视网膜色素变性遗传规律
视网膜色素变性

疾病解读
• 佳学基因的基因解码专家给李先生进行了细致的报告解读, KLHL7基因突变导致的视网膜色素变性为常染色体显性遗传,其 后代遗传风险为50%。
色素视网膜色素变性的遗传方式
• 二是要考虑年龄因素,一般医学上界定35岁以上为高龄,高龄女 性怀孕,胎儿染色体异常风险增加,妊娠期并发症风险也会增加。
• 不过,女性年龄并非是一个绝对的限制因素,一些40多岁的女性, 如果身体健康,有强烈生育要求,也可以尝试。
• 3、性连锁遗传型视网膜色素变性的异常基因位于X染色体上,因 此主要影响男性,如果父亲是患者,母亲正常,则儿子全部正常, 女儿全部是异常基因携带者。
• 从概率上来看,由于带有异常基因和不带异常基因的常染色体是 随机的传给下一代的,因此下一代受影响的概率不是全或无,也 不是绝对的一半对一半,而是从无到全部都有可能发生,大约是 50%,与性别无关。
色素视网膜色素变性的遗传方式
• 因此在有些家庭,常染色体显性遗传型视网膜色素变性可存在于 连续的许多代,有的家庭只存在于连续的两代。
• 一家人辗转看过很多大医院,寻求治疗的同时,还想知道为什么 会患上这个病,如何让后代不遗传?终于,在他们了解到视网膜 色素变性致病基因鉴定的时候,才解开这个家族三代遗传的谜团。
分析结果
• 佳学基因对李先生的血液样本进行了全面的视网膜色素变性致病 基因分析,基因解码显示,李先生KLHL7基因发生杂合致病性突 变(c.449G>A,p.Ser150Asn)。
视网膜色素变性
前言
什么是视网膜色素变性会带来哪些症状

什么是视网膜色素变性会带来哪些症状在我们的眼睛这个精妙的器官中,视网膜扮演着至关重要的角色。
然而,有一种疾病会悄悄地侵袭视网膜,给我们的视力带来严重的影响,那就是视网膜色素变性。
视网膜色素变性,这是一种遗传性的眼部疾病,它会导致视网膜中的感光细胞逐渐受损和死亡。
简单来说,视网膜就像是我们眼睛内的“底片”,而感光细胞则是这张“底片”上的关键元素。
当这些感光细胞出现问题时,我们的视力也就会随之出现障碍。
那么,视网膜色素变性会带来哪些具体的症状呢?夜盲往往是视网膜色素变性最早出现的症状之一。
在昏暗的环境中,比如夜晚或者光线较暗的地方,患者会发现自己的视力明显下降,难以看清周围的物体。
这是因为视网膜中的视杆细胞,它们对于光线较弱的环境特别敏感,而在视网膜色素变性中,视杆细胞往往是最先受到损害的。
所以,当夜幕降临,其他人能够正常行走和活动时,患者可能会感到步履维艰,甚至需要依靠他人的帮助。
随着病情的进展,患者的视野会逐渐缩小。
想象一下,我们的视野原本就像一个宽阔的全景画面,但由于视网膜色素变性,这个画面好像被一只无形的手从四周向中心挤压。
患者可能会发现自己只能看到正前方的一小部分物体,而周围的景象则变得模糊不清,甚至完全看不见。
这种视野的缩小会严重影响日常生活,比如过马路时无法看到两侧来车,在房间里容易碰撞到家具等等。
视力下降也是不可避免的症状之一。
一开始,可能只是轻微的视力模糊,但随着时间的推移,视力会越来越差,最终可能导致失明。
这对于患者的生活和工作无疑是巨大的打击,读书、看报、看电视等日常活动都会变得异常困难。
除了上述这些主要的症状,视网膜色素变性还可能引起其他一些问题。
例如,患者对颜色的感知能力可能会下降,原本鲜艳的色彩在他们眼中变得暗淡无光。
还有,眼睛对强光的敏感度可能会增加,在阳光明媚的日子里,患者可能会感到眼睛刺痛、不适。
视网膜色素变性的症状通常是逐渐加重的,而且这个过程可能非常缓慢,以至于患者在早期很难察觉到明显的变化。
视网膜色素变性的遗传方式与早期症状

视网膜色素变性的遗传方式与早期症状*导读:视网膜色素变性病因不明,是一种视网膜神经上皮的原发性遗传性疾病。
由于视网膜外层组织,特别是视细胞层的原发性进行性营养不良及逐渐退变和消失,使视网膜色素上皮细胞及神经胶质增生所致。
随着病情的进展,视网膜血管外壁增厚,以致发生闭塞性血管硬化。
……
视网膜色素变性是一种遗传性疾病。
有三种遗传方式,即常染色体隐性遗传、常染色体显性遗传以及性连锁隐性遗传。
临床上以常染色体显性遗传与常染色体隐性遗传较为多见,性连锁隐性型少见,但损害最为严重。
临床上也有不少病例没有遗传证据。
视网膜色素变性患者早期自觉症状为夜盲,而且夜盲每多发生在视网膜出现色素病变之前。
此时检查可发现暗适应减退,视野可表现为部分的或环状暗点,这主要是由于视网膜的赤道部首先出现环状退行变性之故。
患者中心视力早期不受影响。
199视网膜色素变性的发病原因是什么,多发生于什么年龄?
视网膜色素变性病因不明,是一种视网膜神经上皮的原发性遗传性疾病。
由于视网膜外层组织,特别是视细胞层的原发性进行性营养不良及逐渐退变和消失,使视网膜色素上皮细胞及神经胶质增生所致。
随着病情的进展,视网膜血管外壁增厚,以致发生闭塞性血管硬化。
中医学认为本病的发生为先天禀赋不足,肝肾脾虚血滞,血脉枯
涩,目失所养,以致神光衰微,夜不见物,视野缩窄。
本病的发病年龄,一般多发生于10岁左右或10~20岁的青年,也有的发生较早。
视网膜色素变性症状
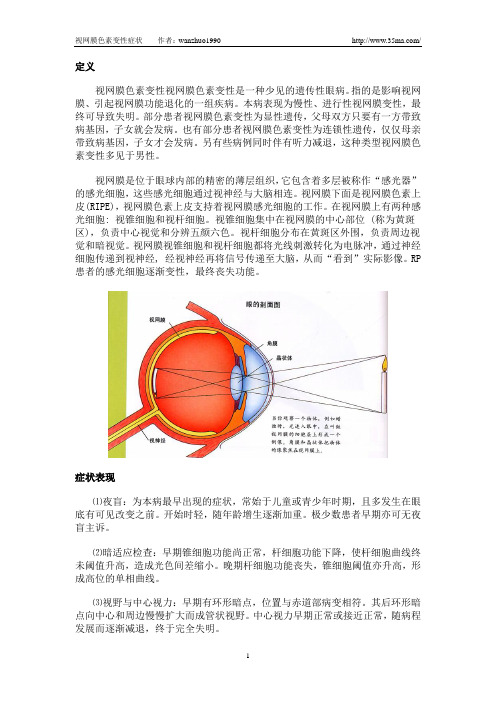
定义视网膜色素变性视网膜色素变性是一种少见的遗传性眼病。
指的是影响视网膜、引起视网膜功能退化的一组疾病。
本病表现为慢性、进行性视网膜变性,最终可导致失明。
部分患者视网膜色素变性为显性遗传,父母双方只要有一方带致病基因,子女就会发病。
也有部分患者视网膜色素变性为连锁性遗传,仅仅母亲带致病基因,子女才会发病。
另有些病例同时伴有听力减退,这种类型视网膜色素变性多见于男性。
视网膜是位于眼球内部的精密的薄层组织,它包含着多层被称作“感光器”的感光细胞,这些感光细胞通过视神经与大脑相连。
视网膜下面是视网膜色素上皮(RIPE),视网膜色素上皮支持着视网膜感光细胞的工作。
在视网膜上有两种感光细胞: 视锥细胞和视杆细胞。
视锥细胞集中在视网膜的中心部位 (称为黄斑区),负责中心视觉和分辨五颜六色。
视杆细胞分布在黄斑区外围,负责周边视觉和暗视觉。
视网膜视锥细胞和视杆细胞都将光线刺激转化为电脉冲,通过神经细胞传递到视神经, 经视神经再将信号传递至大脑,从而“看到”实际影像。
RP 患者的感光细胞逐渐变性,最终丧失功能。
症状表现⑴夜盲:为本病最早出现的症状,常始于儿童或青少年时期,且多发生在眼底有可见改变之前。
开始时轻,随年龄增生逐渐加重。
极少数患者早期亦可无夜盲主诉。
⑵暗适应检查:早期锥细胞功能尚正常,杆细胞功能下降,使杆细胞曲线终未阈值升高,造成光色间差缩小。
晚期杆细胞功能丧失,锥细胞阈值亦升高,形成高位的单相曲线。
⑶视野与中心视力:早期有环形暗点,位置与赤道部病变相符。
其后环形暗点向中心和周边慢慢扩大而成管状视野。
中心视力早期正常或接近正常,随病程发展而逐渐减退,终于完全失明。
⑷视觉电生理:ERG无反应,尤其b波消失是本病的典型改变,其改变常早于眼底出现改变。
EOG LP/DT明显降低或熄灭,即使在早期,当视野、暗适应、甚至ERG等改变尚不明显时,已可查出。
故EOG对本病诊断比ERG更为灵敏。
⑸色觉:多数患者童年时色觉正常,其后渐显异常。
rp遗传规律

RP遗传规律简介RP(Retinitis Pigmentosa),又称视网膜色素变性症,是一种遗传性眼疾,主要影响视网膜中的感光细胞。
它是一组异质性疾病,具有复杂的遗传规律。
本文将详细介绍RP的遗传规律及相关机制。
遗传模式RP可通过不同的遗传模式传递给后代,主要包括常染色体显性遗传、常染色体隐性遗传、X连锁遗传等。
常染色体显性遗传常染色体显性遗传指患者只需从一个患有RP的父母那里继承到一个异常基因就能表现出疾病。
每个患者都有50%的机会从一个患有RP基因突变的父母那里继承到该突变基因。
常染色体隐性遗传常染色体隐性遗传指患者需要从父母双方都继承到异常基因才能表现出疾病。
通常情况下,两个健康人士作为携带者,他们各自携带一个突变基因。
每个子代患RP 的几率约为25%。
X连锁遗传X连锁遗传是指RP基因位于X染色体上,男性只需要继承一个异常基因就能表现出疾病,而女性需要继承两个异常基因。
因为男性只有一个X染色体,所以男性患RP的几率更高。
基因突变RP可以由多种基因突变引起,目前已经发现了50多种与RP相关的基因。
这些基因编码着视网膜中的关键蛋白质,在感光细胞的正常功能中起着重要作用。
当这些基因突变时,会导致感光细胞受损甚至死亡,最终导致视力丧失。
遗传咨询与诊断由于RP具有复杂的遗传规律和多样化的致病基因,对于家族中有RP患者或可能成为携带者的人群来说,进行遗传咨询和遗传检测非常重要。
遗传咨询遗传咨询是指通过向家族成员提供相关信息和建议,帮助他们了解遗传疾病的风险和遗传规律,以便做出更好的决策。
遗传咨询可以提供有关家族史、遗传风险评估、遗传测试等方面的信息。
遗传检测遗传检测是通过分析个体的DNA样本,寻找与RP相关的基因突变。
目前已经开发出多种遗传检测方法,包括基因测序、基因芯片等。
通过遗传检测,可以确定是否携带RP相关的基因突变,并为患者和家族提供更具体的风险评估和建议。
治疗与研究进展目前尚无治愈RP的方法,但一些治疗措施可以延缓其进展并改善患者的生活质量。
视网膜色素变性的原因

视网膜色素变性的原因文章目录*一、视网膜色素变性的简介*二、视网膜色素变性的原因*三、视网膜色素变性的危害*四、视网膜色素变性的高发人群*五、视网膜色素变性的预防方法视网膜色素变性的简介原发性视网膜色素变性(RP),也称为毯层视网膜变性视网膜变性,是一种进行性、遗传性、营养不良性退行性病变,主要表现为慢性进行性视野缺失,夜盲,色素性视网膜病变和视网膜电图异常,最终可导致视力下降。
视网膜色素变性的原因本病为遗传性疾病。
其遗传方式有常染色体隐性、显性与X性连锁隐性三种。
以常染色体隐性遗传最多;显性次之;性连锁隐性遗传最小。
双基因和线粒体遗传也有报道。
生物化学研究发现氨基乙磺酸缺乏可引起猫产生视网膜色素变性,氨基乙磺酸是一种含硫氨基酸,在视网膜中含量很高,有可能是一种神经介质因子,起保护作用。
铜、锌和维生素A异常也可能与RP发病有关。
也有越来越多的证据表明体液免疫和细胞免疫参与RP的发病。
Brinkman发现视网膜色素上皮变性患者的淋巴细胞可被牛的视网膜视杆细胞外节盘和人类可溶性视网膜抗原激活。
其他的免疫系统异常包括T淋巴细胞减少,免疫抑制能力下降及血清免疫球蛋白水平升高。
Hooks发现RP患者外周淋巴细胞缺乏产生γ-干扰素的能力。
因此认为RP可能是一种自身免疫性疾病。
视网膜色素变性的危害视网膜色素变性可引发青光眼,多为宽角,闭角性少见。
有人从统计学角度研究,认为青光眼是与本病伴发而非并发症。
约有50%的病例伴有近视。
近视多见于常染色体隐性及性连锁性隐性遗传患者。
亦可见于家族中其他成员。
一般从儿童青少年期就发病,有些到成年才发病,临床上有夜盲盲,视野逐渐缩小,视力逐渐下降,眼底可见视盘蜡黄,视网膜污浊,血管变细,骨细胞样色素沉着,晶体后囊浑浊,玻璃体浑浊,视神经萎缩等并发症,最后导致失明。
视网膜色素变性的高发人群遗传性疾病有显性遗传和隐性遗传之分。
显性遗传的特点是患者的父母至少有一方是该病患者,所生子女至少有1/2出现该病。
视网膜色素变性遗传规律

视网膜色素变性遗传规律 Prepared on 22 November 2020视网膜色素变性遗传规律石家庄眼底病治疗中心赵井志主任视网膜色素变性(retinalpigmentosa,RP)属于光感受器细胞及视网膜色素上皮(retinalpigmentepithelium,RPE)营养不良性退行性病变,是一种最常见的遗传性致盲眼病。
其临床主要特征:夜盲,进行性视野缩小,色素性视网膜病变,视盘呈蜡黄色萎缩及光感受器功能不良。
典型表现为视网膜色素沉着呈骨细胞样改变。
该病有多种遗传方式,可为性连锁隐性遗传,常染色隐性遗传或显性遗传,也可散发。
一般在30岁以前发病,最常见于儿童或青少年期起病,至青春期症状加重,到中年或老年时因黄斑受累视力严重障碍而失明。
此病在我国的患病率为三千七百八十四分之一,到目前为止还是一个“不治之症“。
RP的发病机制较为复杂,目前主要是基因突变。
基因突变的研究已成为当今生命科学研究的热点之一,检测方法也随之迅速发展。
对于基因突变的检测,经典方法是根剧基因的分离规律,更进一步是在分子水平:印迹法,脱氧核苷酸序列测定分析法,现在主要是运用聚合酶链反应。
随着对人类基因组研究的深入,基因芯片的出现使突变检测变得规模化和程序化。
视网膜色素变性有明显的遗传异质性,报道遗传方式主要有常染色体显性遗传、常染色体隐性遗传、X---连锁遗传和双基因型4种遗传方式。
临床上多见的是前三种类型。
在我国的隐性遗传站91-95%。
显性和性连锁比较少,特别是显性遗传只占3-5%。
视网膜色素变性患者都知道遗传,但是怎样能粗略的了解遗传,赵井志给大家介绍一下患者自己判断的方式:一、视网膜色素变性三种遗传的特点①显性遗传:父母一方有显性基因,一经传给下代就能发病,即有发病的亲代,必然有发病的子代,而且世代相传。
父母之一有病时,子代约有50%发病,父母正常时,子代不会发病,两性发病比率相等。
②隐性遗传:之所以称隐性遗传病,是因为患儿的双亲外表往往正常,但都是致病基因的携带者。
视网膜变性

视网膜变性一概述视网膜变性(retinal degeneration), 属于视锥、视杆营养不良,是一组遗传病,以夜盲、视野缩小、眼底骨细胞样色素沉着和光感受器功能不良为特征。
性连锁隐性遗传、常染色体隐性或者显性遗传均可以见到,也有散发的。
目前包括药物、手术在内的治疗手段对视网膜变性均只有改善、缓解作用。
二病因部分患者视网膜色素变性为显性遗传,父母双方只要有一方带致病基因,子女就会发病。
也有部分患者视网膜色素变性为性连锁遗传,仅仅母亲带致病基因,子女才会发病。
三鉴别诊断1.白点状视网膜变性视力正常或稍减退,可有周边视野向心性缩小,眼底特点是在视网膜上布满大小一致的白点,这些白点多位于视网膜血管后深层。
2.梅毒性脉络膜视网膜炎有梅毒病史或临床表现,可有视物变形,玻璃体混浊,眼底可见色素的分布和形态不规则,且位于视网膜血管下,看不到骨细胞样色素形态,可见视网膜脉络膜萎缩斑。
3.结晶样视网膜变性本病多在20~40岁发病,其眼底在较长时间里视神经乳头颜色正常。
视网膜血管正常,病史长者动脉可变细,后极部网膜污灰色,散布着很多结晶样,闪辉的亮晶,愈近黄斑区越密集,视网膜色素为暗褐色和不规则形,晚期脉络膜血管绽露,呈硬化状,部分患者有色觉障碍。
四检查1.视网膜色素沉着2.视网膜血管改变血管一致性狭窄,随病程进展而加重,尤以动脉为显著。
3.荧光血管眼底造影所见背景荧光大片无荧光区,提示脉络膜毛细血管层萎缩。
视网膜血管可有闭塞,有时还可见到后极部或周边部斑驳状荧光斑。
五治疗原则1.有试用血管扩张剂、维生素A及B1、组织疗法、各种激素、中草药、针灸等方法,或可避免视功能迅速恶化。
2.检查视网膜色素变性和黄斑变性患者的视网膜发现,他们的视网膜叶黄素含量比正常人低得多,实验证明,通过补充叶黄素可以增加视网膜的色素密度。
叶黄素除了可以保护视网膜免受光线和自由基的损害,还具有血管扩张的功能,打断血管狭窄进程,软化和扩张血管,改善眼底微循环。
什么是视网膜色素变性如何预防和治疗

什么是视网膜色素变性如何预防和治疗在我们的眼睛这个神奇的器官中,视网膜起着至关重要的作用,它就像是一部超级相机的底片,负责接收和处理外界的光线信息,让我们能够看清这个五彩斑斓的世界。
然而,有一种疾病却会悄悄地侵蚀视网膜的功能,那就是视网膜色素变性。
视网膜色素变性是一种遗传性、进行性的视网膜退行性病变。
简单来说,就是视网膜中的一些细胞逐渐失去正常的功能,导致视力逐渐下降,甚至最终失明。
这种疾病通常在儿童或青少年时期就开始出现症状,但早期可能并不明显。
最初,患者可能会感觉到在黑暗环境中视力变差,比如走夜路时看不清。
随着病情的发展,周边视野会逐渐缩小,就像通过一个狭窄的管道看东西,只能看到正前方的一小部分,而周围的景象变得模糊或看不见。
再往后,中心视力也会受到影响,阅读、看电视等日常活动变得越来越困难。
那么,为什么会发生视网膜色素变性呢?这主要与遗传因素有关。
目前已经发现了许多与视网膜色素变性相关的基因,如果家族中有人患有这种疾病,那么其他亲属患病的风险就会增加。
此外,环境因素、免疫因素等也可能在疾病的发生和发展中起到一定的作用。
既然视网膜色素变性如此可怕,我们能不能采取一些措施来预防它呢?遗憾的是,由于其主要由遗传因素决定,目前还没有特效的预防方法。
但是,如果您的家族中有视网膜色素变性的患者,建议您尽早进行眼科检查,尤其是基因检测,以便早期发现和诊断。
对于已经确诊为视网膜色素变性的患者,治疗是至关重要的。
虽然目前还无法完全治愈这种疾病,但通过一些治疗方法,可以延缓病情的进展,提高生活质量。
药物治疗是常见的方法之一。
一些营养神经、改善视网膜血液循环的药物,可能有助于保护视网膜细胞的功能。
例如,维生素 A 棕榈酸酯可以在一定程度上减缓病情的恶化,但使用时需要注意剂量,过量可能会引起中毒。
基因治疗是近年来研究的热点。
通过修复或替代有缺陷的基因,有望从根本上治疗视网膜色素变性。
虽然这项技术还处于实验阶段,但已经取得了一些令人鼓舞的成果。
视网膜色素变性初期如何判定

• 郑女士的医生告诉她,典型的视网膜色素变性最早的症状是夜盲, 白天在户外活动时和正常一样,但是到黄昏时在户外和室内光线 暗淡时则看不清,寸步难行,多数始于少年时期。
视网膜色素变性
前言
• 视网膜色素变性是一种遗传性很高的眼科疾病,早期会出现夜盲 的症状,但是并不是所有患者都会出现夜盲,随着病情的发展, 暗适能力退化,怕光,视力悄然下降,视野缩小,最后呈管状视 野甚至失明。对于有家族史的人群来说,该如何早期明确自己是 否遗传到了该病?
临床表征
• (1)夜盲 为本病最早出现的症状,常始于儿童或青少年时期,且多发生在眼底有可见改 变之前。开始时轻,随年龄增生逐渐加重。
• (2)暗适应检查 早期锥细胞功能尚正常,杆细胞功能下降,使杆细胞曲线终末阈值升高, 造成光色间差缩小。晚期杆细胞功能丧失,锥细胞阈值亦升高,形成高位的单相曲线。
• (3)视野与中心视力 早期有环形暗点,位置与赤道部病变相符。其后环形暗点向中心和 周边慢慢扩大而成管状视野。中心视力早期正常或接近正常,随病程发展而逐渐减退,一 般照明下,当周边视野全部丧失后,中心视野尚存5°~10°,患者处于管视状态。最后中 心视野也渐丧失,终于完全失明。RP自然病程中,视野每年损失4.6%。
分析患病原因
• MERTK基因是常染色体隐性遗传疾病视网膜色素变性38型的致病基因,其典 型症状是夜盲,视野逐渐缩小,视力减退。经验证,郑女士未遗传到该致病 性突变,因此不会发病,由于郑女士没有携带致病基因,她的后代也不会 隔代遗传到该致病基因。如此消除了一家人一直以来对于可能遗传这一难治 疾病的恐惧。
视网膜色素变性汇总

视网膜色素变性的药物治疗研究进展视网膜色素变性(RP)是一组视网膜进行性营养不良性退行病变,大部分是遗传性疾病。
病理过程首先是视杆细胞的损害,其后是视锥细胞亦受到损害。
感光细胞的损害主要是通过凋亡进行的,细胞神经营养因子的缺乏是凋亡发生的一个重要原因。
RP的实质是基因缺陷导致的感光细胞及视网膜色素上皮的变性,从而引起夜盲、进行性视野缩小、视力下降、眼底特征性改变及视网膜电图波形振幅减低或熄灭。
RP是发达国家最主要的致盲原因之一,根据世界各地的调查资料,全球人口群体患病率约为1/3000~1/5000(1990年爱尔兰第六届RP会议报道)。
据此估计,全球的RP患者约有150万人,在中国约有40万人。
RP多于幼年或青春期发现,常双眼发病,也有病变仅发生在单眼者,具有遗传倾向。
本病遗传学分型可分为常染色体显性遗传、常染色体隐性遗传、X性连锁隐性遗传及散发型。
临床上以常染色体隐性遗传与常染色体显性遗传较为多见。
RP的目前主要治疗方法可分为:药物治疗(包括神经营养因子)、基因治疗、视网膜移植手术治疗等。
视网膜移植手术治疗有视网膜感光细胞、干细胞、色素上皮细胞及虹膜色素上皮细胞的移植,其治疗的技术不成熟,容易产生严重并发症,疗效亦不确切。
十余年来,数个国家的一百多例人类临床(手术)移植研究却发现术后效果并不明显[1-2],这使国际临床眼科界对用视网膜(细胞)移植手术治疗视网膜色素变性的兴趣大减,从而将研究重点转向如何延缓光感受器细胞变性、延长病人有用视力时间的临床方法研究。
基因治疗在理论上应该是最理想的治疗方法,但由于RP具有遗传异质性及多效性,因此基因改变与视网膜色素变性临床表现之间的关系非常复杂。
目前还难以阐明这种关系[3],使对该病的基因治疗难以找到切入点,因此对该病的基因治疗难以得到有关机构的批准,何况已有临床基因治疗导致病人死亡的报导[4-5],因此在解决基因治疗的切入点及安全性问题以前,临床上对视网膜色素变性的基因治疗还不现实。
视网膜色素变性分布情况回顾性文献分析_董玉萍

视网膜色素变性(Retinitis Pigmentosa,RP)于1857年由Donder[1]首次发现并命名,RP患病率在全球范围内呈逐年上升趋势。
据报道,2001年全世界患病人数约100万人,近五年来,患病人数已达150万余人。
目前已成为威胁全世界中青年人群视觉功能的主要眼部疾病。
随着分子生物学技术的发展,对与RP相关易感基因的研究不断深入,目前已发现70余个,其中,SAG、TULP1、RDS、PRPF31基因与RP的关系已成为分子遗传学领域的研究主要关注点。
通过查阅文献对就RP的分布情况进行汇报。
1RP患者的人群分布特点由于X性连锁遗传(XLRP)的系谱特征[2]为:家系中至少两代男性患者,父母都健康,父亲不携带致病基因,母亲是携带者,母亲的致病基因只能遗传给儿子,而常染色体隐性、显性遗传的系谱特征之一为男女患病比例均等[3],所以RP患者在统计学上男性多于女性[4],男女比例为1.03∶1[5]。
XLRP 决定男性多于女性,发病特点为发病年龄早,进展速度快,是青年男性致盲的主要原因之一[6]。
近亲结婚导致RP为常染色体隐性遗传性RP(ARRP),即遗传特点为隔代遗传,患者双亲没有临床症状,但是携带致病基因,患者同胞中患病比例约各占25%,致病基因携带者占50%,与性别无关,近亲结婚子代易发病,发病率高达1/2000[7]。
本病隐性遗传者,其先辈多有近亲联烟史,禁止近亲联烟可使本病减少发生约22%。
常染色体显性遗传RP (ADRP)者,其子女发生本病约50%。
遗传性RP 患者一般在青少年时期出现夜盲症状,40~50岁时出现视力的损害[8]。
出现RP明显症状的一般集中在30~40岁年龄层,至50岁左右接近全盲,可能与基因表达时间有关。
2RP患者基因突变分布特点由于中国和西方人群在遗传背景、生活环境上的存在着一定的差异,加之种族及地域的不同,不同民族、不同地区的人群可能具有完全不同的突变类型。
视网膜色素变性与遗传因素的关系研究

视网膜色素变性与遗传因素的关系研究视网膜色素变性(Retinitis Pigmentosa,RP)是一组遗传性眼病的统称,其特征是视网膜神经节细胞和视杆细胞的进行性死亡和退化。
RP是一种非常罕见的疾病,但它的遗传性质使得它在某些人群中有较高的发生率。
本文将探讨视网膜色素变性与遗传因素的关系,并介绍一些相关的研究成果。
视网膜色素变性是一种遗传性疾病,目前已经发现与RP相关的遗传突变共有1000余种。
大多数RP病例呈现出常染色体显性遗传的模式,但也存在其他遗传模式,如常染色体隐性遗传、X连锁遗传等。
这些遗传突变可以导致视网膜色素变性相关基因的异常表达或功能缺陷,进而引发视网膜细胞的死亡和退化。
通过近年来进行的大量研究,已经鉴定出许多视网膜色素变性相关基因。
其中最常见的基因突变包括RHO、RPGR、PRPH2等。
这些基因突变会影响视网膜细胞的正常功能,导致视网膜逐渐丧失对光信号的感知和处理能力,从而造成患者在视觉上的受限。
研究表明,RP的发生率在不同的人群中有着显著的差异。
欧洲和北美等地的人群中RP的发生率相对较低,约为1/4000至1/5000,而亚洲地区,尤其是日本、中国和韩国等东亚国家,RP的发生率较高,约为1/1000至1/2000。
这种地域差异可能与不同人群中的遗传突变有关。
除了地域差异外,RP的遗传规律也显示出一定的复杂性。
虽然多数RP病例呈现出常染色体显性遗传的模式,但也存在其他遗传模式。
例如,常染色体隐性遗传模式的RP发生率相对较低,约占RP病例的15%至20%。
此外,X连锁遗传模式的RP主要发生在男性患者中,而女性患者则往往是带有突变基因的携带者。
对于RP的遗传机制的研究已经取得了一些进展,但仍有许多问题需要进一步研究。
例如,尽管已经鉴定出许多与RP相关的基因,但仍有一部分RP病例无法找到明确的遗传突变。
这表明还存在其他未知的遗传因素参与了RP的发生和发展。
此外,研究人员还研究了RP的遗传变异对疾病临床表现的影响。
视网膜色素变性是怎么回事?

视网膜色素变性是怎么回事?*导读:本文向您详细介绍视网膜色素变性的病理病因,视网膜色素变性主要是由什么原因引起的。
*一、视网膜色素变性病因本病为遗传性疾病。
其遗传方式有常染色体隐性、显性与性连锁隐性三种。
以常染色体隐性遗传最多;显性次之;性连锁隐性遗传最小。
目前认为常染色体显性遗传型至少有两个基因座位,位于第一号染色体短臂与第三号染色体长臂。
性连锁遗传基因位于X染色体短壁一区一带及二区一带。
关于发病机制,近20~30年中,有了一些瓣的线索。
根据电镜、组织化学、电生理、眼底血管荧光造影等检查资料推测,认为本病的发生,主要由于视网膜色素上皮细胞对视细胞外节盘膜的吞噬、消化功能衰退,致使盘膜崩解物残留、规程形成一层障碍物,妨碍营养物质从脉络膜到视网膜的转动,从而引起视细胞的进行性营养不良及逐渐变性和消失。
这个过程已在一种有原发性视网膜色素性的RCS鼠视网膜中得到证实。
至于色素上皮细胞吞噬消化功能衰竭的原因,目前还不清楚。
可能与基因异常,某种或某些酶的缺乏有关。
在免疫学方面,近年研究发现本病患者体液免疫、细胞免疫均有异常,玻璃体内有激活的T细胞、B细胞与巨噬细胞,视网膜色素上皮细胞表达HLA-DR抗原,正常人则无此种表现。
同时也发现本病患者有自身免疫现象,但对本病是否有自身免疫病尚无足够依据。
在生化方面,同时也发现本病患者有自身免疫现象,但对本病是否有自身免疫病尚无足够依据。
在生化方面,发现本病患者脂质代谢异常,视网膜中有脂褐质的颗粒积聚;锌、铜、硒等微量元素及酶代谢亦有异常。
综上所述,本病可能存在着多种不同的发病机理。
*温馨提示:以上就是对于视网膜色素变性病因,视网膜色素变性是由什么原因引起的相关内容叙述,更多有关视网膜色素变性方面的知识,请继续关注疾病库,或者在站内搜索“视网膜色素变性”找到更多扩展内容,希望以上内容可以帮助到您!。
视网膜色素变性的病因

视网膜色素变性的病因
眼睛是我们人类主要的感觉器官之一,如果我们的眼睛出现了问题,那么将给我们带来多方面的麻烦,所以大家在日常的生活中一定要保护好自己的眼睛才行,视网膜色素变性是眼睛疾病里面的一种,视网膜色素变性容易导致我们出现夜盲等症状,所以大家一定要了解一下视网膜色素变性的病因才行。
原发性视网膜色素变性(RP),也称为毯层视网膜变性视网膜变性,是一种进行性、遗传性、营养不良性退行性病变,主要表现为慢性进行性视野缺失,夜盲,色素性视网膜病变和视网膜电图异常,最终可导致视力下降。
本病为遗传性疾病。
其遗传方式有常染色体隐性、显性与X 性连锁隐性三种。
以常染色体隐性遗传最多;显性次之;性连锁隐性遗传最小。
双基因和线粒体遗传也有报道。
生物化学研究发现氨基乙磺酸缺乏可引起猫产生视网膜色
素变性,氨基乙磺酸是一种含硫氨基酸,在视网膜中含量很高,有可能是一种神经介质因子,起保护作用。
铜、锌和维生素A异常也可能与RP发病有关。
也有越来越多的证据表明体液免疫和细胞免疫参与RP的发病。
Brinkman发现视网膜色素上皮变性患者的淋巴细胞可被牛的视网膜视杆细胞外节盘和人类可溶性视网膜抗原激活。
其他的免疫系统异常包括T淋巴细胞减少,免疫抑制能力下降及血清免疫球蛋白水平升高。
Hooks发现RP患者外周淋巴细胞缺乏产生γ-干扰素的能力。
因此认为RP可能是一种自身免疫性疾病。
在上面的文章里面我们介绍了眼睛对于人体的重要作用,我们建议读者朋友在日常的生活中一定要做好眼睛的护理工作,视网膜色素变性是一种眼睛疾病,上文为我们分析了视网膜色素变性的病因。
视网膜色素变性的分子遗传学研究进展

c r c e ie y g a ha a t rz d b r dua ge r to fpho or e p o c is Comm o ci c lf a ur s i l d o e s v os f lde ne a n o i t e e t r e l. n l a e t e nc u e a pr gr s i e l s o ni
A w t d o r s f M olc a n tc n Au o o Ne S u y Pr g e s o e ul r Ge e i s i t s mal
D o iLeabharlann a tR e i ts Pi m e t s m n n tnii g noa
TEN G t t W AN G t Ya + r Ha t
t p mm n fMe i l e eis T n ”Mp ia olg t De a e t o d c n tc . o g a G d c l l e H ̄ z o g U ie st C e h n nv r i y o ce c n e h oo y, , a 3 0 0 C ia fS in ea d T c n lg W t n 4 0 3 , h n ) h
视 网 膜 色 素 变性 (e nt ime ts, P 是 由 于 光 感 rd ispg no& R ) [ 受 器 ( 杆 细胞 和 视锥 细 胞 ) 视 网 膜 色素 上 皮 的异 常 而 导 视 或 致 的进 行 性 失 明 的一 组 遗 传 性 疾 病 患 者 首 先 表 现 暗 适 应 障碍 或夜 盲 , 后 出 现 视 野 由 外 周 向 中 心 逐 谇 雏 小 直 至 失 随 i 明 。RP的 发 病 率 为 13 0 ~ 17 0 , 我 国 发 病 率 为 l /0 0 / 0 0 在
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
视网膜色素变性遗传规律
石家庄眼底病治疗中心赵井志主任
视网膜色素变性(retinal pigmentosa,RP)属于光感受器细胞及视网膜色素上皮(retinal pigment epithelium,RPE)营养不良性退行性病变,是一种最常见的遗传性致盲眼病。
其临床主要特征:夜盲,进行性视野缩小,色素性视网膜病变,视盘呈蜡黄色萎缩及光感受器功能不良。
典型表现为视网膜色素沉着呈骨细胞样改变。
该病有多种遗传方式,可为性连锁隐性遗传,常染色隐性遗传或显性遗传,也可散发。
一般在30岁以前发病,最常见于儿童或青少年期起病,至青春期症状加重,到中年或老年时因黄斑受累视力严重障碍而失明。
此病在我国的患病率为三千七百八十四分之一,到目前为止还是一个“不治之症“。
RP的发病机制较为复杂,目前主要是基因突变。
基因突变的研究已成为当今生命科学研究的热点之一,检测方法也随之迅速发展。
对于基因突变的检测,经典方法是根剧基因的分离规律,更进一步是在分子水平:印迹法,脱氧核苷酸序列测定分析法,现在主要是运用聚合酶链反应。
随着对人类基因组研究的深入,基因芯片的出现使突变检测变得规模化和程序化。
视网膜色素变性有明显的遗传异质性,报道遗传方式主要有常染色体显性遗传、常染色体隐性遗传、X---连锁遗传和双基因型4种遗传方式。
临床上多见的是前三种类型。
在我国的隐性遗传站91-95%。
显性和性连锁比较少,特别是显性遗传只占3-5%。
视网膜色素变性患者都知道遗传,但是怎样能粗略的了解遗传,赵井志给大家介绍一下患者自己判断的方式:
一、视网膜色素变性三种遗传的特点
①显性遗传:父母一方有显性基因,一经传给下代就能发病,即有发病的亲代,必然有发病的子代,而且世代相传。
父母之一有病时,子代约有50%发病,父母正常时,子代不会发病,两性发病比率相等。
②隐性遗传:之所以称隐性遗传病,是因为患儿的双亲外表往往正常,但都是致病基因的携带者。
③性链锁遗传又称伴性遗传发病与性别有关,如血友病,其母亲是致病基因携带者。
又如红绿色盲是一种交叉遗传儿子发病是来自母亲,是致病基因携带者,而女儿发病是由父亲而来,但男性的发病率要比女性高得多。
二、视网膜色素变性遗传的判定方法
①“无中生有为隐性,女儿患病为常隐”
②“有中生无为显性,女儿正常为常显”
③“母患子必患,女患父必患,男性患者多于女性”――最可能为“X隐”(女病男必病)
④“父患女必患,子患母必患,女性患者多于男性”――最可能为“X显”(男病女必病)
⑤“父传子,子传孙,子子孙孙无穷尽,无女性患者”――最可能为“伴Y”(男性全为患者)
视网膜色素变性患者遗传复杂,其中每个家系的遗传方式也可能不同,目前在国际上已经明确视网膜色素变性基因位点有38个,其中常染体显性遗传有17个,常染体隐性遗传有16个。
性连锁遗传有5个。
已经克隆了视网膜色素变性致病基因27个,其中常染色体显性遗传有14个,常染体隐性遗传11个,性连锁遗传你有两个
也就是说已经克隆的致病基因在专业的基因检测机构是可以检测的,这样给很多RP解除困惑,即使不能全面的检测,但一样是科技的进步,至少为患者的病程改善提供机会。