唯一医疗器械标识(UDI)系统及GUDID账户的获取精编版
深度解读医疗器械唯一标识附医疗器械分类名录

深度解读医疗器械唯一标识附医疗器械分类名录医疗器械作为支持医务人员进行医疗诊断和治疗的重要工具,扮演着至关重要的角色。
然而,由于医疗器械的多样性和复杂性,对其管理和监管提出了严峻的挑战。
为了更好地管理和追溯医疗器械的使用情况,许多国家和地区引入了医疗器械唯一标识制度。
本文将深入解读医疗器械唯一标识以及附带的医疗器械分类名录,帮助读者全面了解这一系统的运作机制和意义。
医疗器械唯一标识(Unique Device Identification, UDI)是指为每个医疗器械分配的具有独特识别信息的代码。
UDI由一系列特定的标识符组成,包括设备识别码、版本标识、制造商标识和生产日期等信息。
通过这一制度,医疗机构可以准确地追溯和识别每个医疗器械的来源、生产批次、有效期等关键信息。
UDI的引入有效地增强了医疗器械的可追溯性,提供了更加精准的安全监管手段,为患者的用药安全和医疗机构风险管理提供了有力支持。
为了更好地利用UDI系统,许多国家和地区制定了医疗器械分类名录。
医疗器械分类名录是按照功能和风险等级将医疗器械进行分类的清单。
它为医疗器械的备案、监管和使用提供了便利,有助于加强对不同类别医疗器械的针对性管理。
医疗器械分类名录通常包括品名、功能、适应症、风险等级等信息,使得相关部门和医疗机构能够更加全面地了解和管理医疗器械。
UDI系统和医疗器械分类名录在医疗器械管理中的作用不可忽视。
首先,UDI系统和医疗器械分类名录有助于提高医疗器械的安全性。
通过精确地识别和追溯医疗器械,可以及时发现和处理可能存在的质量问题,减少患者用药事故的发生。
其次,UDI系统和医疗器械分类名录有助于减少假冒医疗器械的流通。
通过对唯一标识的认证和验证,可以有效地防止假冒医疗器械的进入市场,保障患者的用药安全和治疗效果。
第三,UDI系统和医疗器械分类名录有助于提高医疗器械供应链的管理效率。
通过准确记录和追踪医疗器械的运输和使用情况,可以更好地预防和处理供应链中的问题,确保医疗器械供应的及时性和可靠性。
医疗器械唯一标识(UDI)系统必知要点

医疗器械唯一标识(UDI)系统必知要点医疗器械UDI医疗器械FDA化妆品FDAVCRP化妆品CPNP 通报1、医疗器械唯一标识,并不是医疗器械行业近几年新诞生的要求,而在ISO 13485体系中本身就有可追溯性要求的要素,但从行业发展的观点看,UDI是全球性的医疗器械唯一标识系统,也是全球医疗器械市场国际化发展的必然趋势。
2、企业实施UDI系统是一项战略,必先有专业人员懂的UDI相关内容,及如何实施,更让老板不爽的是,要投入巨额的money!我们医疗器械从业人员学习为先!3、企业要一步达到UDI的要求有难度,但可以根据质量管理体系及UDI的相关规则,做好全面落实的基础。
转变思想,积极应对是良策,也是上上策。
4、据医疗器械从业者的了解,国内某些大医院已趋向GS1产品唯一标识编码,而不是HIBCC编码。
5、本文是超长文,需要有点耐心才能读完。
本文从UDI的定义、UDI的范围和要求、FDA的授权机构、UDI的重要性、UDI在各国的进展、FDA实施UDI的时间表,及实施UDI的困难与挑战,七个方面进行综述,供医疗器械从业人员的爱好者学习,借鉴。
-----------------------------一、什么是UDI?Unique device identification (UDI) 是美国食品药物管理局FDA 建立的”特殊医疗器械的识别系统”,该注册码的实施是为了有效识别在美国市场上销售并使用的医疗器械,无论其生产地在哪里。
一旦实施,NHRIC和NDC标签将废止,所有的医疗器械都需要将这个新的注册码作为标识贴在产品的外包装上。
除了满足可见(visible)之外,UDI必须满足纯文本形式和自动识别技术(automatic identification and data capture, AIDC). 器械的标签负责人员也必须将每个产品的确实信息发送到“FDA国际特殊医疗器械识别库UDID”内,使公众可以通过访问该数据库查询并下载相关数据(包括从生产,分销到客户使用情况的信息等),但该数据库不会提供器械使用者的信息。
医疗器械FDA UDI 实施规定以及指南教程

中国制造商如何实施美国FDA UDI 唯一医疗器械标识码要求1.美国FDA UDI 医疗器械标识编码医疗器械标识编码要求综述要求综述2013年,FDA 发布了建立医疗器械独特识别符系统的法规,目的是确保医疗器械在分销和使用的全流程都可加以识别追溯。
大多数医疗器械产品的标签以及包装上应包括可供肉眼及机器识别的唯一器械识别符(UDI )。
美国食品药品管理局(FDA )对唯一器械标识UDI 实行分阶段执行政策,依据相关规定在2016年9月24日前所有进入美国市场II 类医疗器械的标签和包装必须带有一个UDI ;且必须在全球唯一器械标识数据库(GUDID)中录入II 类医疗器械的数据。
FDA 规定的实施时间期限如下表: 强制强制日期日期执行要求 2014年9月24日开始- 所有FDA Class III 第三类医疗器械;FDA PHS Act 法案管制的器械,都必须在器械的标签和包装上标注FDA UDI 信息,采用FDA 规定的日期格式并及时报送FDA GUDID 数据库; - 如果特定企业需要申请延期执行UDI ,务必在2014年6月23日向FDA 递交申请; - FDA Class III Stand-alone Software 被FDA 作为第三类别管制的独立软件必须提供UDI 信息 2015年9月25日开始- Implantable, life-supporting, and life-sustaining devices 植入式医疗器械,用于生命支持和维持的医疗器械的标签和包装都必须标注UDI 信息,并采用FDA 规定的日期格式; - 如果上述器械为重复使用器械并在使用前可被再处理,那么必须进行永久性UDI 标识; - 用于生命支持或维持器械的独立软件必须标识UDI ; - 上述器械UDI 数据必须报送FDA GUDID 数据库; 2016年9月24日开始- UDI 管制的Class III 可重复使用并且在使用前可以被再处理的医疗器械都需要进行永久性UDI 标识; - FDA Class II 二类器械的产品标签和包装必须标识UDI ,并采用FDA 规定的日期格式;FDA Class II Stand-alone Software 被FDA 作为第二类别管制的独立软件必须提供UDI 信息; - FDA Class II 二类器械的UDI 数据以及产品关键数据必须报送FDA GUDID 数据库; 2018年9月24日开始 - FDA Class II 可重复使用的并且使用前可以被再处理的FDA 管制的二类器械必须进行永久性UDI 标识;- FDA Class I 一类医疗器械和未被划分级别Class I, II 或III 的器械都必须标识UDI ;所有这些器械,包含豁免UDI 的器械,日期标注都必须符合UDI 法规;- 上述器械的UDI 信息必须及时报送FDA GUDID 数据库;- FDA Class I 一类的Stand-alone software 独立软件必须提供UDI;2020年9月24日开始 - 所有FDA Class I 一类器械和未被分类为Class I, II, III 的器械,如果可以重复使用或使用前可以被再处理,都必须在产品上进行永久性UDI 标注;注意:在这个强制日期前生产以及贴好标签的器械, 可以不需要执行FDA UDI 要求,这个豁免只能在生效强制日期后三年内采纳。
医疗器械唯一标识(UDI)管理制度

医疗器械唯一标识(UDI)管理制度本规范的目的是为了明确医疗器械唯一标识的管理和使用,确保公司对产品的管理和使用符合法规规定。
适用范围为以公司品牌生产销售的医疗器械产品。
医疗器械唯一标识(UDI)是医疗器械在其整个生命周期中赋予的身份标识,是其在产品供应链中的唯一“身份证”。
产品标识是UDI的固定和强制性部分,包含贴标者信息和产品型号。
生产标识是UDI的可变和非强制性部分,根据监管和实际应用需求可包括医疗器械序列号、生产批号、生产日期、失效日期。
UDI的数据载体主要有三种形式:一维条码、二维条码、射频标签(RFID)。
无论采用哪种形式,UDI的结构与编制方法不变。
人工可识文本HRI是对符合标准结构和格式的数据所采用的文本表示,是对数据载体中载入信息的人工可识读解释,其内容完全嵌入在数据载体中。
人工可识补充文本Non-HRI与HRI类似,但表达的文本信息嵌入在GS1数据载体中,不局限于基于GS1标准的结构和格式。
通常展示的文本信息是对GS1数据载体内信息的补充。
条码标签一般包括三个部分,即条码数据载体、HRI和Non-HRI。
如图1所示,展示了包含这三个部分的二维码标签。
提供公司营业执照副本原件及复印件;提供公司组织机构代码证原件及复印件;提供公司税务登记证原件及复印件;提供公司授权委托书及授权人身份证原件及复印件;提供公司产品清单及相关资料。
5.1.2.2申请续展的资料要求:填写完整的《中国商品条码系统成员证书续展申请表》1份并加盖公司公章;提供有效期内的《中国商品条码系统成员证书》原件及复印件;提供公司产品清单及相关资料。
5.1.2.3申请变更的资料要求:填写完整的《中国商品条码系统成员证书变更申请表》1份并加盖公司公章;提供有效期内的《中国商品条码系统成员证书》原件及复印件;提供公司产品清单及相关资料。
5.1.2.4申请增码的资料要求:填写完整的《中国商品条码系统成员证书增码申请表》1份并加盖公司公章;提供公司产品清单及相关资料。
《医疗器械唯一标识数据接口规范》解读

《医疗器械唯一标识数据接口规范》解读一、标准起草的背景(一)背景2021年10月14日,深圳市市场监督管理局举行深圳市医疗器械唯一标识(以下简称“UDI”)追溯平台启动仪式,这是全省首个UDI追溯平台,具有重要示范意义。
UDI是医疗器械的“身份证”,可实现全生命周期医疗器械通查通识,是目前医疗器械追溯管理最好的方法,在国际上被广泛使用。
国家药监局联合国家卫健委、国家医保局于2019年开始启动UDI试点工作,近两年来,陆续进行了制定UDI系统规则、建立UDI数据库等框架性工作,逐步明确UDI 实施工作将扩展到所有第三类医疗器械产品(含体外诊断试剂),并鼓励其他品种医疗器械实施UDI。
另外,一些省市已针对第二类医疗器械产品开展UDI推进工作,依据产品类别分步实施UDI的进程又向前迈了一步,将对医疗器械全生命周期各环节质量监管带来深远影响。
为进一步推进我市UDI实施工作,深圳市市场监督管理局依托智慧监管的良好基础,在深圳市标准技术研究院的技术支持下搭建了“深圳市医疗器械唯一标识(UDI)追溯平台”,为UDI实施提供技术支撑。
平台通过UDI编码串联各环节信息,直观展示了深圳市生产经营使用医疗器械产品的追溯链条,从生产厂家到产品供应商,直至使用到患者身上,都可一一动态呈现,清晰直观地展现了UDI的全生命周期流向。
在前期推进UDI 工作中,深圳先行先试,积极贯彻和落实相关文件和指示精神要求,在国家要求的基础上,扩大实施品种和生产企业数量,同时选取信息化建设完善的医院作为试点,打造生产到使用端的数据闭环。
在平台运行期间,已经采集了我市257家生产企业的61103条产品信息,对接了我市13家三甲医院超27万条人次的患者使用信息,工作取得阶段性成效。
(二)标准意义为更好促进其他平台建立与“深圳市医疗器械唯一标识(UDI)追溯平台”的互联互通,在深圳市卫生健康委员会、深圳市医疗保障局以及相关试点医疗机构等单位的支持下,拟研制《医疗器械唯一标识数据接口规范》地方标准,用来指导第三方(生产经营企业、医疗机构等)平台如何开展与“深圳市医疗器械唯一标识(UDI)追溯平台”的数据对接,未来平台还将实现与深圳阳光采购平台以及医保耗材编码映射等数据对接工作。
医疗器械唯一标识(UDI)专题培训的资料

FDA
UDI UDI 基础
二维码举例
注* 本页信息来自GS1 https:///sites/d efault/files/docs/healthcare /UDI-Label-Samples.pdf
2021/2/2
FDA UDI
2021/2/2
FDA UDI 时间 节点
美国已认可了3 家发证机构: 国际 物品编码协会( globe standard 1, GS1) 、保健业商务通信委员会 ( Health Industry Business Communications Council , HIBCC) 、国际血库 自动化委员会( International Council for Commonality in Blood Bank Automation , ICCBBA) , 首次认可期限为3 年。
医疗器械唯一标识 (UDI)专题培训的资料
2021/2/2
唯一器械标识(Unique Device Identification,缩写UDI)是对医疗器械在其 整个生命周期赋予的身份标识,是 其在产品供应链中的唯一“身份证”。全球 采用统一的、标准的UDI有利于提高供应链透明度和运作效率;有利于实现信息 共享与交换;有利于不良事件的监控和问题产品召回,提高医疗服务质量,保 障患者安全。
MDR于2017年5月26日正式生效,并于2020年5月26日正式取代MDD (93/42/EEC)和AIMDD(90/385/EEC)。
MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并 保持证书的有效性。过渡期内NB签发的CE证书继续有效至2024年5月27日 失效。
2021/2/2
FDA 系统中。 q GUDID 中大部分数据属性均标注在了医疗器械标签上,两者表述应一致。 q GUDID 数据元素医疗器械识别联系人应该给FDA 提供关于贴标者和产品标签上的16 项
实施医疗器械唯一标识(UDI)系统

FDA提议实施医疗器械唯一标识(UDI)系统医疗器械上市后,尤其是高风险的医疗器械的追溯管理、监督是全球性的难题。
为消除隐患,从患者的安全着想,用全球统一的医疗器械命名和唯一标识对医疗器械进行跟踪和追溯成为必要。
医疗器械唯一标示(Unique Device Identification, UDI)的定义是指,根据国际或等同转换的国家物品编码标准系统,采用数字或文字数字表示的代码。
这个代码按照医疗器械追溯的要求构成,在全球范围以内,是一个特定的医疗器械的唯一标识,用于识别上市后需要追溯的医疗器械产品,并可以作为进入相关数据库的钥匙,获取与之关联的特定医疗器械信息。
UDI是目前全球各国协调作为解决上市后特定医疗器械有效追溯,保障病人利益的有效途径。
为了积极推动UDI,GHTF于2008年5月建立了UDI问题的特别工作组织(Ad Hoc Working Group UDI, AHWG)并发布了相关的协调指导文件。
2012年7月,FDA提议对美国境内的大部分医疗器械采用UDI系统。
FDA 认为医疗器械的安全性是其最优先的考虑对象,UDI系统能够提高医疗器械不良事件报告的效率,使FDA更快速地鉴别产品的问题、更有针对性地解决医疗器械的召回从而保护病人的安全。
目前,FDA针对该项提案已经开展了一些先期研究,并且正在与业界、临床机构、病人以及消费团体进行紧密地合作以确保该项提案能够顺利通过。
在FDA的该项提案中,一项UDI应包括:* 一个器械的识别码,该识别码针对特定的器械模组是独有的数字或字母编码;* 一个产品识别码,对某一器械来说,该识别码包括了当前的产品信息。
UDI含有了器械的基本识别信息,例如制造商的名字、器械的类别,同时也可能提供其他的特定信息,如过期日期和批号等。
信息将被保存在可供公众查询的UDI数据库中,但数据库并不包括可识别的病人信息。
FDA提出UDI系统应基于风险管理、在实施步骤上逐步递进,范围从高风险医疗器械逐渐扩大到低风险医疗器械。
《医疗器械唯一标识系统规则》解读

《医疗器械唯一标识系统规则》解读一、什么是医疗器械唯一标识?医疗器械唯一标识由产品标识和生产标识组成,产品标识是识别注册人/备案人、医疗器械型号规格和包装的唯一代码,是从数据库获取医疗器械相关信息的“关键字”,是唯一标识的必须部分;生产标识包括与生产过程相关的信息,包括产品批号、序列号、生产日期和失效日期等,可与产品标识联合使用,满足医疗器械流通和使用环节精细化识别和记录的需求。
唯一标识具备唯一性、稳定性和可扩展性的原则。
唯一性是首要原则,是确保产品精确识别的基础,是唯一标识发挥功能的核心原则。
由于医疗器械产品的复杂性,唯一性应当与产品识别要求相一致,对于相同特征的医疗器械,唯一性应当指向单个规格型号产品;对于按照批次生产控制的产品,唯一性指向同批次产品;而对于采用序列号生产控制的医疗器械,唯一性应当指向单个产品。
稳定性是指唯一标识一旦分配给医疗器械产品,只要其基本特征没有发生变化,产品标识就应该保持不变。
当医疗器械停止销售、使用时,其产品标识不得用于其他医疗器械;重新销售、使用时,可使用原产品标识。
可扩展性是指唯一标识应当与监管要求和实际应用不断发展相适应,“唯一”一词并不意味着对单个产品进行序列号化管理,在唯一标识中,生产标识可以和产品标识联合使用,实现规格型号、批次和单个产品三个层次的唯一性,从而满足当前和未来对医疗器械的识别需求。
二、为什么要建设医疗器械唯一标识系统?医疗技术、药品、医疗器械是构成医疗服务体系的三大支柱,医疗器械涉及声、光、电、磁、图像、材料、力学等行业和近百个专业学科,是国际公认的高新技术产业,具有高技术密集、学科交叉广泛、技术集成融合等特点,代表着一个国家高新技术的综合实力。
近年来,医疗器械产业发展迅猛,新技术、新产品层出不穷,产品多样性、复杂性程度不断提升,医疗器械在流通使用环节无码或者一物多码现象普遍,严重影响了医疗器械生产、流通、使用等各环节对医疗器械的精准识别,难以实现有效监督和管理。
唯一医疗器械标识系统及GUDID账户的获取

唯一医疗器械标识系统及GUDID账户的获取唯一医疗器械标识(Unique Device Identification,UDI)系统是由美国食品药品监督管理局(FDA)引入的一项标准,旨在确保医疗器械的唯一性和可追溯性,进一步提高医疗器械的安全性和有效性。
UDI系统要求制造商为其产品分配一个唯一的标识码,并将该标识码与关键信息存储在全球唯一设备标识数据库(GUDID)中,以供各方访问和查询。
获取UDI系统和GUDID账户的步骤如下:1.了解UDI系统:在开始注册UDI系统和GUDID账户之前,首先需要了解UDI系统的基本原理、法规要求和使用方法。
可以通过FDA的官方网站、相关培训课程或指南来获取必要的信息。
3.创建FDA账户:在进行UDI系统和GUDID账户注册之前,首先需要在FDA的官方网站上创建一个账户。
填写必要的信息,确认账户的有效性。
6.GUDID账户激活:FDA审核通过审核GUDID账户申请后,会发送一封包含GUDID账户激活链接的电子邮件。
点击链接,按照指引设置GUDID账户的密码和安全信息。
8.添加产品信息:在GUDID账户中,可以通过手动方式或者利用GUDIDXML导入功能添加产品信息。
确保填写的产品信息准确、完整、符合FDA的要求。
9.提交产品信息:在添加产品信息后,需要验证和审查信息的正确性。
确认无误后,点击提交按钮,将产品信息存储至GUDID数据库。
10.维护产品信息:定期检查GUDID账户中的产品信息,并根据需要进行必要的更新和维护操作。
如更改产品规格、添加新的产品版本等。
总之,要获取UDI系统和GUDID账户,需要按照相关的要求准备材料,创建FDA账户,注册UDI系统,申请GUDID账户,并经过审核和激活等流程。
获取账户后,可以通过GUDID账户管理产品信息,确保医疗器械的唯一性和可追溯性。
《医疗器械唯一标识系统规则》解读
2019年10月1日起实施
制定依据
为规范医疗器械唯一标识系 统建设,加强医疗器械全生 命周期管理,根据《医疗器 械监督管理条例》,制定本 规则。
使用范围
在中华人民共和国境内销售、 使用的医疗器械,其唯一标 识系统应当符合本规则。
定义
本规则所称医疗器械唯一标 识系统,由医疗器械唯一标 识、唯一标识数据载体和唯 一标识数据库组成。
缺点:载体成本和识读设 备成本相较于一维码和二 维码要高
数据载体使用要求
责任主体:注册人/备案人 要求: ① 选择与其创建的医疗器械唯一标识相适应的数据
载体标准。 ② 对以其名义上市的医疗器械最小销售单元和更高
级别的包装赋予唯一标识数据载体。 ③ 或者医疗器械产品上赋予唯一标识数据载体。 ④ 确保在医疗器械经营使用期间唯一标识数据载体
态维护 每年1月31日前向国家药品监督管理局提交按照其
标准创建的唯一标识上一年度的报告
唯一标识数据载体的要求
应当满足自动识别和数据采集技术以及人工识读的 要求。如空间有限或者使用受限,应当优先采用符 合自动识别和数据采集技术的载体形式。
自动识别和数据采集技术 (常用数据载体)
一维码
可将产品标识和生产标识串 联,也可多行并联
二维码
射频标签(RFID)
应当同时具备一维码或者二 维码
如何选择数据载体
注册人/Байду номын сангаас案人可根据产品的特征、 价值、主要应用场景等因素选择 适当的医疗器械唯一标识数据载 体。
选择适当的数据载体
一维码
优点:只在一维方向上表 示信息的条码符号,应用 多年已经很成熟,成本低, 能很好兼容市面上现有的 扫码设备。
国际医疗器械监管者论坛《唯一医疗器械标识系统》介绍及对我国启示

国际医疗器械监管者论坛《唯一医疗器械标识系统》介绍及对我国启示中国科技论文在线////0>.国际医疗器械监管者论坛《唯一医疗器械标识系统》介绍及对我国启示*刘清峰5 (上海医疗器械高等专科学校管理系,上海市 200093)摘要: 文章在阐述唯一医疗器械标识的概念及其重要性的基础上,介绍了国际医疗器械监管者论坛《唯一医疗器械标识拟定规则》的主要内容,提出了我国应尽快制定 UDI系统建设规划,指出建立我国的 UDI 系统应结合国际医疗器械监管者论坛倡导的《UDI System forMedical Device》拟定规则,并与医疗器械监管的其他职能结合起来。
10 关键词:卫生事业管理;UDI;介绍;启示中图分类号:R19International Medical Device Regulators Forum "UniqueDevice Identification Proposed Rule"15 and Its Enlightenment toChinaLIU QingfengManagement Department,Shanghai Medical Instrumentation College,Shanghai 200093Abstract: In this paper, based describing on the concept and its importance of unique medical device20 identification,it introduced the main content of the proposed rule about UDI System for MedicalDevice from International Medical Device Regulators Forum. The plan of the UDI system in ourcountry should be proposed as soon as possible. It pointed out that the establishment of UDI system inChina should be combined with UDI System for Medical Device from International Medical DeviceRegulator Forum and with other functions of the medical device regulatory affaires25 Key words: Health Management; UDI, introduce, enlightenment0 引言UDI是英文 Unique Device Identification的缩写,通常中文翻译为“医疗器械唯一标识”。
医疗器械唯一标识UDI知识分享

大多数医疗器械企业选择GS1标准
1、中国物品编码中心,GS1
项目 DI
PI
代码 01 11 17 10 21
GS1应用标识
含义 产品标识 生产日期 失效日期 批号 序列号
格式 数字 数字(年月日) 数字(年月日) 字母+数字 字母+数字
数字长度 14位 6位 6位 不固定 不固定
2. 医疗器械唯一标识组成
UDI
DI
产品标识
生产商根据标准规定的编码 规则生成,以确保全球范围 内的唯一性
数据库维护
PI
生产标识
生产商的唯一标识符,它是 由全球唯一的组织机构分配 的代码
可选项
2、UDI 的特点
发展史
3、推进历程
第三批,
第二批, 2022年6月1日
2024年6月1日 部分第二类医疗器械
1.监管加强:有利于构建智慧监管体系,提升监管能力。 2.产业升级:助力医疗器械产业转型升级和高质量发展。 3.公众受益:为公众提供更安全、高效的医疗服务,增强获得感。 4.全球合作:有助于国际间医疗器械监管的合作与互认。
谢
Zhu
da
谢
!
jia
haihaipipi
3、价值与意义
实施医疗器械唯一标识(UDI)管理,不仅满足了相关法规的严格要求,从而显著降低了潜在的法 律风险,而且还通过赋予每个医疗器械产品独一无二的识别码,实现了对产品从生产到使用全过 程的精准追踪。这种强大的追踪管理能力极大地增强了医疗器械的质量控制和风险管理能力,确 保了医疗器械的安全性和可靠性
3、阿里健康科技(中国)有限公司,AHM
标识符
业务含义
MF
医疗器械唯一标识udi查询
医疗器械唯一标识UDI从2021年正式实施至今,所有第三类医疗器械已全部纳入,并于2023年逐步向第二类医疗器械覆盖,医疗器械赋码已成为必然。
那什么是医疗器械唯一标识码UDI?有什么编码规则呢?医疗器械唯一标识数据库有什么作用?在哪里可以实现UDI码在线查询?很多人对此不甚了解,今天借此机会盘点一下医疗器械唯一标识码UDI 的相关知识。
一、医疗器械唯一标识码(UDI码)是什么意思?有什么含义?医疗器械唯一标识、医疗器械唯一标识数据载体和医疗器械唯一标识数据库共同组成医疗器械唯一标识系统。
官方定义来说,医疗器械唯一标识(unique device identification,UDI)是指在医疗器械产品或者包装上附载的,由数字、字母或符号组成的代码,具有唯一标志性。
简单来说,医疗器械产品包装上除了常见的商品条码以外的另外一个标志UDI的编码符号(一维码、二维码或射频识别RFID标签),就是医疗器械唯一标识码,也称UDI码,用于编号与产品的配对识别。
医疗器械唯一标识数据载体,是指医疗器械唯一标识码的媒介表现形式,目前以一维码、二维码、射频标签三种形式为主。
1.医疗器械唯一标识数据库则是由NMPA建设直管的,用于储存医疗器械唯一标识及相关信息的数据库,所有的UDI码都需上传至数据库内进行动态备案维护。
数据信息面向公众开放,支持查询下载。
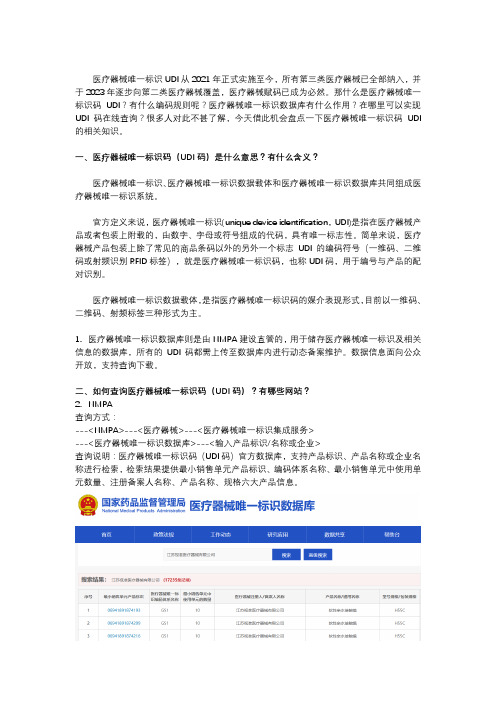
二、如何查询医疗器械唯一标识码(UDI码)?有哪些网站?2.NMPA查询方式:---<NMPA>---<医疗器械>---<医疗器械唯一标识集成服务>---<医疗器械唯一标识数据库>---<输入产品标识/名称或企业>查询说明:医疗器械唯一标识码(UDI码)官方数据库,支持产品标识、产品名称或企业名称进行检索,检索结果提供最小销售单元产品标识、编码体系名称、最小销售单元中使用单元数量、注册备案人名称、产品名称、规格六大产品信息。
basic udi di和udi di的编码规则

basic udi di和udi di的编码规则基本UDI-DI(Unique Device Identification - Device Identifier)和UDI-DI的编码规则是医疗器械行业提出的一系列标识编码的要求和规则。
这些编码规则旨在确保医疗器械能够被精确地标识和追踪,以便在全球范围内提高患者的安全性和医疗质量。
本文将详细介绍基本UDI-DI和UDI-DI的编码规则,包括其定义、标准和实施步骤。
首先,我们来了解一下基本UDI-DI和UDI-DI的定义。
基本UDI-DI是指唯一标识设备的基本识别字符,其由制造商根据一系列规则和标准进行编码。
UDI-DI是基本UDI-DI的组成部分,包括标记设备的产品代码、制造商代码和一位校验和码。
这些标识编码可以是数字、字母或符号的组合,以确保每个医疗器械都具有唯一的标识。
接下来,我们将介绍UDI-DI的编码规则。
UDI-DI的编码规则由国际卫生技术标准组织(ISO)制定的标准和指南来指导。
根据ISO 15459标准,UDI-DI应遵循以下规则:1. 制造商代码:由Global Unique Device Identification Database (GUDID)分配给制造商的唯一代码,通常由FDA(美国食品和药物管理局)负责分配。
2. 产品代码:用于标识特定型号、规格或变种的设备,通常由制造商自主定义。
3. 校验和码:用于验证UDI-DI的正确性,以避免录入错误或误读。
校验和码是根据UDI-DI中其他字符生成的,以确保其准确性。
UDI-DI的编码规则确保了每个医疗器械都具有独一无二的标识,使其可以在全球范围内准确地进行追踪和识别。
这对于药物库存管理、质量控制、产品召回以及患者安全都非常重要。
最后,我们来看一下UDI-DI的实施步骤。
在实施UDI-DI之前,制造商需要先获得GUDID分配的制造商代码。
然后,根据ISO 15459标准,制造商可以自主定义产品代码,并使用校验和算法生成校验和码。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3月22日,由医保商会和美国FDA驻华办公室共同主办的“美国FDA医疗器械UDI规程专题研讨会”在北京举办,来自中国国家食品药品监督管理总局和出口美国的医疗器械企业代表
参加了研讨会。
会议由医保商会蔡天智副秘书长主持,许铭副会长和美国FDA驻华办公室主任古丽分别致辞,介绍了中美在医疗器械领域的合作,探讨如何加强监管机构和产业的对话交流,共同促进质量体系、产品标准、编码分类等相关工作,从而为公众提供安全有效的医疗产品。
FDA驻华办公室助理主任倪可,FDA总部交流教育办公室行业与消费者教育处副处长威廉∙
萨顿就输美的II类医疗器械产品如何进行UDI注册进行了详细解读。
根据要求,输美II类医疗器械须在2016年9月24日之前完成网上UDI登记系统,有关准备工作包括研究UDI网站上的相关内容、选择UDI签发机构并将UDI加印在医疗器械上、选择主要的数据提交方式、对数据进行收集、了解GUDID账户的结构、找到/获取邓氏码、获取GUDID账户、提交器械标识数据、订阅GUDID系统邮件及时获知申请状态等工作。
日前,FDA驻华办专门约见我会,就如何帮助中国企业了解政策最新变化和要求进行了讨论。
本次会议为监管机构和产业提供了很好的面对面交流的机会,会议PPT我们在此向大家公布。
同期录像后续也将为大家提供在线链接以供观看,敬请关注!企业如果在操作过程中遇到具体问题,欢迎随时联系商会。
联系人:陈婧婧:md@。