我们要把它种到原核生物这个小花园里。
找目的基因有时候就像大海捞针,在基因的海洋里翻来翻去,直到找到那一颗闪闪发光的“金种子”。
然后呢,要有合适的“地盘”,这就是原核表达载体。
这个载体就像是一块神奇的土地,不过它可不是随随便便的土块,而是经过精心设计的,上面有各种功能区域,就像土地上划分好了不同的功能区一样,有启动子区,这就像是房子的大门开关,决定着基因能不能开始工作,还有终止子区,那是工作结束的信号,就像下班的铃声。
接下来要把目的基因和载体连接起来,这过程就像是用超级胶水把种子粘到土地上。
这个胶水就是连接酶啦,它特别神奇,能准确地把基因和载体紧紧地连在一起,就像把两根细细的线完美地缝起来一样。
在这之前,还得对载体和目的基因进行处理,就像给土地松松土,给种子去去壳。
我们要用限制酶在载体和目的基因上切出合适的口子,这限制酶就像一把超级小但无比锋利的剪刀,精确地剪出想要的形状。
构建好之后,还得检查一下有没有问题呀。
这就像是房子盖好了要验收一样。
要看看基因有没有正确地连接,就像检查房子的结构有没有稳固。
有时候如果出了差错,那就像盖歪了房子,一切就得重新来啦。
把构建好的原核表达载体送进原核细胞这个小家园,就像把种好种子的花盆放到温室里。
原核细胞会像勤劳的小园丁一样照顾这个载体,让目的基因开始表达,生产出我们想要的蛋白质。
这蛋白质就像是花朵或者果实,是我们精心构建载体的最终收获。
整个过程充满了各种惊喜和挑战,有时候一个小失误就像在蛋糕里放错了盐,整个味道就全变了。
但当一切顺利的时候,就像是在微观世界里创造了一个小奇迹,那些小小的分子按照我们的意愿开始工作,感觉自己就像一个微观世界的大魔法师呢。
原核表达载体构建虽然复杂又繁琐,但就像一场充满乐趣的微观冒险,每一步都充满了未知和期待。
原核表达原理

原核表达原理及应用一、目的基因获取目的基因的获取是原核表达的第一步,通常通过基因克隆技术从基因文库或PCR扩增中获得。
克隆技术包括质粒载体克隆、噬菌体载体克隆、染色体重组克隆等。
PCR技术则包括常规PCR、巢式PCR、反转录PCR等。
二、载体构建载体构建是将目的基因连接到原核表达载体上的过程,该载体含有原核生物可以识别的启动子、多克隆位点以及筛选标记等必要元件。
构建载体时,需要确保目的基因插入后的正确阅读框架和终止子。
常用的原核表达载体有pET系列、pGEX系列、pBAD等。
三、转化转化是将目的基因导入到受体细胞的过程,受体细胞通常是原核生物。
常用的转化方法有电击转化法、热激转化法、化学转化法等。
转化成功后,目的基因会整合到受体细胞的染色体上或质粒上,进而实现基因的表达。
四、诱导表达诱导表达是目的基因在受体细胞内表达的过程,通常在特定的诱导条件下进行。
常用的诱导方法有温度诱导、IPTG 诱导、金属离子诱导等。
诱导表达后,目的基因在受体细胞内转录和翻译,生成相应的蛋白质。
五、表达产物检测表达产物检测是通过特定的方法检测目的基因表达产物的过程。
常用的检测方法有SDS-PAGE电泳、Western blot、ELISA等。
这些方法可以用来检测表达产物的分子量、含量以及纯度等信息,为后续的优化和纯化提供依据。
六、表达优化表达优化是提高目的基因在受体细胞内表达水平的过程,通过调整诱导条件、宿主菌种类和培养基成分等方法实现。
优化后的目的基因表达水平更高,有利于提高产物的产量和纯度。
七、产物纯化产物纯化是从表达产物中分离和纯化目标蛋白质的过程。
常用的纯化方法有离心、过滤、沉淀、离子交换层析、凝胶过滤层析等。
这些方法可以根据目标蛋白质的理化性质和分子量等进行分离和纯化,获得高纯度的蛋白质。
八、应用研究原核表达技术在应用研究方面具有广泛的应用价值。
它可以用于蛋白质结构与功能的研究,通过同源建模和晶体结构解析等方法研究蛋白质的结构和功能;可以用于蛋白质药物的开发,如抗体药物、细胞因子药物等;可以用于疫苗的研制,如细菌性疫苗和病毒性疫苗等;可以用于酶工程领域,如固定化酶、酶抑制剂等;可以用于基因工程领域,如基因诊断和基因治疗等。
Cys C的原核表达载体构建和鉴定

a pie i T P R h D A f g e tw s c n d i o p T 2 ( ) v c r w ih W o f m d w t sq e c g a d m l d w t R - C .T e e N r m n a l e n E 3 a + i f h a o t et , hc a c n r e i e u n i n o s i h n e c o h rs .R s / T e rsl o l t p oei so e h t C s C h d b e l e i o p T 2 ( ) vc r a d t l t p o i e u t h eut f e cr h rs h w d ta y a en c n d n E 3 a + er es s: e o s o t et ,n e o h
d t.C ncu in: e po ayt x rsin v co fc s t C( y a a o ls o T rk roi e pe s e tro y t i h c o a n C s C)Wa b ie po iig a b ss o u ic t n o s s o t n d,rvdn ai fp r ai fCy a i f o
WANG 帆 , t l e a
( e K y L b r oy o a oa r dclDi nsi , ns y o d c in te F c l a oao e a oa r f L r oy Me ia a ot sMiir f E u a o , aut o L r r t b t g c t t h yf b t y
【 关键词 】 胱抑素 c; 原核表达系统 பைடு நூலகம் 中国图书分类法分 类号 】 3 016 R 2 、10 【 文献标识码 】 A 【 收稿时间 】07 0 — 3 2 0 — 9 1
pet28a原核表达载体的表达原理

pet28a原核表达载体的表达原理Pet28a原核表达载体是一种常用于原核生物中蛋白质表达的载体。
它的表达原理基于其构建的特点和作用机制。
Pet28a原核表达载体的构建特点是将多个功能元件组合在一起,以实现高效的蛋白质表达。
主要包括启动子、编码序列、标签序列和终止子等。
Pet28a的启动子是一段能够识别并与细菌RNA聚合酶结合的序列,用来启动基因的转录。
细菌RNA聚合酶在启动子的识别下,能够将DNA的信息转录成mRNA,作为蛋白质合成的模板。
编码序列是Pet28a中最重要的部分,它包含了目标蛋白质的编码信息。
编码序列是由一串三个核苷酸组成的密码子序列,每个密码子对应一个氨基酸。
在细胞内,mRNA会被核糖体识别并通过翻译作用将其转化为氨基酸序列,从而合成出目标蛋白质。
Pet28a载体中还包含了标签序列,用于方便对目标蛋白质进行检测和纯化。
常用的标签序列有His标签和GST标签。
His标签是一段连续的组氨酸序列,能够与金属离子亲和层析柱结合,实现目标蛋白质的纯化。
GST标签是谷胱甘肽S-转移酶标签,能够与谷胱甘肽结合,实现目标蛋白质的纯化。
Pet28a载体的终止子是一段能够识别并终止转录的序列。
在RNA聚合酶到达终止子时,转录过程会终止,mRNA链被释放出来,进一步被核糖体翻译为蛋白质。
总结起来,Pet28a原核表达载体的表达原理是通过启动子识别和RNA聚合酶的转录作用,将编码序列转录为mRNA。
然后,mRNA通过核糖体的翻译作用,合成目标蛋白质。
最后,通过标签序列的存在,可以对目标蛋白质进行检测和纯化。
Pet28a原核表达载体的表达原理使其成为研究人员在原核生物中高效表达蛋白质的重要工具。
它的构建特点和作用机制为研究人员提供了方便、快捷和可靠的蛋白质表达平台。
通过对Pet28a载体的合理设计和选择,可以实现目标蛋白质的高效表达,为生物学研究和工业生产提供了有力支持。
人TLR1胞外段原核表达载体的构建、蛋白表达及纯化

di1 .99jin 10—8X.00 1.1 o:0 36/ .s .0044 2 1 .20 1 s
・
免 疫 学技 术 与方 法 ・
人 T R1胞 外 段原 核 表达 载体 的构 建 、 白表达 及 纯化① L 蛋
年 四季 李 祥② 黄 黎 曹 新梅 周 云 刚 袁
o u iid p oen wi — f nt ou sa o t6 D y S - AGE . e W e tr lta d EH S rs lss o d t tt x ese r— fp rfe rti t Niaf i c l mn wa b u 8 k b DS P h i y h T se n bo n A e u t h we ha hee pr s d po
T R1 w ss c e s l o srce hc rv d b C a l c t na d s q e cn . e s eo mpi e e e sa o t 0 p a dt a L a u c sf l c n t td w ih po e y P R mpi ai n e u n i g T i fa l d g n s uy u i f o h z i f wa b u 7 0 b t 1 n h
pe e . o L 1 T eepesdp t n a p r e di n f db s r l . eut: er o bnn l m dp T 0( ) r sd i E cl B 2 . x rs r e s u f da e t e yWet b t s l T cm iat a i E 3 a +/ s n i h e o iw i n d i i i e n oR sh e p s
Abd-Bcr/Abl原核表达载体的构建和表达

a lc b DNA NM
一
通信作者简介 : 梁英民 , 第四军 医大学 , 唐都 医院 血液科 陕西 西
安 7 0 3 : - i:a gm@ f u eu c o 10 8 Emalln y i mm .d .n
维普资讯
第 8卷
第 9期
20 0 8年 5月
科
学
技
术
与
工
程
⑥
Vo . N . Ma 0 8 18 o9 y2 0
17 —89 20 ) -3 10 6 11 1 (0 8 92 3 —4
S e c c noo y a d Engn e i cin e Te h lg n i e rng
研究 表 明 , 蛋 白 对 白 血 病 的 生 成 具 有 促 进 作 该
用 , 国外 大 量研 究 表 明 A d在 B rA l T 介 导 b c/ b P K
的 白血 病 生 成 中起 重 要 作 用 j c/ b P K介 ,B rA l T
导 的 白 血 病 生 成 活 性 依 赖 于 碳 末 端 的 Ad 存 在 , b
占菌体 总蛋 白的 6 , % 纯化后达到 7 .% ,Wet nb t 测为特异性表达 , 43 s r l 检 e o 为进 一步研究该 蛋 白对慢 性 白血病 细胞 的作用 奠
定 了分 子 基 础 。
关键词
慢性髓 细胞 性 白血 病
B rA l 因 c b基 /
肌 动蛋 白结合域 ( b ) A d
载体 p T2 ,构 建 p T2 —b 重 组 表 达 质 粒 , 酶 切 、 序 鉴 定 , 列 、 码 框 均 正 确 。 通 过 转 化 E cl B21( E ) 经 E 3a E 3 aA d 经 测 序 读 oi I D3 ,
原核生物和真核生物的载体构建

本实验用内切酶将T-GFP质粒的GFP cDNA切割下来,与用同样内切酶消化的pcDNA3.1连接,构建真核表达载体pcDNA3.1-GFP。
ቤተ መጻሕፍቲ ባይዱ
【试剂】
1.菌种含绿色荧光蛋白重组子T-GFP质粒
2.LB液体培养基称取胰蛋白胨3.0g,酵母提取物1.5g,NaCl3.0g,加双蒸水200ml溶解,用5mol/LNaOH调至pH7.4,定容至300ml,转移至三角烧瓶中,以103.4Kpa高压灭菌20min。
【原理】
引物中设计入限制酶位点:由于PCR引物的5'末端可以增加一些非互补碱基,因此可以在两引物的5'末端设计单限制酶或双限制酶切位点。这样得到的PCR产物用限制酶消化产生粘性末端,即可与有互补粘端的载体DNA重组。这种克隆方法效率较高,且当两引物中设计不同酶切位点时,可有效地定向克隆PCR产物。其缺点是需要加长PCR引物,除限制酶识别序列外,还需要在其5'端多合成2—3个碱基以利于限制性内切酶与PCR产物末端的稳定结合。
15.DNA分子量标准根据需要购买,一般浓度为0.5μg/μl。
16.其他试剂氯仿、异丙醇、无水乙醇。
【操作步骤】
1.将下列试剂缓冲液2μl,T-GFP质粒或pcDNA3.1质粒(1μg),KpnI和ApaI各5~10U加至0.5mlEP管中,加双蒸水至20μl,加盖,混匀后稍离心,37℃水浴反应1h。
9.10mg/mlRNaseA称取RNaseA10mg溶于10mmol/LTris-HCl(pH7.5)、15mmol/LNaCl中,于100℃加热煮沸15min灭活DNase,缓慢冷却至室温,分装成小份保存于-20℃。如为Sigma公司产品,一般则不需加热煮沸。
MAGE_3目的基因原核表达载体的构建和表达
-----------------------------------Docin Choose -----------------------------------豆 丁 推 荐↓精 品 文 档The Best Literature----------------------------------The Best Literature第16卷第21期中国现代医学杂志Vol.16No.212006年11月ChinaJournalofModernMedicineNov.2006收稿日期:2006-03-31*基金项目:河南职工医学院重点科研项目2004-2肿瘤特异性免疫治疗由于在早期诊断、预防、控制微小残瘤、预防术后复发以及延长患者生存期等方面较传统疗法相比具有其独特优势而逐渐成为研究的热点。
该疗法的前提是如何发现和利用肿瘤抗文章编号:1005-8982(2006)21-3264-04·论著·MAGE-3目的基因原核表达载体的构建和表达*裴瑞1,陈洁1,杨红梅1,陈晓玲1,杨萍1,赵国强2(1.河南职工医学院,河南郑州451191;2.郑州大学医学院微免教研室,河南郑州450052)摘要:目的构建原核表达载体pGEX-4T-1-MAGE-3并检测其在大肠杆菌(E.coli)BL21中的表达,为制备以黑色素瘤抗原-3(MAGE-3)基因片段为基础的肽疫苗及特异诊断试剂提供抗原打下基础。
方法通过逆转录-聚合酶链反应(RT-PCR)法制备MAGE-3目的基因,以pGEM-TEasy为克隆载体,pGEX-4T-1为原核表达载体,将MAGE-3目的基因克隆至pGEM-TEasy载体和亚克隆至pGEX-4T-1载体上。
筛选、鉴定阳性克隆并将其转化E.coliBL21.经IPTG诱导,12%SDS-PAGE电泳分离和WesternBlot表达鉴定。
结果正确构建了原核重组表达质粒pGEX-4T-1-MAGE-3;在E.coliBL21中检测到含该重组表达质粒的转化菌表达出分子量35kD的融合蛋白;基因测序结果表明,阳性克隆中的MAGE-3目的基因序列与GenBank公布的已知序列完全一致。
表达载体的构建方法及步骤

表达载体的构建方法及步骤一、载体的选择及如何阅读质粒图谱目前,载体主要有病毒和非病毒两大类,其中质粒 DNA 是一种新的非病毒转基因载体。
一个合格质粒的组成要素:(1)复制起始位点 Ori 即控制复制起始的位点。
原核生物 DNA 分子中只有一个复制起始点。
而真核生物 DNA 分子有多个复制起始位点。
(2)抗生素抗性基因可以便于加以检测,如 Amp+ ,Kan+(3)多克隆位点 MCS 克隆携带外源基因片段(4) P/E 启动子/增强子(5)Terms 终止信号(6)加 poly(A)信号可以起到稳定 mRNA 作用选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。
如果构建的目的是要表达一个特定的基因,则要选择合适的表达载体。
载体选择主要考虑下述3点:【1】构建 DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。
【2】.载体的类型:(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。
如<10kb 选质粒。
(2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。
【3】载体 MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。
综上所述,选用质粒(最常用)做载体的5点要求:(1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载体);(2)一般使用松弛型质粒在细菌里扩增不受约束,一般 10个以上的拷贝,而严谨型质粒<10个。
(3)必需具备一个以上的酶切位点,有选择的余地;(4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位 Ampr(试一试)。
(5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。
缬氨酸转氨酶基因原核表达载体构建及表达

t i me 8 h .
Ke y wo r d s : v Mi n e - p y uv r a t e t r a n s a mi n a s e ; E s c h e r i c h i a c o l i ; g e n e c l o n i n g ; p r o t e i n e x p r e s s i o n
中 图分类 号 : Q 9 3 9 . 9 7 文献 标 志码 : A D O I : 1 0 . 3 9 6 9 / j . i s s n . 2 0 9 5— 4 7 6 X. 2 0 1 3 . 0 1 . 0 0 2
Co ns t r u c t i o n a nd e x pr e s s i o n o f pr o k a r y o t i c v e c t o r o f v a l i n e ・ py r u v a t e t r a ns a mi na s e g e ne
e x p r e s s i o n w a s d i s p l a y e d . T h e o p t i ma l c o n d i t i o n s w e r e p e p t o n e 1 2 g / L , I P T G 0 . 4 mm o l f L a n d i n d u c e d
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
二 实验步骤
1、将鉴定为阳性的重组质粒pGEMT-NP、pGEMT-P、 pGEMT-M、pGEMT-F和pGEMT-HN与原核表达载体pET32a分别用相应的限制性内切酶进行双酶切。以 pGEMT-NP为例,双酶切体系(30μL体系)如下:
ddH2O 10×buffer H pGEMT-NP EcoRI Sal I 21 µL 3 µL 4 µL 1 µL 1 µL ddH2O 10×buffer H pET-32a EcoRI Sal I 22 µL 3 µL 3 µL 1 µL 1 µL
第四组
ddH2O 10×buffer H pGEMT-F EcoRI Sal I 21 µL 3 µL 4 µL 1 µL 1 µL ddH2O 10×buffer H pET-32a EcoRI Sal I 22 µL 3 µL 3 µL 1 µL 1 µL
第五组
ddH2O 10×buffer H pGEMT-HN Sal I Xhol I 21 µL 3 µL 4 µL 1 µL 1 µL ddH2O 10×buffer H pGEMT-HN Sal I Xhol I 22 µL 3 µL 3 µL 1 µL 1 µL
新城疫病毒NP、P、M、F、HN 原核表达载体的构建
一 实验所需材料
原核表达载体pET-32a(+);
Ecoli DH5α菌株;
限制性内切酶EcoRⅠ、SalⅠ、Hind Ⅲ、XhoⅠ; LB液体培养基(未加Amp+和加Amp+); LB固体培养基(含Amp+); 0.1mol/L CaCl2溶液; 凝胶回收试剂盒。
2、双酶切反应条件:37 ℃ 3 h。进行1%琼脂糖凝 胶电泳鉴定,然后在紫外灯下切取目的条带,用凝 胶回收试剂盒回收目的片段和原核表达载体片段。 3、目的基因片段与原核表达载体pET-32a的连接, 连接体系为10μL:
ddH2O 10×T4 Buffer 目的基因片段 载体片段 T4 DNA Ligase 2 µL 1 µL 5 µL 1 µL 1 µL 4℃连接过夜
NP基因两端带有的酶切位点为EcoRI和SalI,通用buffer为10×buffer H P基因两端带有的酶切位点为EcoRI和HindⅢ,通用buffer为10×buffer M M基因两端带有的酶切位点为EcoRI和SalI,通用buffer为10×buffer H F基因两端带有的酶切位点为EcoRI和SalI,通用buffer为10×buffer H HN基因两端带有的酶切位点为SalI和XhoI,通用buffer为10×buffer H
ddH2O 10×buffer O pET-32a-NP EcoRI Sal I 15.5 µL 2 µL 1.5 µL 0.5 µL 0.5 µL 37℃酶切3h
8、酶切产物经1%琼脂糖凝胶电泳鉴定,将阳性质 粒送去测序,序列分析正确的重组原核表达载体因两端酶切位点及双酶切通用Buffer
4、制备大肠杆菌DH5α感受态细胞,冰上放置4h以 上备用。 5、连接产物转化DH5α感受态细胞,将转化的细菌 涂于Amp+ LB平板,37℃倒置培养过夜待菌落长出。 6、从Amp+ LB平板上随机挑取生长的单菌落,于3mL LB液体培养基(含Amp+)中培养12-16h。
7、碱裂解法提取质粒,然后用相应的限制性内切酶 进行双酶切鉴定。以待鉴定的pET-32a-NP为例,双 酶切体系为20μL:
EcoR Ⅰ
Sal Ⅰ
Xhol Ⅰ
Hind Ⅲ
第一组
目的基因酶切体系
ddH2O 10×buffer H pGEMT-NP EcoRI Sal I 21 µL 3 µL 4 µL 1 µL 1 µL
原核表达载体酶切体系
ddH2O 10×buffer H pET-32a EcoRI Sal I 22 µL 3 µL 3 µL 1 µL 1 µL
谢谢! 谢谢!
Thanks!
二 实验步骤
1、将鉴定为阳性的重组质粒pGEMT-NP、pGEMT-P、 pGEMT-M、pGEMT-F和pGEMT-HN与原核表达载体pET32a分别用相应的限制性内切酶进行双酶切。以 pGEMT-NP为例,双酶切体系(30μL体系)如下:
ddH2O 10×buffer H pGEMT-NP EcoRI Sal I 21 µL 3 µL 4 µL 1 µL 1 µL ddH2O 10×buffer H pET-32a EcoRI Sal I 22 µL 3 µL 3 µL 1 µL 1 µL
第二组
ddH2O 10×buffer M pGEMT-P EcoRI HindⅢ 21 µL 3 µL 4 µL 1 µL 1 µL 22 µL 3 µL 3 µL 1 µL 1 µL
ddH2O 10×buffer M pET-32a EcoRI HindⅢ
第三组
ddH2O 10×buffer H pGEMT-M EcoRI Sal I 21 µL 3 µL 4 µL 1 µL 1 µL ddH2O 10×buffer H pET-32a EcoRI Sal I 22 µL 3 µL 3 µL 1 µL 1 µL