手性药物高效液相色谱拆分
液相色谱法分离手性药物

第一讲手性分离色谱手性药物常用的色谱分离方法有:高效液相色谱、气相色谱、毛细管电泳、超临界流体色谱。
手性药物给人类曾经带来过空前的灾难——反应停事件。
概念手性:指一种化合物分子由于其三维空间结构的原因所显示出的相互不能重合,但互为镜像关系,它形象的比喻为人的左右手,这叫手性。
对映体: 由于手性中心连接的四个基团在空间三维排列的不同,对偏振光产生的旋转方向不同,从而产生不能重叠的互为镜像的光学异构体,称对映体。
旋光性:手性药物对映体之间对偏振光的偏转程度相同,但偏转方向相反,即旋光性。
右旋体:能使偏振光按顺时针方向旋转的对映体称为右旋体以d-或(+)-表示。
左旋体:按逆时针方向旋转者称为左旋体以l-或(-)-表示。
外消旋体:等量的左旋体和右旋体构成外消旋体,没有旋光性,以(dl)或(±)表示。
内消旋体:分子中含有手性碳原子,但作为分子整体来说是非手性的。
内消旋化合物是纯净物。
外消旋体与内消旋体的共同之处是:二者均无旋光性。
外消旋体:是混合物,可拆分出一对对映体。
内消旋体:是化合物,不能拆分。
手性药物:是指由具有药理活性的手性化合物组成的药物。
手性药物的表示方法1.dl-或(±)-表示能使偏振光的偏振面按顺时针方向旋转的对映体称为右旋体(dextrotatory),在药名前用d-或(+)-表示;反之,称为左旋体(levorotatory),在药名前加l-或(-)-表示。
外消旋体(racemate)则是由等量的左旋体和右旋体构成,没有旋光性,在其药名前用dl-或(±)-表示。
2. D/L标记法(相对构型)1951年前,人们还无法确定化合物的绝对构型。
费歇尔(Fischer)人为地选定(D)-甘油醛为标准物,以标准参照物来确定药物的立体化学构型,相对构型。
由于D/L构型表示法它只适用与甘油醛结构类似的化合物,对多个手性碳的化合物使用不方便。
与表示旋光方向的d和l容易混淆,目前多限于糖和氨基酸的立体化学命名。
手性药物色谱拆分法研究发展

5.1 间接拆分法
[1]Zukowski J,De Biasi V,Berthod A. Chiral
等特点,并具在手性分离方面与高效液相色谱、
间接拆分法[8]虽需进行衍生化反应,但生 separation of basic drugs by capillary elec-
气相色谱相互补充,在光学纯药物的制备方面 成的非对映体异构体,物化性质不同,可用常规 trophoresis with carboxymethylcyclodextrins [J].J
的技术,它以高压电场为驱动力,以毛细管为分 - NHCO- 基团。苯环的取代基的性质,数目及位 [11]LI Bing,SHI Jie -hua,YANG Gen -sheng,.
离通道,依据样品中各组分间电荷及质量的差 置对手性化合物的拆分影响很大[11]。蛋白质类 Cellulose-based chiral stationary phase in high
副作用。因此手性药物拆分近年来引起人们的 D- 10- 樟脑磺酸胺作为手性离子对试剂添加到 是很广泛;GC 法对于药物的沸点要求严格,故
广泛关注。目前,手性药物的拆分主要有化学拆 流动相中,在硅胶 GF254 薄层板上分离了两种芳 GC 应用范围有限;CE 法和 TLC 法检测灵敏度
分法、结晶法、生物拆分法和色谱法等等,其中 香醇胺类药物对映体拉贝乐尔和倍它乐克,并 较低,有待研究提高发展;HPLC 法因手性固定
也有其局限性,如检测灵敏度不足,重现性差等 磺酰基 - 1,2- 二苯基乙二胺,研究了流动相中 对甲基苯磺酰基-1,2-二苯基乙二胺在卵类粘
[6]。
有机调节剂的种类和含量等色谱条件对拆分结 蛋白柱上的手性拆分[J].色谱,2003, 21(4): 407.
手性药物的拆分

*
COOH
COOH
COOH
酶
H2N CH3
H
+H
CH3
NHCOCH3
消旋丙氨酸
NH2
消旋乙酰丙氨酸
NHCOCH3
L-丙氨酸(溶于乙醇)D-乙酰丙氨酸 (不溶于乙醇)
三.色谱法
1. 高效液相色谱法(HPLC)
HPLC法包括直接法和间接法 直接法的分离原理:手性药物对映体之 一与手性固定相或手性流动相之间发生 分子间的三点作用,同时另一对映异构体 则发生两点作用,形成暂时的非对映异构 体的结合物质,前者较后者稳定,通过洗 脱使两对映异构体分离
优先结晶法是一种高效、简单而又快捷 的拆分方法,晶种的加入造成2个对映异 构体具有不同的结晶速率是该动态过程 控制的关键。 利用循环优先结晶方法进行拆分的实例: 抗高血压药物L-甲基多巴的拆分[5],见 图(三)。
HO HO H3C COOH NH2
图(三)L-甲基多巴
1.3 逆向结晶法
在外消旋体的饱和溶液中加入可溶性某 一种构型的异构体(如R-异构体),添 加的(R)-异构体就会吸附到外消旋溶 液中的同种构型异构体结晶的表面,从 而抑制了这种构造体结晶的继续生长, 而溶液中的(S)-异构体结晶速度就会 加快,从而形成结晶析出。
奥沙西泮新戊酸酯
2.气相色谱法(GC)
在气相色谱仪中选择适当的吸附剂作固 定相(通常是手性固定相),使之选择性地 吸附外消旋体中的一种异构体,可以快速 分离手性化合物。 手性化合物的直接气相色谱分离,其关键 问题是必须找到一个合适的手性固定相, 如高聚物固定相、均三氮苯型固定相、 菊酰胺型固定相、光学活性金属络合物 固定相等。
手性药物的拆分
浅谈高效液相色谱(HPLC)在目前手性药物分析领域中的应用

体进行分离。铜和锌等都是常用的配位金属。氨基酸及衍生物、 多巴胺、氧氟沙星等均可用此类方法分离[4]。由于目前为止还未 发现任何一种试剂可以作为通用型试剂,所以在选择手性试剂时 可能会经过多次尝试,选择分离效果最好的手性添加剂。
2.3 手性衍生化试剂法(CDR) 当满足以下条件时可以使用手性衍生化试剂法:①手性化 合物对映体中有氨基、羟基或羧基等基团,其容易发生衍生作 用;②反应产物具有稳定的化学性质,手性试剂具有稳定的手 性性质,以及较高的光学纯度,不易发生变化,不会在色谱条 件下发生消旋化反应[5]。根据手性化合物对映体中有氨基、羟 基或羧基等基团以及分离效率之间的差别,将反应产物进行分 离。胺类试剂、酰化试剂氯甲酸酯类等均是目前常用的衍生化 试剂。因为该类方法是使用普通色谱柱,因此成本较低,分离 的灵敏度较高。
过去常使用酶消化法、分布结晶法等非色谱方法对手性药 物进行拆分,拆分过程耗时、烦琐,具有较大的不可控性,近 年来随着色谱技术的不断发展,在对手性药物进行拆分方面有 了较为广泛的应用[2]。 目前在手性药物进行拆分时较为常用的 方法有气象色谱、毛细管电泳和毛细管色谱以及高效液相色谱 法等,其中高效液相色谱法(HPLC)以其反应速度快、效率 高、准确性强等特点被广泛应用。
究进展[J].药物分析杂志,2015,35(7):1127-1133. [2] 刘丽敏.高效液相色谱在中草药和抗生素类药物分析中的应用
[D].成都:西南大学,2008. [3] 潘永玉.手性药物的对映体分离方法与药物动力学研究[D].沈阳:
沈阳药科大学,2007. [4] 康自华,阳小成,陈婷.高效液相色谱法在药物分析中的应用[J].广
高效液相色谱(HPLC)是一种在近年来被广泛应用的色 谱分析方法,其与传统色谱法相比,具有效率高、灵敏性高和 分析分离速度快等优点。高效液相色谱法原理上可对所有的 热稳定性差、沸点高和相对分子质量大的有机物进行分离和分 析,其不仅可用于对手性药物的定量分析,而且可用于制备分 离,在手性药物分析领域具有较为广泛的应用。
高效液相色谱法手性固定相拆分手性药物研究进展

高效液相色谱法手性固定相拆分手性药物研究进展李雪;李优鑫;张勇【摘要】As the most common method used for drug analysis,HPLC has been widely used for enantio separating of chiral drugs,and CSP is a key factor of isolation effect. Reviewed the advances in enantio separating of chiral drugs by CSPs latest years and predict for the future developments.%高效液相色谱法(HPLC)作为最常用的药物分析方法对手性药物的拆分具有广泛的应用,而手性固定相(CSP)则是拆分效果的关键因素。
介绍了近几年手性固定相在手性药物拆分中的研究进展,并展望其发展前景。
【期刊名称】《应用化工》【年(卷),期】2014(000)006【总页数】4页(P1125-1127,1132)【关键词】手性药物;高效液相色谱;手性固定相【作者】李雪;李优鑫;张勇【作者单位】天津市现代药物传递及功能高效化重点实验室天津大学药物科学与技术学院,天津 300072;天津市现代药物传递及功能高效化重点实验室天津大学药物科学与技术学院,天津 300072;天津市现代药物传递及功能高效化重点实验室天津大学药物科学与技术学院,天津 300072【正文语种】中文【中图分类】TQ460;R917手性,指化合物具有结构上镜像对称而又不能完全重合的分子,作为自然界生命基础的生物大分子都具有手性,人们所使用的药物绝大多数也具有手性。
手性分子两对映体具有完全相同的物理、化学性质,但具有不同的药理活性,手性药物进入人体往往一种对映体有效而另一种无效甚至具有毒性。
20世纪60年代,一种称为反应停的孕妇使用的镇定剂,上市后导致1.2万名婴儿的生理缺陷,因为反应停的右旋体为止吐药,而左旋体具有强烈的致畸作用。
高效液相色谱手性拆分中的配体交换色谱手性固定相

Ξ高效液相色谱手性拆分中的配体交换色谱手性固定相张占辉(河北师范大学化学学院,河北石家庄 050091)摘 要:综述了高效液相色谱配体交换色谱手性固定相的发展、制备及其在手性拆分中的应用,讨论了洗速、进样量、中心金属离子及其浓度、流动相p H 值、柱温、有机改性剂等对对映体分离的影响,阐述了对手性识别机理的认识.关键词:高效液相色谱;手性拆分;手性固定相;配体交换色谱中图分类号:O 658 文献标识码:A 文章编号:100025854(2005)03202842070 前 言手性是自然界的普遍现象,在药物化学领域尤其突出,已知的药物中约有30%~40%是手性的.已发现的许多手性药物的对映体在药理、毒理和代谢过程存在显著差异,基于此情况,1992年美国食品和药品管理局规定:今后凡研制具有不对称中心的药物,必须对其各个对映体进行测定和评价.欧共体也采取了相应的措施,因此,手性分离手段越来越受关注,各种分离和测定方法得到发展[1].与气相色谱、毛细管电泳分离对映体比较,高效液相色谱不会发生被分离物质高温构性转化或破坏生物活性等,它已成为现代合成化学、生物医学、农业化学、地球化学、天然有机化学等领域对对映体分离必不可少的工具.具有不对称中心或手性识别能力的手性固定相(CSP )的开发与研制,是手性色谱发展的前沿领域.CSP 的研制始于20世纪70年代,发展异常迅速,现在用于色谱分离的手性固定相已有100多种[2].目前所研究和使用的高效液相色谱手性固定相(HPLC CSP )主要可分为下列几类:1)“刷型”CSP ;2)聚合物CSP ,包括天然的多糖(纤维素、淀粉等)和合成的手性聚合物制备的CSP ;3)蛋白质CSP ;4)大环CSP ,包括经修饰的环糊精、手性冠醚和大环抗生素作为手性选择剂的CSP ;5)分子印迹CSP ;6)配体交换色谱CSP 等.手性配体交换色谱(chiral ligand 2exchange chromatography ,CL EC )技术是1961年由Helfferich 首次提出,并通过Rogozhin 和Davankov 等的发展,使其成为一种有效的手性色谱分离法[3].CL EC 要求在色谱系统中引入某种金属离子和某种手性配体,待测对映体与配位体可形成2个非对映的三元络合物,经色谱过程实现光学异构体的立体选择性分离,它已成为拆分未衍生化氨基酸、羟基酸、二胺及其衍生物以及生物体小分子最简便有效的方法.它有3种类型[4]:1)手性键合固定相(chiral bonded stationary phases ,CSPs );2)手性涂渍或覆盖固定相(chiral coated stationary phases ,CCSPs );3)手性流动相(chiral mobile phases ,CMPs ).本文中,笔者对CL EC 固定相的发展、制备、影响拆分的因素及拆分机理进行简单综述.1 手性配体交换色谱固定相的发展、制备和应用1.1 发 展Davankov [5]首次将脯氨酸键合到苯乙烯二乙烯苯树脂上,通过流动相引入铜离子形成铜离子配合Ξ收稿日期:20040928;修回日期:20041117基金项目:国家自然科学基金资助项目(20477009)作者简介:张占辉(1965),男,河北省深泽县人,河北师范大学副研究员,现为南开大学博士研究生,从事有机合成与分析研究.第29卷第3期2005年 5月河北师范大学学报(自然科学版)Journal of Hebei Normal University (Natural Science Edition )Vol.29No.3May.2005物,用L EC 分离了氨基酸对映体.后来将氨基酸和N ,N 二苯基1,2丙二胺键合到聚苯乙烯和聚丙烯酰胺树脂上,以Cu (Ⅱ)和Ni (Ⅱ)作为络合金属,这些材料尽管表现出良好的对映选择性,但拆分效率不高.此后,许多聚合物如交联聚甲基丙烯酸酯、球形酚醛树脂、交联聚乙烯酰胺等被用作L EC 的载体,但由于机械强度差而难以满足高效液相色谱对填料的要求.适用于HPLC 的硅胶键合手性配体交换色谱固定相(L EC CSP )是由G übitz 等[6]发展起来的,键合的配体分别为L 脯氨酸、羟脯氨酸、组氨酸、缬氨酸、苯丙氨酸、酒石酸、氮杂环丁烷酸、哌啶酸、丙二胺、麻黄碱等,与以聚合物为载体的手性固定相相比,它具有亲水性、高选择性和高强度等优势.1994年G übitz 等又以S 脯氨酸为手性选择因子制备了2个新的手性固定相CSP1和CSP2.CSP1中在手性选择因子和硅胶连接臂之间引入了一个更刚性的环,增强了手性识别能力;CSP2中在连接臂中插入氨基形成一个乙二胺结构,氨基有可能参与配位,这2个手性固定相对氨基酸、丹磺酰氨基酸、二肽和羟基酸进行了很好的分离,CSP2还应用于巴比妥酸盐的拆分,取得了满意的结果[7].1998年Wachsmann 等[8]报道通过三嗪连接臂将L 脯氨酸或L 赖氨酸键合到氨基丙基硅烷上制备了2个新的手性固定相CSP3和CSP4,适合于未衍生的氨基酸和N (2,4二硝基苯基)氨基酸的拆分,CSP4还对N 丹磺酰氨基酸具有很好的拆分能力.G ebber 等[9]在键合配体中插入键合非配体烷基,如正丁基、正癸基,形成具有疏水性的混合型L EC CSP ,改进了手性分离的选择性和柱效.Saigo 等[10]为提高分离选择性合成了结构更为复杂的L EC CSP.2000年Vidyasankar 等报道了第1个分子印迹配体交换聚合物,该聚合物由Cu (Ⅱ)N (4乙烯基)苯基亚氨基二乙酸作单体,氨基酸作模板分子,连接到衍生化的硅胶上制得,它以配体交换为机制,直接拆分未衍生化的氨基酸,对DL 苯丙氨酸的选择因子可达1.65,但对脂肪氨基酸拆分效果不好[11].我国在研制L EC CSP 方面也取得了很大进展:马建标等[12]将亲水性间隔臂插入聚苯乙烯与固定配体之间,合成了具有亲水性质的高强度有机固定相;袁直、何炳林合成了亲水性手性配体铜络合聚乙烯胺树脂,并以此树脂作为高效液相色谱固定相,对一系列氨基酸进行了拆分,并探讨了拆分机理[13];阎虎生等合成了薄壳型手性配体交换HPLC 填料,并用于DL 氨基酸的拆分[14];黄天宝课题组合成了(S )1,2,3,4四氢3异喹啉羧酸[(S )THIQCA ]硅胶键合手性配体固定相,并应用于氨基酸的拆分,对映选择性在1.11~1.51之间[15];李永民、陈立仁课题组合成了L 羟基脯氨酸、L 脯氨酸和L 苯丙氨酸3种键合手性配体固定相,并用于α羟基酸的直接拆分,取得了较为满意的结果[3];王俊德课题组将β(3,4环氧环己基)乙基三甲氧基硅烷与硅胶反应,得到环氧化硅胶中间体,然后与L 异亮氨酸反应,再与铜离子配位,得到一种新型手性配体交换固定相CSP5[16].1980年Davankov 等[17]发展了另一种新型涂渍手性配体交换色谱固定相,他们把N n 烷基L 羟基脯氨酸涂渍在反相色谱柱上直接用于氨基酸的拆分,7个消旋的氨基酸在10cm 长的柱上得到很好的拆分,这种涂覆相可获得与硅胶键合相类似的结果.N 癸基L 羟基脯氨酸涂渍在RP C 18柱上制成的CCSP ,能在0.5~3min 对氨基酸进行基线分离.另一个涂渍手性配体交换色谱柱Chirapak MA (+)是把N ,N 二辛基L 丙氨酸涂渍在3μm RP C 18材料上制成,它能有效拆分羟基羧酸[4].2002年Remelli 等合成了1个新的手性配体———N τn 癸基菠菜素(1),该化合物中氨基和咪唑环之间含有1个亚甲基桥,把该配体涂渍在RP C 18柱上,它能拆分外消旋的氨基酸和甘氨酸等[18].N ,S 二辛基D 青霉胺(2)涂渍在反相硅胶柱上制成的涂渍手性配体交换色谱柱在德国Penomenex 公司商品化,这个手性选择子的分子结构中含有3个可用于配位的杂原子N ,O 和S ,它能拆分氨基酸、酰胺羧酸、羟基酸、氨基乙酰二肽和三肽、氨基醇、1氨基乙基磷酸和3氨基吡咯烷等[19].由日本Sumichiral Chem 2ical Analysis Service 提供的OA 6000是以单(R )1(α萘乙基胺)(R ,R )酒石酸(3)作为手性配体制成的涂渍手性配体交换色谱柱,用它定量测定了尿中的对羟基扁桃酸对映体[20].十八烷酰L 肉碱(4)涂渍在RP 硅胶柱上,在L EC 条件下拆分了一系列氨基酸和羟基酸[21].582第3期张占辉:高效液相色谱手性拆分中的配体交换色谱手性固定相 除了上面RP 硅胶作为支载剂外,多孔性石墨最近被用作固载N 烷基化氨基酸手性选择子,Knox 等用N 2萘磺酸基苯丙氨酸的铜络合物吸附在多孔性石墨上制备的CSP ,亦可拆分非衍生化的氨基酸对映体[22].Wang 等[23]通过L 脯氨酸烷基化或芳基化合成了含有C7,C9,C12、萘甲基和蒽甲基的手性选择子,并把它们涂覆在多孔性石墨上制备了5个手性固定相,用这些固定相拆分了一系列氨基酸,研究了N 上的烷基链长度对保留行为和对映选择性的影响.王群标等[24]合成了2(2羟基3烷氧基)丙基(S )1,2,3,4四氢3异喹啉羧酸[C 8(S )THIQCA ]手性选择子,制备了一种新型CCSP ,拆分了一些氨基酸.1.2 制 备手性配体交换色谱固定相制备法主要有2种.1)涂渍法:将含有手性选择子的水有机溶剂混合溶液通过C 18键合固定相色谱柱或其他支载剂,然后用含有金属离子的溶液平衡柱子使固定相表面预饱和.2)键合法:最简便的制备方法是用氨基酸钠盐,如L 异亮氨酸钠与含环氧基的硅胶键合相反应[16],亦可在柱上进行开环缩合反应,如图1所示.图1 手性配体交换色谱固定相CSP5的合成1.3 应 用自从手性配体交换色谱固定相研制并得到迅速发展以来,已经拆分了大量的化合物.大多数拆分的化合物为游离α氨基酸、β氨基酸、单酰氨基酸、羟基酸、二肽、三肽、二胺及其衍生物以及生物上重要的氨基醇,包括儿茶酚胺、丙醇胺、羟基乙胺等.如用Chirapak WH 手性柱拆分ACE 抑制剂盐酸咪唑普利(Imidapril Hydrochloride )[25],抗真菌剂益康唑(econazole )、咪康唑(Miconazole )和硫康唑(sul 2conazole )[26],用MCI gel CRS 10W 柱手性拆分2,6二胺庚二酸[27]、泛解酸和泛酸钙,用Shodex Orpac CRX 453B 柱测定人脑脊髓液中的L和D 乳酸[28].用N 正十二烷基(1R ,2S )降麻黄碱涂渍在ODS 柱上制备的固定相,含有巴比妥的铜(Ⅱ)溶液作流动相,在L EC 条件下拆分了未衍生化的β氨基醇,巴比妥的加入有利于改善对映选择性[29].2 影响手性拆分的因素配体交换色谱的分离过程受多方面因素的影响,如洗速、进样量、中心金属离子及其浓度、流动相p H 值、柱温、有机改性剂等均影响其对映体分离选择性.2.1 流速的影响随着流动相流速的降低,对映体在柱上的保留时间会增加,有利于对映体的配体交换,从而有利于682河北师范大学学报(自然科学版)第29卷2种对映异构体的分离.分离物与手性配体进行配位交换的能力不同,对于那些配位能力差的分离物,在高流速下分离物与手性配体无法充分实现配位交换,对拆分不利;对于配位能力较强的分离物,在高流速下也可实现分离物与手性配体的充分作用:因而流速对其分离影响不大.流速对塔板高度也有影响,随着流速的降低,塔板高度(h )也降低[30].丁国生等[31]在研究中发现,一般对中性氨基酸来说,降低流速,选择性(α)和分离度(Rs )均有一定程度的提高,而对碱性氨基酸,降低流速虽可提高α,但由于色谱峰扩展严重,结果造成Rs 降低.2.2 进样量的影响在分离物进样量过载的情况下,样品的分离效果较差,这是因为配体交换色谱的分离过程是配位交换.在进样量过载的情况下,没有足够的手性配体与分离物进行配位交换,因而对映体的分离就不会充分;当进样量没有过载的情况下,进样量对分离的影响不大.2.3 中心金属离子的影响 不同的中心金属离子所产生的拆分结果差别较大,常见中心金属离子拆分能力的顺序为Cu (Ⅱ)>>Ni (Ⅱ)>Zn (Ⅱ)>Cd (Ⅱ),故Cu (Ⅱ)常作为络合金属.金属离子的浓度对分离也有重要影响,一般金属离子浓度增加有利于手性分离.金属离子的引入是为了使被分离的溶质与配体交换色谱固定相中的手性选择子能够形成络合物,依据2种非对映络合物稳定常数的不同来实现手性分离,金属离子浓度增加,体系中可以有更多的金属离子手性配体(M CL )络合物与分离物进行配位交换形成混配物,从而保证了对映体分离;但当金属离子浓度超过一定值使体系达到饱和后,这种变化不再明显.2.4 流动相p H 值的影响 一般情况下,随着p H 值的增加,分离物的容量因子(k ′),α和Rs 都会增加,当增加至碱性时,k ′和α增加更为显著,但往往会引起保留时间过长,峰形变差,同时过高的p H 值对柱寿命有不利影响.pH 值对分离的这种影响,是基于固定相所键合的手性配体在与中心金属离子形成二齿配体时,α氨基酸以未质子化氨基和阴离子的形式存在;同样地,α羟基酸对映体分离时,配体主要发生在羟基酸的α羟基非质子基团和羧基阴离子与金属离子之间,以形成非对映配合物.流动相酸度的降低有利于这些非质子基团和阴离子的存在,从而有利于配合物的形成.流动相离子强度增加,一般k ′减小,但塔板数(N )会增加,Rs 基本不变,在同一pH 值下,采用较高的离子强度既可使分析时间缩短,又可达到相同的分离度.2.5 固定相键合量的影响 祝馨怡等[32]研究了硅胶键合手性配体交换色谱固定相键合量对α氨基酸拆分的影响.研究结果表明:对于不同种类氨基酸的手性拆分,应该选择不同键合量的手性固定相,因为不同种类的氨基酸与手性配体形成的配合物的稳定性各不相同.对DL 天冬氨酸、DL 丝氨酸、DL 天冬酰胺、DL 甲硫氨酸、DL 苏氨酸和DL 缬氨酸等,它们与手性配体所形成的配合物稳定性较弱,因此应该选择键合量较大的长色谱柱进行分离,以增强柱保留能力;而对于DL 酪氨酸、DL 色氨酸、DL 苯丙氨酸和DL 异亮氨酸等,它们与手性配体形成的配合物稳定性相对较强,这时应选择键合量较小的短色谱柱进行分离,以避免保留过强所引起的峰拖尾现象,以及由于过强的柱保留而不能实现手性分离.2.6 温度的影响 在拆分过程中,温度对拆分结果的影响比较明显.一般在亲水性强的聚乙烯胺体系上,保留值随柱温升高而增大,而在疏水性的聚苯乙烯体系上,保留值随柱温的升高而减小.这是因为聚乙烯胺骨架的亲水性强,柱温升高加强了高分子链的活动性,表现为保留值增大;对于疏水性的聚苯乙烯骨架,温度升高,并不能明显地改变其活动性,在这种情况下,升高温度使配体交换反应加快的作用为主,表现为保留值减小:所以在分离过程中,应根据不同的固定相选择适宜的温度,以便使样品得到良好的分离.2.7 有机改性剂的影响 一般情况下,有机改性剂的加入会缩短保留时间.Hyun 等[33]合成了2个手性配体:N ((S )1羟甲基3甲基丁基)N 十一烷基氨基乙酸钠和N ((R )2羟基1苯乙基)N 十一烷基氨基782第3期张占辉:高效液相色谱手性拆分中的配体交换色谱手性固定相乙酸钠,并把它们键合到硅胶上制得2个手性固定相CSP6和CSP7,详细研究了有机改性剂在上述2个手性固定相上对分离α和β氨基酸的影响.研究结果表明:当亲油性的分析物在亲油性的CSP6上拆分时,k ′随有机改性剂甲醇在流动相浓度的增加而降低,当亲水性的分析物在上述固定相上拆分时,k ′随甲醇浓度的增加而增加;相反,仅仅强亲油性的分析物在相对低亲油性的CSP7上拆分时,k ′随有机改性剂在流动相浓度的增加而增加.在2种不同CSP 上的保留行为是由于分析物在极性流动相和亲油性的固定相之间分配竞争不同引起的.3 分离机制 手性配体交换色谱固定相拆分机理是基于固定相手性配体、金属离子与被分离溶质对映体形成一对非对映的配合物,二者的热力学稳定性差异导致了色谱分离.适当的对映体配体能给出电子到过渡金属的d 轨道,且占据有力空间,形成一定构型的配合物:因此要求被分离的溶质对Cu (Ⅱ)或其他金属离子必须具有双配位基,主要是游离氨基酸、羟基酸、二胺及其衍生物.除配合物形成外,一些附加力,如氢键、偶极、疏水作用亦对分离有影响.分离过程如下: [CL ]n M +L CS K 1CL ]n -1M[L CS]+CL (n ≥2). [CL ]n M +D CS K 2[CL ]n -1M[D CS]+CL (n ≥2).CL 与M 形成[CL ]n M 并与对映体(CS )发生交换作用,对映体D 和L 型与手性配体固定相所形成的配合物稳定性不同,K 1≠K 2,从而决定了它们在洗脱液作用下的保留时间或保留体积不同,这种稳定性的差异是由于2种配合物的空间构型不同所决定的.形成的配合物越稳定,其保留时间越长,后洗脱,因而可以进行DL 对映异构体的拆分.图2描述了L 异亮氨酸作为手性选择因子的CSP5和D ,L 氨基酸之间形成配合物的可能结构.从图2可以看出,手性选择因子中的N 原子与羧基中的O 原子和Cu 2+共处于一个平面而进行配位,环氧键开环以后生成的—OH 处于平面上方的轴向方向,它与另一处在平面下方轴向方向的H 2O 共同参与配位,形成六配位的配合物.同时,D 构型氨基酸的侧链R 处于平面上方,与刚性环环己基产生较大空间位阻,造成D 氨基酸与手性选择因子产生的配体作用减弱;相比之下,L 构型氨基酸的侧链R 处于平面下方,未产生空间位阻作用,因此所形成的配合物较稳定,2种配合物稳定性之间的差异成为对映体拆分的基础,D 氨基酸总比L 氨基酸先洗脱下来.不同的手性配体对立体差异的识别能力也不同,一般具有刚性构象的手性配体,特别是那些包含环状结构的配体,如L 羟基脯氨酸和L 脯氨酸,它们与2种构型的对映体所形成的配合物的立体位阻差异较大,因而具有较好的对映选择性;相反,那些构象可弯曲型配体如苯丙氨酸,当立体构型不利时,可以通过构象弯曲来弥补,结果导致2种对映体所形成的配合物的立体构型差异不明显,因而显示出较差的对映选择性.另一方面,如果手性配体上带有羟基等基团,在进行配位交换时,往往可以增加手性配体与分离物之间的作用力,使分离物与手性固定相的作用更充分,分离效率会更高.此外,配位体和金属离子所形成的络合物必须易于断裂和再生,这样才有利于配体交换的进行.图2 以L 异亮氨酸为手性选择子的CSP5在拆分D (a )和L 氨基酸(b )时形成混合配合物的可能结构882河北师范大学学报(自然科学版)第29卷对映体的出峰顺序还受化合物极性、酸碱性、手性选择剂键合到载体上的方式等因素的影响,Chilmonczyk 等[34]对Davankov 的对映体出峰顺序模型进行了修饰,提出了用ZINDO 半经验量子力学理论和密度功能理论(DF T )对配合物的稳定性进行计算,从而更准确地预测D ,L 型异构体之间的洗脱顺序,根据其洗脱顺序还可以确定对映体的绝对构型[35].综上所述,手性配体交换色谱经过30多年的发展,已取得了很大进展,现已成为一种非常有用的手性色谱法,特别是在拆分氨基酸、羟基酸等多基团化合物方面体现了独特的优越性,从而推动了生物无机化学的发展.可以预见,随着新手性配体的合成和对手性识别机理的深入研究,将会有更多的新手性配体交换色谱固定相设计出来,此类手性固定相的应用范围也将不断扩大.参考文献:[1] WARD T J ,HAMBUR G D M.Chiral se parations [J ].Anal Chem ,2004,76(16):463524644.[2] Y ASHIMA E ,Y AMAMO TO C ,O K AMO TO Y.Polysaccharide 2based chiral LC colums [J ].Synlett ,1998,(4):3442360.[3] 祝馨怡,陈立仁,柳春辉,等.α2羟基酸在不同的手性配体交换色谱固定相上分离性能的考察[J ].分析化学,2003,31(6):6502654.[4] DAVAN KOV V A.Enantioselective ligand exchange in modern separation techniques [J ].J Chromatogr (A ),2003,1000(122):8912915.[5] DAVAN KOV V A ,RO G OZHIN S V.Ligand chromatography as a novel method for the investigation of mixed complex 2es :Seteroselective effects in α2amino acid copper (Ⅱ)complexes [J ].J Chromatogr ,1971,60(2):2802283.[6] G ΒBITZ G ,J ELL ENZ W ,SAN TI W.Se paration of the optical isomers of amino acids by ligand 2exchange chromato 2graphy using chemically bonded chiral phases [J ].J Chromatogr (A ),1981,203:3772384.[7] G ΒBITZ G ,MIHELL YES S ,KOBIN GER G ,et al .New chemically bonded chiral ligand 2exchange chromatography sta 2tionary phase [J ].J Chromatogr (A ),1994,666(122):91297.[8] WACHSMANN M ,BR ΒCKN ER H.Ligand 2exchange chromatography separation of DL 2amino acids on aminopropyl 2silica 2bonded chiral s 2triazines [J ].Chromatographia ,1998,47(11212):6372642.[9] GEBBER L R ,K AR GER B L ,N EUMEYER J L ,et al .Ligand exchange chromatography of amino alcohols :Use of Schiffbases in enantiomer resolution [J ].J Am Chem S oc ,1984,106(25):772927731.[10] SAIG O K ,YU KI Y ,KIMO TO H ,et al .A novel chiral stationary phase for optical resolution of amio acids and theirderivatives by ligand exchange high 2performance liquid chromatography [J ].Bull Chem S oc Jpn ,1988,34(1):3222324.[11] V ID Y ASAN K AR S ,RU M ,ARNOLD F H ,et al .Molecularly imprinted ligand 2exchange adsorbents for the chiral sepa 2ration of underivatized amino acids [J ].J Chromatogr (A ),1997,775(122):51263.[12] 马建标,何炳林.接有亲水性间隔臂和L 2脯氨酸功能基的交联聚苯乙烯树脂的合成[J ].科学通报,1989,34(24):2232226.[13] 袁直,何炳林.亲水性手性配体树脂对氨基酸的拆分[J ].离子交换与吸附,1995,11(1):68272.[14] 阎虎生,王东,李文兰,等.薄壳型手性配体交换HPLC 填料的合成及用于DL 2氨基酸拆分[J ].离子交换与吸附,1999,15(1):86289.[15] 龙远德,王群标,黄天宝.一种新型手性配体交换色谱键合固定相[J ].高等学校化学学报,2001,22(4):5662568.[16] 黄晓佳,丁国生,王俊德,等.新型手性配体交换色谱固定相的合成及其对DL 2氨基酸的拆分[J ].色谱,2003,21(3):2302232.[17] DAVAN KOV V A ,BOCHKOV A S ,KURANOV A A ,et al .Dealing with the ligand 2exchange chromatography 13:Separation of unmodified α2amio 2acid enantiomers by reverse phase HPLC [J ].Chromatographia ,1980,13(11):6772685.[18] REMELL I M ,TROMBIN D ,CONA TO C.Chiral ligand 2exchange chromatography on an RP HPLC column coated witha new chiral selector derived from L 2spinacine [J ].Chromatographia ,2002,55(526):301.[19] :i N ,KITAHARA H ,KIRA R.Direct se paration of enantiomers by high 2performance liquid 2chromatography on a newchiral ligand 2exchange phase [J ].J Chromatogr ,1992,592(122):2912296.[20] ARAI K ,J IN D ,KUSH F ,et al .Determination of p 2hydroxymandelic acid enantiomers in urine by high 2performanceliquid chromatography with electrovhemical detection [J ].J Pharm Biomed Anal ,1997,15(9210):150921514.982第3期张占辉:高效液相色谱手性拆分中的配体交换色谱手性固定相092河北师范大学学报(自然科学版)第29卷[21] K AMIMORI H,KON ISHI M.Evaluation and a pplication of liquid chromatographic columns coated with‘intelligent’ligands(I):Acylcarnitine column[J].J Chromatogr(A),2001,929(122):1212.[22] KNOX J H,WAN Q H.Chiral chromatography of amino2and hydroxy2amino on surface modified porous graphite[J].Chromatographia,1995,40(122):9214.[23] WAN G Qian2heng,SHAW P N,DAV IES M C,et al.Role of alkyl and aryl substituents in chiral ligand exchange chro2matography of amino acids:Study using porous graphitic carbon coated with N2substituted2L2proline selectors[J].J Chromatogr(A),1997,786(2):2492257.[24] 王群标,龙远德,黄天宝.新型手性配体交换色谱固定相的制备及应用[J].色谱,2000,18(2):1122114.[25] N ISHI H,Y AMASA KI K,KO KUSEN Y A Y,et al.Optical resolution of imdapril hydrochloride by high2performanceliquid2chromatography and application to the optical purity testing of drugs[J].J Chromatogr(A),1994,672(122):2492 257.[26] ABOUL2EN EIN H Y,AL I I.Enantiomeric resolution of some imidazole antifun gal agents on chiralpak WH chiral sta2tionary phase using HPLC[J].Chromatographia,2001,54(324):2002202.[27] NA G ASAWA T,L IN G J R,ONODERA R.Chiral high2performance liquid2chromatography separation of the threestereoisomers of2,62diaminopimelic acid without derivatisation[J].J Chromatogr(A),1993,653(2):3362340.[28] O KUBO S,ASHIGE F,OMORI M,et al.Enantiomeric determination of L2and D2lactic acid in human cerebros pinalfluid by chiral ligand exchange high2performance liquid chromatography[J].Biomed Chromatogr,2000,14(7):4742 477.[29] Y AMAZA KI S,SSITO K,TANMURA T.Enantiomeric se paration of underivatized aliphaticα2amino alcohols by lig2and2exchange chromatography using N2n2dodecyl2(1R,2S)2norephedrine as a coating reagent for reversed2phase column [J].J High Resol Chromatogr,1998,21(10):5612564.[30] REMELL I M,FORNASARI P,PUL IDORI F.Stud y of retention,efficiency and selectivity in chiral ligand2exchangechromatography with a dynamically coated stationary phase[J].J Chromatogr(A),1997,761(122):79289.[31] 丁国生,黄晓佳,王俊德,等.硅胶键合手性配体交换色谱固定相拆分α2氨基酸[J].分析测试学报,2003,22(6):33235.[32] 祝馨怡,韩小茜,齐邦峰,等.硅胶键合手性配体交换色谱固定相键合量对α2氨基酸拆分的影响[J].色谱,2002,20(3):2232226.[33] HYUN M H,WHAN G BO S H,CHO YJ.E ffect of analyte lipophilicity on the resolution ofα2andβ2amino acids on liq2uid chromatographic ligand exchange chiral stationary phases[J].J Sep Sci,2003,26(18):161521622.[34] CHILMONCZYK Z,KSYCINSK A H,CY BUL SKI J,et al.Direct separation of a2hydroxy acid enantiomers by ligandexchange chromatography[J].Chirality,1998,10(9):8212830.[35] ROUSSEL C,RIO A D,PIERRO T2SANDERS J.Chiral liquid chromatography contribution to the determination of theabsolute configuration of enantiomers[J].J Chromatogr(A),2004,1037(122):3112328.Ligand2exchange Chromatography Stationary Phase forChiral R esolution in High Performance LiquidZHAN G Zhan2hui(College of Chemistry,Hebei Normal University,Hebei Shijiazhuang 050091,China)Abstract:Developments of liquid chromatographic ligand exchange chiral stationary phases for the di2 rect separation enantiomers in high performance liquid chromatography are reviewed.Factors affecting enan2 tiomeric resolution including flow2rate,sample size,metal ions,concentration of metal/complex,eluent p H, column temperature,organic modifier are discussed.The views on the chiral recognition mechanism are also introduced.K ey w ords:high performance liquid chromatography;chiral resolution;chiral stationary phase;ligand2 exchange chromatography(责任编辑 邱 丽)。
高效液相色谱手性流动相添加剂法拆分佐匹克隆对映体

高效液相色谱手性流动相添加剂法拆分佐匹克隆对映体叶祥喜;宋燕西;陈旭涛;季有知;孙璐璐;吴小倩;杜春燕【摘要】以β-环糊精作为手性流动相添加剂,研究了佐匹克隆对映体在反相高效液相色谱系统中的拆分情况.探讨了β-环糊精浓度、流动相中甲醇含量、流动相pH 值、流动相流速及温度等因素对佐匹克隆对映体拆分的影响.通过采用单一变量法对实验条件进行考察,最终得到了佐匹克隆对映体手性拆分的最佳色谱条件.结果表明:当采用Inertsil ODS-SP(5μ.m,4.6 mm×250 mm)色谱柱,甲醇为有机相,流动相为20mmol/L磷酸二氢钾缓冲液(含15 mmol/Lβ-环糊精)-甲醇(85∶15),流速为0.8 mL/min,pH值为5.5,温度为35℃,紫外检测波长为305 nm,进样量为20 μL 时,佐匹克隆对映体得到良好分离,其对映体的保留时间分别为67.42 min和74.48 min,分离度(R)为1.62.该方法操作简单,灵敏度高,拆分效果理想,且比手性固定相法成本低,从而为佐匹克隆对映体的进一步深入研究提供了一种新方法.【期刊名称】《分析测试学报》【年(卷),期】2014(033)004【总页数】4页(P471-474)【关键词】高效液相色谱法;手性流动相添加剂;佐匹克隆;β-环糊精;对映体分离【作者】叶祥喜;宋燕西;陈旭涛;季有知;孙璐璐;吴小倩;杜春燕【作者单位】东华大学环境科学与工程学院,上海201620;东华大学环境科学与工程学院,上海201620;东华大学环境科学与工程学院,上海201620;东华大学环境科学与工程学院,上海201620;东华大学环境科学与工程学院,上海201620;东华大学环境科学与工程学院,上海201620;东华大学环境科学与工程学院,上海201620【正文语种】中文【中图分类】O657.7;TQ460.72佐匹克隆(Zopiclone)是一种安全性较高的速效催眠药,有催眠、镇静、抗焦虑和抗惊厥等作用,以及延长睡眠时间、提高睡眠质量、减少夜间觉醒和早醒次数等效果,且对呼吸系统的抑制作用极小,因而具有次晨残余作用低的特点[1]。
如何应用高效液相色谱法进行手性药物对映异构体拆分

如何应用高效液相色谱法进行手性药物对映异构体拆分对于市场上越来越多对目标物准确定性的要求,而手性产物是其中较为重要的一部分,因此对于手性目标物的检测就显得尤为重要。
一、什么是手性导构和对映异构体?当药物分子中碳原子上连接有4个不相同的基团时,该碳原子被称为不对称碳或手性碳(中心),会导致药物分子存在异构体,如果两个异构体之间的关系如同一个物体的立体结构在照镜子,这个立体结构和它在镜子中的像互为对映异构体(对映体)。
图1是手性对映异构体的图示。
图1 手性对映异构体图示对映体具有相同的物理性质(如熔点,沸点,溶解度,折射率,酸性,密度等),热力学性质(如自由能,焓、熵等)和化学性质。
除非在手性环境(如手性试剂,手性溶剂)中才表现出差异。
对映体对偏振光的作用不同,它们的比旋光度数值相同,但方向相反。
对映体的生物活性不相同,化学反应中表现出等速率。
等量的左旋体与右旋体的混合物构成外消旋体。
从对映体中分离出单纯一个光学异构体的方法称手性拆分。
最普通的手性拆分方法是消旋旋体与光学活性相反的离子(称拆分剂)作用生成非对映体。
手性药物对映体拆分的方法主要有非色谱法和色谱法。
非色谱法(主要包括结晶法、微生物消化法等)耗时长,过程繁琐不能制备高纯度对映体,色谱法是基于把对映体的混合物转换成非对映异构体,然后利用它们在化学或物理性质上的差异进行分离。
主要包括气相色谱(GC)、超临界流体色谱(SFC)、毛细管电泳(CE)和毛细管电色谱(CEC)等。
表1罗列了色谱手性拆分的发展史。
其中高效液相色谱(HPLC)因其独特的优势成为手性分析领域最常用的一种技术。
表1 色谱手性拆分发展史二、HPLC手性拆分方法手性药物拆分法通常分为直接法和间接法两大类。
间接法和直接法的共同特点是均以现代技术为基础并引人不对称中心或光活性分子;不同的是间接法是将其引入分子溶质内,而直接法则是引人分子间。
引人手性环境使对映异构体间呈现物理特征的差异是手性进行光学异构体拆分的基础。
手性药物拆分技术及分析

手性药物拆分技术及分析手性药物(chiral drugs)是指分子内部有一个或多个不对称碳原子的药物,即具有手性结构的药物。
手性药物由于具有左右旋异构体,使得其药理学效应、药效学性质、药代动力学以及安全性能等方面出现差异。
因此,手性药物的拆分技术及分析对于药物的研发、生产和应用具有重要意义。
手性药物的拆分技术主要有下述几种方法:晶体化学方法、酶法、化学拆分、色谱法和光学活性检测。
首先是晶体化学方法,该方法是利用手性药物晶体的对称性差异完成拆分。
通过晶体中的尖、刃、拱等特征差异,将手性药物分离为晶体异构体。
其次是酶法,手性药物的拆分可以通过酶的催化作用实现。
酶是具有高选择性、高催化效率和高效底物转化率的催化剂。
通过选择合适的酶,可以将手性药物转化为对应的手性异构体或原生态精细化靶化合物。
化学拆分是指通过特定的化学反应将手性药物分解为不对称碳原子具有相反手性的产物。
该方法较为常用,但对于存储稳定性较差的手性药物较不适宜。
色谱法是利用不同手性列进行手性分离,如手性HPLC(高效液相色谱)和手性毛细管电泳等。
这些方法主要是利用手性固定相对手性药物进行分离,可达到手性药物的拆分效果。
光学活性检测是通过光学活性的手性试剂或手性染料,以手性化合物的吸光性能差异检测手性药物的拆分效果。
根据手性分析原理,通过手性分析仪器对手性药物进行检测和分析。
手性药物的分析对于药物研发、生产和应用非常重要。
分析手性药物的关键是确保其纯度和药效学性质,并且有助于合理掌握手性药物在体内的吸收、分布、代谢和排泄的信息。
以下是手性药物分析的一些常用方法。
首先是纳米液相色谱法,该方法是将分离的手性药物样品通过微量泵输送到纳米柱中,在极小的流速和流体容量下进行分离。
该方法对于手性药物样品的需求量很小,因此可以减少手性药物样品的消耗。
其次是循环偏振负压电流法,该方法通过测量手性药物样品对光的旋光性质,直接反应其手性结构。
该方法准确、快速,适用于灵敏度高的手性药物分析。
手性高效液相色谱法

9
手性HPLC法
直接法和间接法异同点 均以现代色谱分离技术为基础,引入手性 环境(不对称中心),使药物对映体间呈现理化 性质的差异而实现分离,不同的是间接法是将 其引入分子内,而直接发引入分子间。
10
10
手性衍生化试剂法
对映异构体与手性试剂反应,产生相应的非对 映异构体
( R) SA ( R) SE ( R) SA ( R) SE (S ) SA ( R) SE (S ) SA
16
基本原理
在HPLC流动相中加入光学纯反离子可与流动 相中的对映体生成非对映体复合物,离子对复合物 之间具有不同的稳定性和分配性质,并可与固定相 发生不同的静电,疏水和氢键作用,进而差速迁移 得以分离。
17
17
3.配基交换型手性添加剂
配基交换原理: 在流动相缓冲溶液中加入金属离子和配位 体交换剂形成二元络合物,药物对映体再与其 形成稳定性不同的三元络合物而达到手性分离。 常用的手性配合试剂:氨基酸及其衍生物 如L-苯丙氨酸,L-脯氨酸等配位金属有 Cu2+、 18 Zn2+、Ni2+、Cd2+等。
色谱柱:Aglient Zorbax C18 (250mm×4.6mm,5μm) 流动相:乙腈-0.02mol/L磷酸二氢钾缓冲 盐(55:45,用磷酸调pH至4.5) 流速:0.75 ml/min 24 检测波长:224nm 进样量:20μl 柱温:室温
24
艾司洛尔
应用实例
样品处理和手性衍生化方法:
常用手性添加剂有: 环糊精类 手性离子对试剂 13 配基交换型等
13
1.环糊精类手性添加剂
环糊精的手性识别主要 来自环内腔对芳烃或脂肪烃 侧链的包合作用以及环外壳 上的羟基与药物对映体发生 氢键作用。
药物研究中手性分离分析方法及技巧

药物研究中手性分离分析方法及技巧药物研究中手性分离分析是指将手性药物中的手性异构体(也称为对映体)分离出来,并进行定量分析。
由于手性异构体具有不对称的结构,其物理化学性质和药理活性可能差异巨大,因此手性分离分析对于药物研究具有重要意义。
以下将介绍几种常用的手性分离分析方法及技巧。
1.气相色谱法(GC法):GC法是通过在手性固定相柱上进行气相色谱分析,分离手性异构体。
该方法基于手性碳氢化合物在手性固定相上的不同吸附能力来实现手性分离。
同时,通过合适的手性底物和手性固定相的选择,还可以更好地提高手性分离的选择性和灵敏度。
2.液相色谱法(HPLC法):HPLC法是手性分离分析中最常用的方法之一、常见的手性固定相有手性液相、手性离子对和手性硅胶等。
通过在手性固定相上进行液相色谱分析,利用手性化合物在固定相上的差异相互作用,实现手性分离。
此外,还可以结合负载式手性液相色谱法、手性离子对液相色谱法等技术,提高手性分离效果。
3.毛细管电泳法(CE法):CE法是一种高效、快速的分离技术,特别适用于分析手性药物。
通过在毛细管中施加电场,利用手性化合物在毛细管中的迁移速率差异实现分离。
此外,还可以通过改变运行缓冲液的组成、pH值等条件,调节手性分离的选择性和分离效果。
除了上述主要的手性分离分析方法外,还存在一些辅助技巧和方法,可以进一步提高手性分离的效果:1.共处理:将两个手性化合物混合在一起进行共处理,通过比较混合物中手性峰的相对峰度等信息,来判断手性分离的效果。
2.离子对调整:通过调整分析液中离子对的浓度和种类,来改变手性分离的效果。
一般来说,手性离子对可以提高手性分离的分辨率和选择性。
3.pH调控:通过改变液相色谱系统中溶液的pH值,可以影响毛细管电泳法和液相色谱法中手性分离效果。
pH值的改变可以调节化合物分子的电荷状态,从而影响手性分离的选择性。
总之,手性分离分析方法及技巧在药物研究中起着重要的作用。
通过合理选择合适的手性分离方法,并结合辅助技巧和方法,可以实现对手性异构体的高效、准确的分离和定量分析,从而为药物研究提供有价值的数据。
手性药物的拆分高效液相色谱HPLC

1.3 环糊精手性固定相
OH
HO O OHOHO
OHO OH O
HO
OH
O OH
O
OH O
HO
OH
O OH
O OH
O
OHO OH O OH
HO
?-CD环状构型(俯视图)
环糊精包封药物的立体结构
环糊精分子中每个葡萄糖单元含有5个手性碳原子,如β-CD含有 35个手性碳原子,同时与各种有机分子形成包容配合物,分子 整体上具有光学活性和立体识别能力,是一种理想的手性选择剂
? 此类CSPs还有几个显著的缺点:负载量低, ? 进样量大于 lμg就可观察到过载现象;对周围环境过
于敏感, 0.1单位的 pH值变化就可 ? 能导致容量因子 20%的变化;稳定性差,必须使用
预柱提高寿命;结构选择性差,很容易受干扰。
1.7其它类CSPs
? 1.配体交换类 CSPs
? 当配体给出的电子能够进入过渡金属空的 d轨道, 就能形成金属配位络合物。这种配位络合物具有非 常明确的几何形状,配体在空间只能占据某种给定 的位置,配体交换手性色谱分离的机理就是基于此。
? 随着异丙醇含量的减少,流动相的极性减弱,对映体在CDMPC 上的保留逐渐增强,分离度逐渐增大;但分离因子基本不变,说 明流动相极性的改变不影响手性固定相识别的实质。
? 除托品酸和酮洛芬外,其它溶质都获得了基线分离。
? CDMPC上手性识别主要取决于溶质的结构,包括大小、形状 和环的数目。
? 西替立嗪在固定相上的保留最强,获得了最好的分离;而托品 酸保留虽强,却只获得部分分离,说明溶质保留的强弱并不是 手性识别的关键因素,可能溶质在固定相手性空腔中的包容作 用以及溶质体积大小和立体结构对其适应性才是关键。
高效液相色谱与手性分离

对含有多个手性中心的药物使用含多糖类手性固定相的高效液相色谱法进行手性拆分摘要对含有多个手性中心的药物进行手性分离是一项具有挑战性的工作。
这篇文章介绍了用多糖类手性固定相对含有多个手性中心的药物进行分离。
并且,柱转换技术在这种化合物得分离中也被应用。
关键词: 回顾;对映体分离; 手性固定相, LC;多糖; 纳多洛尔; 吲多洛尔; 奈必洛尔;地尔硫卓目录1.介绍2.含两个手性中心的药物的手性分离实例3. 含多个手性中心(多于两个)的药物的手性分离实例4. 结论5. 参考文献1. 介绍手性是一个显著的生物学过程,一个生物活性分子的对映体通常具有不同的生物学特性。
生物学作用中的对映体选择性现象并不局限于药物学,它是所有生物活性试剂(杀虫剂、除草剂、香精香料、食物添加剂等)的一个共同特征。
来源于自然物质的药物通常是光学活性或纯形式的单一异构体。
然而,那些用化学方法合成的药物通常是根据不对称中心的数目由两个,四个或者更多异构体混合而成。
因此,立体选择性在手性药物的生物利用度、分配、与受体的相互作用、异构体活动中的代谢和消除过程中所产生的差异从不期望的毒性到毫无意义增大活性。
在过去的30年中,通过高效液相色谱法(HPLC)进行手性分离已经显得越来越重要。
这可以通过以下两个方面得出:(a)间接进行手性分离的方法,包括在色谱分析法中通过一个非手性柱用一个手性衍生物试剂合成非对映异构体;(b)直接进行手性分离的方法,包括用手性固定相(CSPs)将外消旋药物拆分成相应的对映体。
基于手性固定相(CSPs)的直接分离方法因其可以快速、合适的用于分离外消旋酸盐而深受分析和制备行业的喜爱。
自然形成和合成的手性固定相(CSPs)存在着广泛的多样性,绝大多数是用于商业(超过120种)。
这些手性固定相(CSPs)中的很多在应用方面具有局限性。
因此,多糖类固定相和其它固定相,如:化学键合的蛋白质、环糊精及其衍生物、Π-型和大环抗生素已经被证明是在高效液相色谱法进行手性药物的分离中最有用的固定相。
高效液相色谱间接拆分法的手性衍生化试剂研究进展

98 0
药 学 学 报 A t h r c uiaSnc 0 2,7( 1 :0 ca P amae t iia2 0 3 1 ) 9 8—9 2 c 1
高 效 液 相 色 谱 间接 拆 分 法 的 手 性 衍 生 化 试 剂 研 究 进 展
孙 贤祥
( 苏 石 油 化 工 学 院 化 【系 ,江 苏 常 州 2 3 1 ) 江 10 6
关键 词 : 性 衍 生 化 试 剂 ;对 图 分 类 号 :R 1 97 文献 标 识 码 :A 文章 编 号 :0 1 5 3—4 7 (0 2 1 —0 0 8 0 2 0 ) 1 9 8—0 5
现 代 药理 学研 究证 明 , 物 的手 性 与 药 效 有 密 药 切关系 , 药物 不 同光 学异 构体 进人 人 ( 或生 物 ) , 体 经 体 内受体 酶 、 载体 等 完 全 不 同 的分 子 处 理 所 引 起 的 药效 或 毒副 作 用 往 往 存 在 着 显 著 的 差 异 ~ 。随 着 人们 对 单一 光学 异构 体 高疗 效新 药 物产 品不 断增 长 的需 求 , 产 具有 高疗 效 低 副作 用 的光 学 异 构 体 新 生
酸对 映体 的手性 衍 生化 试 剂 。邻 苯 二 甲醛 和光 学纯
异 构体 的噻 吩 是拆 分 氨 基 酸 对 映 体 的 C R; 样 单 D 同
一
光学 异构 体 的氨 基 酸和邻 苯 二 甲醛也 可 作 为手性
噻 吩拆 分用 的 C R 这 样既 增 加 了 C R的 品种 , D 。 D 又 扩 大 了 C R 的 应 用 范 围 ;2 荧 光 C R 已 成 为 C R D () D D
足 的 进 展 , 现 有 以 下 特 点 : 1 利 用 衍 生 反 应 中 衍 表 ()
手性药物的拆分——高效液相色谱(HPLC)
SiO2
OH
(CH3O)3Si(CH2)3NH(CH2)2NH2 SiO2
O Si(CH2)3NH(CH2)2NH2
(1)
(OH)2
H3C
SO2Cl
(OTs)2
(2)
SiO2 O
n(NHR)
H2NR
SiO2 O
β-CD固定相改性过程
n(OTs)
1.5 刷型手性固定相
刷型手性固定相(CSP)是通过链烃基将手性有机 小分子链接到硅胶载体上制得,又叫Pirkle型手性固 定相。
刷型手性固定相的合成主要有两种途径,即含端羧 基或异氰酸酯基的手性基团的化合物与氨基键合硅 胶进行缩合反应,分别形成含酰胺型或脲型结构的 手性固定相:
OH
O
+
O
H
OH
OH
n
NCO
OCONH
H
NHOCO
O
O OCONH
n
3.纤维素-三(3,5-二甲基苯基氨基甲酸酯)(CDMPC)
商品名为手性OD柱,具有极高的光学拆分能力,是广泛使用的 手性固定相之一。以微晶纤维素与3,5-二甲基苯基异氰酸酯反 应,将生成的纤维素-三(3,5-二甲基苯基氨基甲酸酯)以 15%的量涂敷在氨丙基硅胶上,制得CDMPC-CSP:
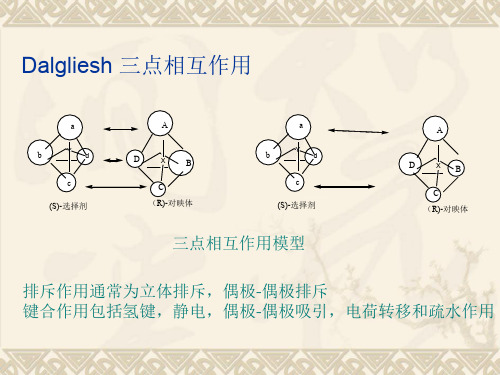
Dalgliesh 三点相互作用
a
b
Y d
c (S)-选择剂
A
D
X
B
C (R)-对映体
a
b
高效液相色谱法手性拆分和测定药片中华法林钠对映体

高效液相色谱法手性拆分和测定药片中华法林钠对映体王惠;王喜萍;双亚洲;李来生【摘要】采用衍生化纤维素手性柱,实现了华法林钠对映体的基线分离.优化的流动相组成为乙腈-0.1%甲酸(4060,v/v),流速为0.5mL·min-1,进样量为10μL,柱温为20℃,检测波长为308nm,拆分时间在15min内.在此基础上,建立了一种定量测定药片中华法林钠对映体含量的新方法.两对映体的浓度与响应信号之间呈良好的线性关系,线性范围均为0.5~250μg·mL-1(r=0.999 2).采用标准添加法测得药片中华法林钠两对映体的平均回收率范围为93.5%~101.1%(n=3),两对映体检出限均小于0.1μg·mL-1.该方法对华法林钠对映体的测定具有较高的选择性、灵敏度和重现性,在手性药物质量检测中有一定的应用前景.%The baseline separation of the warfarin sodium enantiomers was achieved using a derivatized cellulose chiral column.The optimized mobile phase consists of acetonitrile-0.1%formic acid(4060,v/v)at a flow rate of 0.5mL·min-1,a 10μL of injection volume,column temperature at 20°C and detection wavelength at 308nm within 15minutes.Based on the above condition,a new method for determination of the enantiomeric content of warfarin sodium in tablets was established.The good linear relationship between the concentration of enantiomers and the response signal were observed in the linear range of 0.5-250μg·mL-1(r=0.9992).The average recoveries of the two warfarin sodium enantiomers in the tablets ranged from 93.5%to 101.1%(n=3),respectively,and the detection limits of both enantiomers were less than 0.1μg·mL-1.The method has high selectivity,sensitivity andreproducibility,which has certain application prospects for the quality monitoring of chiral drugs.【期刊名称】《南昌大学学报(理科版)》【年(卷),期】2018(042)005【总页数】5页(P436-440)【关键词】高效液相色谱法;纤维素手性固定相;华法林钠;对映体含量;片剂【作者】王惠;王喜萍;双亚洲;李来生【作者单位】南昌大学化学学院,江西南昌 330031;南昌大学化学学院,江西南昌330031;南昌大学化学学院,江西南昌 330031;南昌大学化学学院,江西南昌330031【正文语种】中文【中图分类】O6571 引言手性拆分一直都是色谱领域的研究热点,已发展成为手性药物分析、食品分析及农药分析的重要手段[1]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
C S P 法优缺点
优点:分离时间短,而手性选择性和拆分能力
高,多数药物在分离前都不需要进行衍生化反应, 分 离方法直接。
局限性:色谱柱价格昂贵,部分固定相还存在稳
定性差,柱容量低,柱强度差等缺点,且根据不同手 性药物的性质不同,选用的分析方法也不同。
手性流动相( C M P ) 拆分法
又称手性添加剂法,这种拆分法是在流动相中加 入手性试剂,利用手性试剂与各对映体结合的稳定常 数不同,以及药物与结合物在固定相上分配系数的不 同来进行分离。
局限性:是手性试剂需要有高的光学纯度,各对映体的衍生
化速率及平衡常数应一致, 要求衍生化反应迅速、 彻底, 否 则影响定量结果。衍生化和色谱分析过程中应不发生消旋化, 外消旋药物需要有可被衍生化的基团, 此外衍生化法步骤较烦 琐, 衍生化试剂 绝大多数毒性相当大, 而且该方法难以实现 分析的自动化。
该法要求手性试剂及反应产物在化学性质上和 手性上很稳定,在反应及色谱条件下,试剂、手 性药物和反应产物不发生消旋化反应。手性试剂 应具有紫外或荧光吸收等敏感结构, 使生成物具 有良好的可检测性。
一些手性衍生化试剂和应用
CD F法优缺点
优点:可使用已有的非手性同定相, 花费少,通过选用具有
强烈紫外吸收或荧光吸收的手性试剂, 可提高检测敏感度, 而 且多数的衍生化试剂具有良好的对热及水的稳定性。
扑尔敏的拆分
色谱柱: 2 5 0× 4 .6 m m i .d ,(制备醋酸纤维素 酯类液相色谱固定相,涂渍在修饰硅胶表面), 采用 湿法自行装柱。
色谱条件:以正己烷 一异丙醇的混合溶液( 体积比 为9: 1 ) 作流动相, 流速为 0 . 5 mL / m i n ,柱 温3 0 ℃ ,紫外检测波长为2 5 4 n m。
结果表明:
此柱在分离药物扑尔敏时,随着进样量的增大, 此柱有些过载,分离效果不佳。一般来说,在分 离物进样过载的情况下,样品的分离效果较差, 随着进样量的降低,样品的分离更充分。
这是因为手性分离是 一个吸附和分配的过程, 在进样量过载的情况下, 没有足够的手性基团与 分离物进行作用 , 因而对映体的分离就不充分。
手性药物的高效液相色谱拆分 方法包括薄层色谱、气相色谱、液 相色谱、超临界流体色谱和电泳在 内的几乎所有色谱和准色谱手段都 已应用于对映体拆分。一般分为间 接法与直接法。
直接法
手性固定相( C S P ) 拆分法
手性流动相( C M P ) 拆分法
间接法
手性试剂衍生化法 ( CD F)
柱性能评价:用流动相冲洗该色谱柱, 直到基线稳 定. 以 1 , 3 , 5——叔 丁基苯测定死时间, 以 苯测其理论塔板数为 2 2 9 6 9 m—1 。
药物扑尔敏溶解在流动相溶剂中, 配制的 浓度为3 .2 mg/mL 。
扑尔敏的结构示意图1所示。
在上述色谱条件下,分别以0 . 4和0 .5uL的进样量进样分析
雷诺嗪的拆分
色谱柱:1 5 0 m m×4 .6 m m ,用纤维素 —三 ( 3 ,5 - 二甲基苯基氨基甲酸酯)湿法装柱。
色谱条件:以纯甲醇作流动相,流速为1 mL/min , 紫外检测波长为 2 5 4 n m。
分离效果:雷诺嗪对映体获得比较好的拆分效果, 分离度达到 1 .5 7 9 ,保留时间也比较短 ,7m i n 内即可拆分完毕。
蛋白质CSP 可识别药物对映体在蛋白质的结合 位点而达到手性分离
氨基酸、 乙内酰脲、 内酰脲、 胺类、 醇类及硫醇类药物对映
体 拆分 β一环糊精:适用于大多数药物 α一环糊精:适用于相对分子质
量小于200的药物 γ一环糊精:适用于较大相对分
子质量药物 主要用于分离巴比妥类药物、 麻醉药和抗抑郁药等
可直接分离许多药物,如硫喷 妥因、 心得怡及阻滞剂等
CSP种类
作用机理
应用
Pirkle型 CSP
通过含末端羧基或异氰酸酯基手 性前体与氨基键合硅胶进行缩合反 应, 分别形成含酰胺或脲型结构
CSP
环糊精CSP
环糊精的手性识别主要来自环内腔 对芳烃或脂 肪烃类侧链的包容作 用, 以及环外壳上的羟基与药物对 映体分子发生氢键作用
多糖类CSP
主要来自氨基甲酸酯或酯的部位与 被拆分物之间形成氢键或偶极 ~偶 极作用, 或通过被拆分物分子进入 纤维索网状腔, 导致腔内立体环境 的改变而实现
随着流速的下降, 分离度逐渐升高。
通过控制流速、流动相等方法对雷诺嗪的 拆分进行了下述研究。
通过虽然上法雷诺嗪得到非常好的拆分, 但是拆分时间过短,容量因子k太小(k<1),且 无法根据需要调节。
于是考察醇类系统对雷诺嗪的手性分离的 影响。
优点:此法不需昂贵的手性柱,亦无须进行柱
前衍生,手性添加剂可视要求而更换,使用比较方 便。
其中主要应用的有:配体交换型手性添加剂、环 糊精添加剂、手性离子对添加剂。
手性试剂衍生化法 ( CD F)
该法是药物对映体在分离前与高光学纯度衍生化试 剂( C D A) 反应, 形成非对映体, 再进行色谱分离 测定。
手性固定相( C S P ) 拆分法
手性固定相是由担体键合高光学纯度的手性异构体制作 而成,检测物在手性固定相表面形成非对映体对,根据其稳 定常数不同而获得分离。
用于色谱分离的手性固定相已经被大量研究,已有100多 种液相色谱固定相被商业化。目前所研究的高效液相色谱手 性固定相有7大类。
其中主要应用的有:Pirkle型CSP、环糊精CSP、多糖类 CSP、蛋白质CSP 。