色谱分析中各种图谱现象的判断
色谱测纯度的依据-概述说明以及解释

色谱测纯度的依据-概述说明以及解释1.引言1.1 概述色谱测纯度是一种常用的分析方法,广泛应用于化学、生物、药学等领域。
通过色谱技术,可以快速、准确地分离和分析混合物中的组分,进而评估样品的纯度和质量。
色谱测纯度的依据主要是基于分离原理和分析结果的解读。
色谱技术的基本原理是利用样品中组分在固定相和流动相之间的差异,通过分离过程获得纯度信息。
在色谱柱中,通常有一相是固定的,如固定在柱内壁上的固定相;另一相是流动的,如流经柱内的溶剂。
当样品通过色谱柱时,不同组分会因其与固定相的相互作用力不同而以不同的速率运动,从而达到了分离的目的。
色谱测纯度的方法主要包括定性分析和定量分析两个方面。
定性分析主要通过比对待测样品与已知标准样品的色谱图谱或数据,通过比对特征峰的保留时间和形状来判断样品中是否存在特定的化合物。
而定量分析则是通过测定特定峰的高度或面积,与已知浓度的标准曲线进行对比,从而计算出样品中目标物质的含量。
在色谱测纯度的分析中,一般会引入一些依据进行验证和判定。
常用的依据包括对比样品的单一组分,峰形的对称性和尖峰度,保留时间的稳定性,以及重测结果的一致性等。
这些依据可以帮助分析人员判断样品中是否存在杂质,以及样品的纯度程度。
总之,色谱测纯度是一种基于分离技术的分析方法,通过分析样品中不同组分在色谱柱中的运动差异,实现对混合物的分离和纯度评估。
在分析过程中,我们可以依靠一系列的依据来验证样品的纯度程度,以获得准确可靠的结果。
1.2文章结构文章结构部分的内容可以按照以下方式编写:文章结构部分的目的是介绍整篇文章的组织结构,以帮助读者更好地理解文章的内容和思路。
本文以色谱测纯度的依据为主题,共分为引言、正文和结论三个部分。
引言部分将概述本文的主题和内容,并介绍文章的结构。
首先,我们将对色谱技术的基本原理进行简要介绍,包括不同类型的色谱技术及其原理。
接下来,我们将重点探讨色谱测定纯度的方法,包括色谱图的解读、峰定量和标准曲线的建立等。
高效液相色谱中异常峰分析

异常峰分析异常的色谱峰指的是色谱图中的无峰或出现负峰、宽峰、双峰、肩峰、峰形不对称等情况。
异常峰是色谱实验工作中最棘手的问题。
这些峰严重影响色谱分析的结果。
色谱图中不可能有纯正的高斯对称峰 , 轻微的拖尾是正常的 , 这是由分离系统所决定的。
在此仅对几种异常峰进行分析。
1.峰前或峰后有小峰的分析产生原因大致分为以下几种情形(1) 样品不纯。
可改变不同的流动相和色谱柱 ,对样品进行分离比较 , 选择合适的分离条件。
(2) 分析柱或保护柱柱头塌陷。
此情况较常见 ,可更换分析柱或保护柱后对峰形进行比较。
柱头塌陷时往往所有的峰都会出现峰分裂。
对色谱柱再生和清洗可以改善分离效果。
(3) 色谱柱柱容量下降。
当长时间使用后 , 有一些强保留组分吸附在柱子中 , 不大的进样量往往就会出现峰分裂。
用强洗脱能力的溶剂清洗色谱柱 , 或更换色谱柱可使问题得到改善。
(4) 样品溶剂与流动相不匹配或进样体积过大。
当样品溶剂比流动相极性大时 , 有时即使进样体积很小 , 也容易出现峰变形和裂分现象。
建议用流动相溶解样品。
(5) 流动相不恰当。
此情况较罕见 , 有些样品在特定的色谱条件下可能存在结构的动态平衡 , 而出双峰 , 这种双峰是无法分离完全的 , 改变色谱条件尤其是p H 值会使峰形正常。
(6) 样品分解。
不稳定的样品在色谱分离过程中变成其他物质而出现双峰。
这时需改变样品处理方法或色谱分离条件。
2.负峰分析在色谱分析中有时会出现负峰或倒峰 , 如图 3 中的大峰的左下就有一负峰。
出现这种现象可能是由以下几种原因引起的 , 可针对不同情况进行排除 , 进而使问题得到解决。
(1) 流动相吸收本底值过高。
此时可适当改变检测波长。
(2) 进样过程中进入空气。
进行排气处理 , 直到基线平稳再进样。
(3) 样品组分的吸收低于流动相。
可改变流动相或改变检测波长。
(4) 配制样品的溶液与流动相不一样。
重新配制样品 , 用与流动相一样的溶剂配制或稀释样品。
气相色谱质谱联用谱图解析

依次为:间位、对位、邻位。
质谱解析
由质谱的高质量端确定分子离子峰,求出分子量。
一般来讲,分子离子峰是荷质比最大的一个, 但是对于易断裂的的物质来说,分子离子峰很小或 者没有,需积累经验。
分析同位素峰簇的相对强度比和峰与峰间的差值,
判断化合物是否含有Cl和Br等元素。
质谱解析
由分子离子峰丢失的碎片及主要碎片离子推分子式。 由分子离子峰的相对强度了解分子结构的信息。分
基;若m/z 91或105为基峰或强峰,表明化合物含
有苄基或苯甲酰基。
常见失去离子碎片
M-15(CH3)
,C2H3) M-29(CHO,C2H5) M-31(CH2OH,OCH3) M-35(Cl) M-43(CH3CO,C3H7) M-45(OC2H5,COOH)
子离子峰的相对强度由分子的结构所决定,结构稳 定性大,相对强度就大。 例如:萘分子离子峰m/z 128为基峰,蒽醌分 子离子峰m/z 208也是基峰。分子离子峰弱或不出 现,化合物可能为多支链烃类、醇类、酸类等。
质谱解析
根据特征离子峰及丢失的中性碎片了解可能的结构
信息。 若质谱图中出现系列CnH2n+1峰,则化合物可能 含长链烷基;若出现或部分出现m/z 77,66,65, 51,40,39等弱的碎片离子蜂,表明化合物含有苯
M-18(H2O)
M-26(C2H2) M-28(CO,C2H4) M-30(NO) M-32(S,CH3OH) M-42(CH2CO,CH2N2) M-44(CO2,CS2) M-46(NO2,C2H5OH)
M-79(Br) M-16(O,NH2)
M-127(I)……
谢 谢!
气质联用谱图解析
FID气相色谱仪异常图谱分析

FID 气相色谱仪异常图谱分析张 敏/上海市质量监督检验技术研究院0 引言氢火焰离子化检测器(简称FID)是一个质量型检测器,它具有灵敏度高、检测限小、线性范围广等特点,现已广泛地应用于各大领域,成为分析多组分混合物最为有效的手段之一,但其结构复杂,条件设置多,在使用过程中会出现各种故障,影响正常的检测分析结果,因此,如何迅速、准确地判断故障原因并及时予以排除,是仪器操作人员经常面临和急需解决的问题。
1 FID气相色谱仪原理气相色谱是一种物理分离方法。
利用被测物质各组分在不同两相间分配系数(溶解度)的微小差异,当两相作相对运动时,这些物质在两相间进行反复多次的分配,原来只有微小的性质差异产生很大的效果,而使不同组分得到分离, 进而加以定性和定量测定。
气相色谱仪有气路系统、进样系统、分离系统以及检测和记录系统组成。
在分离分析方面,具有如下特点:1)高灵敏度。
可检出10 mg-10 g的物质,可作超纯气体、高分子单体的痕量杂质分析和空气中微量毒物分析。
2)高选择性。
可有效地分离性质极为相近的各种同分异构体和各种同位素。
3)高效能。
可把组分复杂的样品分离成单组分。
4)速度快。
一般分析只需几分钟即可完成,有利于指导和控制生产。
5)应用范围广。
可分析低含量的气、液体,亦可分析高含量的气、液体,且不受组份含量的限制。
6)所需试样量少。
一般气体样品用几亳升,液体样用几微升或几十微升。
样品和载气经过柱子后进入FID的氢气-空气火焰中发生电离产生离子,极化电压把这些离子吸引到火焰附近的收集极上,产生的电流与燃烧的样品量成正比。
用一个电流计检测电流并转换成数字信号,送到输出装置得到样品浓度。
它只对含有C-H键的化合物有响应。
因此FID灵敏度高,其检测限最小可至1 pg/s,线性范围约为107。
2 异常图谱分析一般在日常使用中,FID气相色谱仪异常色谱图如表1、表2所列。
3 讨论综上所述,FID气相色谱仪作为一种高精密的分析仪器,影响基线与图谱的异常因素有许多,要准确判断出异常现象的所在,就必须完全了解FID气相色谱的各组成部分及工作原理,面对不同的故障现象,既要考虑到局部又要考虑到整体,有“果”必有“因”,逐步排除产生故障的“因”,把范围缩小。
实验三__纸色谱

实验三__纸色谱实验三纸色谱一、实验原理、方法、注意事项1、原理纸层色谱为在纸上将混合物进行分离的色谱方法,分为分析型和制备型纸层色谱。
多数情况下,纸层色谱的原理属于分配色谱原理,色谱滤纸为支持剂,滤纸纤维可以吸附25%,30%的水分,其中6,7%的水分和滤纸结构中的羟基以氢键结合,为固定相。
其他溶剂可自由通过,为流动相。
流动相流经支持物时,与固定相之间连续抽提,使物质在两相间不断分配而得到分离。
物质被分离后在纸色谱图谱上的位置用Rf值(比移值)来表示:R值 = 原点到色谱点中心的距离/ 原点到溶剂前沿的距离 f在一定条件下某种物质的R值是常数,其大小受物质的结构、性质、溶剂系统物质组f成与比例、pH值、选用滤纸质地和温度等多种因素影响。
此外,样品中的盐分、其他杂质以及点样过多均会影响的有效分离。
但由于影响比移值的因素较多,因而一般采用在相同实验条件下与对照物质对比以确定其异同。
作为药品的鉴别时,供试品在色谱中所显主斑点的颜色(或荧光)与位置,应与对照品在色谱中所显的主斑点相同。
作为药品的纯度检查时,可取一定量的供试品,经展开后,按各药品项下的规定,检视其所显杂质斑点的个数或呈色(或荧光)的强度。
作为药品的含量测定时,将主色谱斑点剪下洗脱后,再用适宜的方法测定。
无色物质的纸色谱图谱可用光谱法(紫外光照射)或显色法鉴定,氨基酸纸色谱图谱常用茚三酮显色法鉴定。
纸层色谱适用于极性较大的亲水性化合物或极性差别较小的化合物的分离。
2、实验方法(1) 下行法将供试品溶解于适当的溶剂中制成一定浓度的溶液。
用微量吸管或微量注射器吸取溶液,点于点样基线上,溶液宜分次点加,每次点加后,俟其自然干燥、低温烘干或经温热气流吹干,样点直径为2,4mm,点间距离约为1.5,2.0cm,样点通常应为圆形。
将点样后的色谱滤纸上端放在溶剂槽内并用玻棒压住,使色谱纸通过槽侧玻璃支持棒自然下垂,点样基线在支持棒下数厘米处。
展开前,展开室内用各品种项下规定的溶剂的蒸气使through the "scorecard" on borrowers ' scores. 24th account manager in the personal lending survey returns (4) fill out the survey, along with other loan information to review. And common findings include the borrower repayment of basic information, income 之饱和,一般可在展开室底部放一装有规定溶剂的平皿或将浸有规定溶剂的滤纸条附着在展开室内壁上,放置一定时间,俟溶剂挥发使室内充满饱和蒸气。
色谱指纹图谱评价PPT课件

面取得了显著进步。
毛细管电泳法(CE)
03
CE具有高分离效率和低成本的优势,尤其适合分析复杂生物样
品。
数据库的建立与完善
建立标准色谱指纹图谱库
收集各种样品的标准色谱指纹图谱,建立全面的数据库,为比对 分析提供参考。
数据标准化
对色谱数据进行处理和标准化,确保不同实验条件下的数据具有可 比性。
数据库更新与维护
谢谢观看
根据样品的性质和目标成 分,选择合适的色谱柱, 确保目标成分的有效分离。
流动相的选择
根据样品的性质和目标成 分,选择合适的流动相, 以获得最佳的分离效果。
检测波长的选择
选择适当的检测波长,以 便在指纹图谱中获得准确 的检测结果。
数据处理与分析
数据采集
按照设定的色谱条件进行实验 ,并采集色谱图数据。
色谱指纹图谱评价ppt课件
目录
• 引言 • 色谱指纹图谱的建立 • 色谱指纹图谱的解析 • 色谱指纹图谱的应用 • 色谱指纹图谱的局限性 • 色谱指纹图谱的发展趋势
01
引言
目的和背景
目的
介绍色谱指纹图谱在中药质量评价中的应用。
背景
随着中药产业的快速发展,中药质量评价方法的创新与改进成为行业关注的焦 点。色谱指纹图谱作为一种新型的中药质量评价方法,具有独特的优势和广泛 的应用前景。
数据预处理
对采集的数据进行必要的预处 理,如平滑化、基线校正等, 以提高数据的准确性和可靠性 。
特征峰的确定
通过对比对照品和样品色谱图 ,确定色谱指纹图谱中的特征 峰。
相似度评价
利用相关软件计算样品之间的 相似度,以评估色谱指纹图谱
的一致性和可靠性。
03
色谱指纹图谱的解析
基于小波变换的代谢组色谱指纹图谱的判别分析

据差异变量筛选 , 结合保 留时间对质谱数据进行定位。 本 文将 卵巢癌 患 者与 正常对 照样 品 的色谱 图通 过连 续
小 波 函数进 行预 处理 , 然后 使用 随机 森林 ( r a n d o m f o r .
e s t , R F) 模 型进 行判别 分 析 , 并评 价 其分类 效 果 。
0 2 4 6 8 1 0 1 2 1 4 1 6 1 : TOF M SES+
中 国卫 生 统 计 2 0 1 3年 4月 第 3 O卷 第 2期
基 于小 波变 换 的代 谢 组 色 谱 指纹 图谱 的 判别 分 析
哈尔滨医科大学卫生统计学教研室( 1 5 0 0 8 1 ) 孙凤 宇 李 贞子 侯 艳 李 康
【 提
要】 目的
探索小波变换在代谢组学超高效 液相色谱 信号分析 中的作用 。方法
原理 与方 法 1 . 小 波变换
小波 变换 ( w a v e l e t t r a n s f o r m, wT) 是 一 种 信 号处 理技 术 , 它在数 据压 缩 、 噪声 过滤 、 信号 分析 、 特 征提取 等理 论 和应 用 方 面 都 得 到 发 展 , 具 有 “自适 应 性 ” 和
将 卵巢癌 和卵巢囊 肿 的色
谱数据使用不同 的连续小波基 函数 和不 同尺度变换 , 随机抽取一定数量 的训 练数据 , 采用 随机 森林 ( R F ) 方法进行 特征筛 选建立模型 , 最后对测试数 据集 进行判别分析 , 计算 R O C曲线下面积 A UC值 。结果 经过小 波变换 预处理 后 的数据 建
模分类效果 明显优于原始数据 , 其 中墨西哥帽 ( me x h ) 小波 变换分 类效果 最好 。结论 方法能够 明显提高模 型的判别 能力 , 具有研究价值 。
色谱定性与定量

仪器分析中各分析定量定性的依据定量分析是依据统计数据,建立数学模型,并用数学模型计算出分析对象的各项指标及其数值的一种方法。
定性分析则是主要凭分析者的直觉、经验,凭分析对象过去和现在的延续状况及最新的信息资料,对分析对象的性质、特点、发展变化规律作出判断的一种方法。
1、气相色谱:色谱峰保留值和面积,这样气相色谱可根据这两个数据进行定性定量分析。
色谱峰保留值是定性分析的依据,而色谱峰面积则是定量分析的依据。
2、紫外光谱:最大吸收波长λ、摩尔吸收系数ε及吸收曲线的形状不同是进行物质定性分析的依据。
进行定量分析依据朗伯-比耳定律:A=εbc3、核磁:定量分析以结构分析为基础,在进行定量分析之前,首先对化合物的分子结构进行鉴定,再利用分子特定基团的质子数与相应峰谱的峰面积之间的关系进行定量测定。
定量分析的根据:吸收能量的大小取决于核的多少。
以磁场强度为横坐标提供定性分析所依据的参数,以吸收能量为纵坐标,纵坐标对应于不同H0的出峰面积就是定量分析参数。
4、质谱:利用电磁学原理,对物质气相离子依其质荷比(m/e)进行分离和分析的方法。
被分析的样品首先离子化,然后利用离子在电场或磁场中的运动性质,将离子按质荷比(m/e)分开并按质荷比大小排列成谱图形式,根据质谱图可确定样品成分、结构和相对分子质量。
5、原子吸收:原子吸收光谱法进行定量分析的依据是:试样中待测元素的浓度与待测元素吸收辐射的原子总数成正比,即A=k'C 。
定量分析方法有标准曲线法和标准加入法两种。
6、红外:红外光谱的定性主要根据图谱中的:基团的特征吸收频率红外光谱的定量是根据图谱中的:特征峰的强度7、离子:利用离子交换的原理,连续对多种阴离子进行定性和定量的分析。
保留时间定性,峰高或峰面积定量。
8、荧光:物质吸收的光,称为激发光;物质受激后所发射的光,称为发射光或荧光。
根据荧光的光谱和荧光强度,对物质进行定性或定量测定9、差热:定性分析:定性表征和鉴别物质依据:峰温、形状和峰数目方法:将实测样品DTA曲线与各种化合物的标准(参考)DTA曲线对照。
高效液相色谱中异常峰分析

异常峰分析异常得色谱峰指得就就是色谱图中得无峰或出现负峰、宽峰、双峰、肩峰、峰形不对称等情况。
异常峰就就是色谱实验工作中最棘手得问题。
这些峰严重影响色谱分析得结果。
色谱图中不可能有纯正得高斯对称峰, 轻微得拖尾就就是正常得, 这就就是由分离系统所决定得。
在此仅对几种异常峰进行分析。
1、峰前或峰后有小峰得分析产生原因大致分为以下几种情形(1)样品不纯。
可改变不同得流动相与色谱柱,对样品进行分离比较,选择合适得分离条件。
(2) 分析柱或保护柱柱头塌陷。
此情况较常见,可更换分析柱或保护柱后对峰形进行比较。
柱头塌陷时往往所有得峰都会出现峰分裂。
对色谱柱再生与清洗可以改善分离效果。
ﻫ(3) 色谱柱柱容量下降。
当长时间使用后, 有一些强保留组分吸附在柱子中, 不大得进样量往往就会出现峰分裂。
用强洗脱能力得溶剂清洗色谱柱,或更换色谱柱可使问题得到改善。
(4) 样品溶剂与流动相不匹配或进样体积过大。
当样品溶剂比流动相极性大时,有时即使进样体积很小, 也容易出现峰变形与裂分现象。
建议用流动相溶解样品。
(5)流动相不恰当。
此情况较罕见, 有些样品在特定得色谱条件下可能存在结构得动态平衡,而出双峰, 这种双峰就就是无法分离完全得, 改变色谱条件尤其就就是p H值会使峰形正常。
(6) 样品分解。
不稳定得样品在色谱分离过程中变成其她物质而出现双峰。
这时需改变样品处理方法或色谱分离条件。
2、负峰分析在色谱分析中有时会出现负峰或倒峰, 如图 3 中得大峰得左下就有一负峰。
出现这种现象可能就就是由以下几种原因引起得, 可针对不同情况进行排除,进而使问题得到解决。
(1)流动相吸收本底值过高。
此时可适当改变检测波长。
(2) 进样过程中进入空气。
进行排气处理, 直到基线平稳再进样。
(3)样品组分得吸收低于流动相。
可改变流动相或改变检测波长。
(4)配制样品得溶液与流动相不一样。
重新配制样品, 用与流动相一样得溶剂配制或稀释样品。
3、前沿、拖尾峰分析拖尾:1 干扰峰,优化条件分离;2色谱柱塌陷,更换色谱柱; 3 流动相pH不合适,调节pH值;4 管路没有接好,存在较大得死体积,可以重新接一下。
圆二色谱分析结果怎么看?
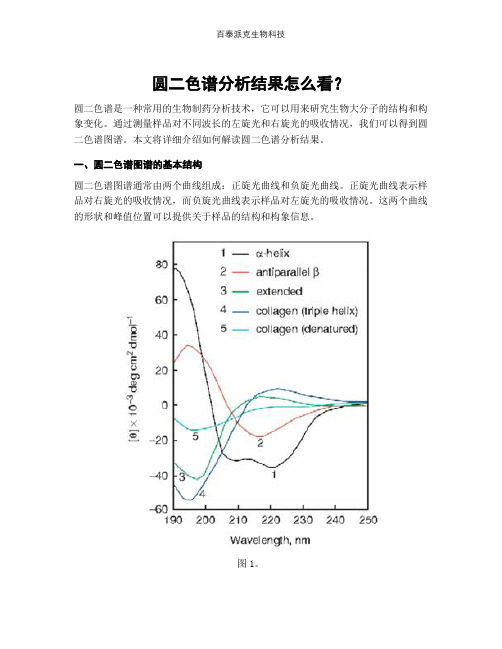
圆二色谱分析结果怎么看?圆二色谱是一种常用的生物制药分析技术,它可以用来研究生物大分子的结构和构象变化。
通过测量样品对不同波长的左旋光和右旋光的吸收情况,我们可以得到圆二色谱图谱。
本文将详细介绍如何解读圆二色谱分析结果。
一、圆二色谱图谱的基本结构圆二色谱图谱通常由两个曲线组成:正旋光曲线和负旋光曲线。
正旋光曲线表示样品对右旋光的吸收情况,而负旋光曲线表示样品对左旋光的吸收情况。
这两个曲线的形状和峰值位置可以提供关于样品的结构和构象信息。
图1。
二、峰值位置的解读圆二色谱图谱中的峰值位置可以告诉我们样品的二级结构信息。
一般来说,α-螺旋结构在190-200 nm处会出现负峰,而β-折叠结构在210-220 nm处会出现正峰。
通过观察峰值位置的变化,我们可以推断样品的二级结构变化。
三、峰值强度的解读除了峰值位置,圆二色谱图谱中的峰值强度也是解读样品结构的重要指标。
峰值强度反映了样品对旋光的吸收程度,强度越高表示吸收越强。
通过比较不同样品的峰值强度,我们可以判断它们的结构差异。
四、色谱图形的解读除了峰值位置和峰值强度,圆二色谱图谱的整体形状也提供了有关样品结构的信息。
例如,如果图谱呈现出对称的双峰形状,那么可能存在一种手性结构。
而如果图谱呈现出单峰形状,那么样品可能是非手性的。
五、结构变化的解读通过比较不同条件下的圆二色谱图谱,我们可以观察到样品结构的变化。
例如,当样品在不同温度下进行测量时,我们可以观察到峰值位置的变化,从而推断样品的热稳定性。
类似地,通过比较不同pH值下的圆二色谱图谱,我们可以了解样品的酸碱稳定性。
圆二色谱分析结果的解读需要考虑峰值位置、峰值强度、峰形、色谱图形和对比分析等因素。
通过综合分析这些指标,我们可以得出关于样品结构特征、纯度和质量变化情况的重要信息。
圆二色谱作为一种重要的分析技术,在生物制药领域具有广泛的应用前景。
百泰派克生物科技——生物制品表征,多组学生物质谱检测优质服务商。
全二维气相色谱/飞行时间质谱对饱和烃分析的图谱识别及特征

Per lu Ex oa ina d De eo me t toe m plr to n v lp ni a;
关 键 词 : 二 维 气 相 色 谱 ; 行 时 间质 谱 ; 和 烃 ; 环 萜 烷 ; 油 地 质 全 飞 饱 三 石 中圈 分 类 号 : 5 . 3 O 6 76 文献 标 识 码 : A 文 章 编 号 :0 42 9 ( 00 0 -0 81 10 —97 2 1 ) 10 1 -0
藿烷类的全二维点阵谱图特征 ; 测到过去 G / 检 C MS分析 中 常被 忽 视 的 C 。三 环 萜 烷 , 石 油 地 质 实 验 s~C s 为 和 研 究 提 供 了 参 考 依 据 。全 二 维气 相色 谱 / 行 时 间 质 谱 相 比于 G / 飞 C MS灵 敏 度 更 高 、 容 量 更 大 , 合 复 峰 适 杂 混 合 物 体 系 的 分 析 , 石 油 样 品的 分 析 有 很 好 的 应用 前 景 。 对
Ch r c e i tc n d ntfc tO f S t r t d Hy r c r O s b a a trsisa d I e iia j n o a u a e d o a b n y
Co p e e s v m r h n i e Two Di e s o lGa - m n i na s Chr m a o r ph u l d o t g a y Co p e
3 .提 高 石 油 采 收率 国家 重 点 实 验 室 , 北京 10 8 ) 0 0 3
示差液相色谱仪空白出峰

示差液相色谱仪空白出峰-概述说明以及解释1.引言1.1 概述示差液相色谱仪是一种常用的分析仪器,广泛应用于化学、药学、环境、食品等领域。
它通过将样品溶液注入色谱柱,在不同条件下进行分离,再通过检测器对不同组分进行分析,从而得到样品的成分及其含量信息。
然而,在使用示差液相色谱仪进行分析时,经常会出现空白出峰的问题。
所谓空白出峰,指的是在样品分析过程中,出现未添加任何化合物的溶剂溶液所产生的峰。
这些峰往往会干扰到对样品中目标成分的分析结果,影响数据的准确性和可靠性。
因此,研究和解决示差液相色谱仪空白出峰的问题具有重要意义。
本文将首先简要介绍示差液相色谱仪的原理和应用,进而详细探讨示差液相色谱仪空白出峰的原因及其可能的解决方法。
通过对空白出峰问题的认识和解决,旨在提高示差液相色谱仪的分析效果和准确性,为相关领域提供更可靠的数据支持。
在下一节中,我们将详细介绍示差液相色谱仪的原理,包括液相色谱技术基本原理、示差液相色谱仪的组成和工作原理。
这将为读者进一步理解空白出峰问题的原因和解决方法提供必要的背景知识。
文章结构是指文章整体内容的组织和布局方式。
本文将按照以下结构展开内容:1. 引言1.1 概述1.2 文章结构1.3 目的1.4 总结2. 正文2.1 示差液相色谱仪的原理2.2 示差液相色谱仪的应用2.3 示差液相色谱仪空白出峰的原因2.4 示差液相色谱仪空白出峰的解决方法3. 结论3.1 对示差液相色谱仪空白出峰问题的认识3.2 总结示差液相色谱仪空白出峰的解决方法3.3 对示差液相色谱仪的应用前景展望在引言部分,首先概述了示差液相色谱仪的基本情况和研究背景。
然后介绍了文章的整体结构,列举了各个部分的标题来展示整体的逻辑框架。
接下来说明了文章的目的,即探究示差液相色谱仪空白出峰问题,并提出解决方法。
最后通过总结,为读者提供整个文章内容的简要概述。
在正文部分,分为四个小节,分别阐述了示差液相色谱仪的原理、应用、空白出峰的原因以及解决方法。
色谱 肩峰 测量 对比 误差

色谱技术在科学研究和工业生产中具有广泛的应用。
其中,色谱肩峰的测量对比和误差分析是色谱技术中非常重要的内容。
本文将对色谱肩峰测量、对比和误差进行深入探讨,以期为相关研究和实践提供参考。
一、色谱肩峰的测量色谱肩峰是指在色谱图谱中出现的两个相邻色谱峰之间的相对宽度较大的区域。
肩峰的形成与物质的分布不均匀、柱效应以及仪器分辨率等因素有关。
测量色谱肩峰的主要方法有多峰拟合法、导数法、峰峰距法等。
在实际测量中,需要结合实验条件和仪器特性选择适当的方法,保证测量结果的准确性和可靠性。
二、色谱肩峰的对比在色谱分析中,往往需要对不同样品的色谱图谱进行对比。
色谱肩峰的对比是对样品之间相似性和差异性进行评价的重要手段。
对比色谱肩峰需要考虑色谱峰的形状、峰面积、峰高、峰宽等参数。
需要注意使用适当的数据处理方法,如相似度分析、主成分分析等,提高对比的准确性和全面性。
三、色谱肩峰测量的误差分析色谱肩峰测量中存在着各种误差,如仪器误差、样品制备误差、环境条件误差等。
对色谱肩峰测量误差进行分析和评价对于保证色谱分析结果的可信度非常重要。
误差分析的主要方法包括误差来源的分类、误差的传递和放大规律、误差的定量评价等。
通过误差分析,能够找出色谱肩峰测量中存在的问题,并采取相应的措施进行修正和改进。
色谱肩峰测量、对比和误差分析是色谱技术中不可或缺的一部分。
通过深入研究和实践,能够提高色谱分析的准确性和可靠性,为科学研究和工业生产提供有力的支撑。
期待在未来的发展中,色谱肩峰测量技术能够得到更好的应用和推广。
四、色谱肩峰在不同应用领域的特点色谱肩峰在不同的应用领域具有一定的特点和应用要求。
在药物分析领域,色谱肩峰的特点可能受到不同成分的相互影响,需要通过精密的测量和对比来确保药物成分的分离和鉴定。
而在环境监测和食品安全领域,色谱肩峰的测量和对比则需要考虑更多的外部干扰因素,如样品的复杂性以及环境条件对色谱分析的影响。
针对不同的应用领域,需要结合具体的实际情况,选择合适的色谱肩峰测量方法和对比策略,以确保分析结果的准确性和可靠性。
中国药典2005年版中药薄层色谱鉴别技术

中国药典2005年版中药薄层色谱鉴别技术中药的真伪优劣直接关系到人民用药的安全和有效。
在检验中药真伪方面通常采用的检测手段有形态鉴别、理化鉴别和光谱色谱分析等。
而作为色谱技术一个分支的薄层色谱由于其独具的长处而被广泛应用于中药分析。
自从《中华人民共和国药典》(1990年版一部)起,薄层色谱鉴别除有化学对照品作为鉴别药材的指标成分对照品以外,鉴于单一化学对照品不能反映药材的整体特征,一些多种植物共存的化学成分没有专属性,以及没有化学对照品鉴别就无法进行的问题,开始增加了“对照药材”,以药材对照品的色谱整体为特征进行对比鉴别,解决了上述存在的问题,并于1993年出版了《中华人民共和国药典1990年版中药薄层色谱彩色图集》,作为中药薄层鉴别的重要参考资料。
在该书的第一章中根据多年的实践经验并参考Friedrich Geiss的《Fundamentals of Thin Layer Chromatography (Planar Chromatography)》一书的内容,从实用的角度叙述了薄层色谱实际操作中的各个环节的注意事项,起到了薄层色谱操作规范化的指导作用。
但是直至中国药典2000年版,中药薄层色谱鉴别基本是以手工点样、实验室自制薄层板为基础的较为粗放的操作进行试验,由于硅胶材料的质量不高和手工操作的个体差异较大,致使色谱质量仍然不高,分离度、重现性均难以达到满意的效果。
尽管自上世纪90年代以来,国外尤其是欧洲国家在薄层色谱技术及相应的仪器开发逐步有了很大发展,目前已经达到单元操作计算机自动化,高质量商品预制薄层板(常规板与高效板)代替了自制薄层板,进入一个高层次的仪器化和微机化的阶段。
美国药典、欧洲药典等对草药(植物药)均采用薄层色谱鉴别,并有规范化的要求,以保证结果的专属性和可重现性,这对药典的实施是非常重要的。
因而我国药典在中药的薄层色谱鉴别的应用和推广也需要在技术层面有明显的提高。
在实验室条件方面。
硫酸二甲酯气相色谱各峰意义

爱慕的反义词
中文发音:[ài mù]
词语解释:爱慕,指喜欢羡慕。
反义词:妒忌、嫌弃、嫌恶
【用爱慕造句】
1.他具有会使人爱慕他的天赋。
2.很多人都爱慕铁哥的才学。
3.他们彼此低声倾吐着爱慕之情。
4.他给她写了信,表述自己的爱慕之情。
5.他的爱慕的性质是一清二白的。
6.不管他多爱慕她,她对他仍冷若冰霜。
7.那位漂亮的女孩走过街时受到男孩子爱慕眼光的注视。
【用爱慕的反义词造句】
妒忌:她被一种疯狂的妒忌所支配着。
嫌弃:他得了传染病,可同学们一点不嫌弃。
嫌恶:他的行为使每个人都嫌恶。
1。
鲜铁皮石斛茎与叶高效液相色谱特征图谱对比分析

鲜铁皮石斛茎与叶高效液相色谱特征图谱对比分析摘要:目的:为更好的比较鲜铁皮石斛茎、叶不同部位的HPLC特征图谱的对比差异,以此来有效的分析鲜铁皮石斛茎叶部位的关联性,进而为铁皮石斛叶色谱柱,的实际应用提供数据参考。
方法:高效液相色谱的基本情况为ZorbaxSBC18体积分数为0.2%的甲酸溶液,对鲜铁皮石斛进行梯度的清洗。
柱温为35度,流速为每分钟1毫升,波长为270nm。
结果:鲜铁皮石斛的叶部分特有峰面的面积要远远大于铁皮石斛的茎。
结论:铁皮石斛的茎叶部位有相似成分,可以将铁皮石斛的叶用于药中,以此来有效的缓解铁皮石斛的药用短缺。
关键词:鲜铁皮石斛;高效液相色谱;特征图谱对比引言:铁皮石斛是一种兰科石斛属植物,对其的古籍记载最早可以追溯到《神农本草经》,是当前药用石斛中的绝佳品种。
铁皮石斛可以用作治疗热病津伤、伤病后虚热不退,眼睛不明等症状。
当前野生铁皮石斛的数量十分稀少,我国早已将铁皮石斛列为国家保护物种,当前用于药品中的铁皮石斛都是人工栽培种植的。
1.高效液相色谱简述高效液相色谱一般也被称为“高压液相色谱”,英文简称为HPLC,是色谱法中的一个重要分支。
使用高压输液系统,液体为流动相,将单一溶剂、混合溶剂、缓冲液等试剂放入固定相的色谱柱中,当试剂在色谱柱中内完全分离后,在将其放置在专业的检测仪器中进行检测。
HPLC具有四高一广的特点,四高分别为高压、高速、高效以及高灵敏,一广是指其应用的范围相对较广。
高压是指其液体主要为流动状态,在其经过色谱柱时,会遭到一定的阻力拦截,为了使其可以快速的通过色谱柱,操作人员就要对其进行高压加载。
高速是指HPLC的分析速度以及载液流动速度相对较快,一个检测样品一般在15到30分钟之内就可以完成,甚至有的载液在5分钟内就可以完成。
高效是指HPLC的分离性能高,在固定与流动状态下,都可以到达样品的最佳分离效果。
高灵敏度是指HPLC的之外检测器可以达到0.01ng,其进样量也是可以保持在μL数量级。
高效液相色谱中异常峰分析
异常峰分析异常的色谱峰指的是色谱图中的无峰或出现负峰、宽峰、双峰、肩峰、峰形不对称等情况。
异常峰是色谱实验工作中最棘手的问题。
这些峰严重影响色谱分析的结果。
色谱图中不可能有纯正的高斯对称峰, 轻微的拖尾是正常的, 这是由别离系统所决定的。
在此仅对几种异常峰进行分析。
1.峰前或峰后有小峰的分析产生原因大致分为以下几种情形(1) 样品不纯。
可改变不同的流动相和色谱柱,对样品进行别离比较, 选择合适的别离条件。
(2) 分析柱或保护柱柱头塌陷。
此情况较常见,可更换分析柱或保护柱后对峰形进行比较。
柱头塌陷时往往所有的峰都会出现峰分裂。
对色谱柱再生和清洗可以改善别离效果。
(3) 色谱柱柱容量下降。
当长时间使用后, 有一些强保留组分吸附在柱子中, 不大的进样量往往就会出现峰分裂。
用强洗脱能力的溶剂清洗色谱柱, 或更换色谱柱可使问题得到改善。
(4) 样品溶剂与流动相不匹配或进样体积过大。
当样品溶剂比流动相极性大时, 有时即使进样体积很小, 也容易出现峰变形和裂分现象。
建议用流动相溶解样品。
(5) 流动相不恰当。
此情况较罕见, 有些样品在特定的色谱条件下可能存在结构的动态平衡, 而出双峰, 这种双峰是无法别离完全的, 改变色谱条件尤其是pH 值会使峰形正常。
(6) 样品分解。
不稳定的样品在色谱别离过程中变成其他物质而出现双峰。
这时需改变样品处理方法或色谱别离条件。
2.负峰分析在色谱分析中有时会出现负峰或倒峰, 如图 3 中的大峰的左下就有一负峰。
出现这种现象可能是由以下几种原因引起的, 可针对不同情况进行排除, 进而使问题得到解决。
(1) 流动相吸收本底值过高。
此时可适当改变检测波长。
(2) 进样过程中进入空气。
进行排气处理, 直到基线平稳再进样。
(3) 样品组分的吸收低于流动相。
可改变流动相或改变检测波长。
(4) 配制样品的溶液与流动相不一样。
重新配制样品, 用与流动相一样的溶剂配制或稀释样品。
3.前沿、拖尾峰分析拖尾:1 干扰峰,优化条件别离;2 色谱柱塌陷,更换色谱柱; 3 流动相pH 不合适,调节pH值;4 管路没有接好,存在较大的死体积,可以重新接一下。
色谱峰20个点

色谱峰20个点色谱峰分析是一种在化学、生物及环境科学中广泛使用的技术,用于分离和鉴定混合物中的不同组分。
在色谱图中,每一个峰都代表着样品中的一个或多个组分,而峰的位置、形状和大小都包含了关于这些组分的重要信息。
在本文中,我们将深入探讨20个色谱峰的分析过程,以及它们所揭示的关于样品组成和性质的信息。
首先,我们需要明确色谱峰分析的基本原理和步骤。
色谱分析基于不同物质在固定相和移动相之间的分配系数差异,从而实现混合物的分离。
在色谱图上,每个峰的位置(即保留时间或距离)反映了该组分的分配系数,而峰的面积则与其浓度成正比。
接下来,我们将逐一分析这20个色谱峰。
每个峰都需要关注其位置、形状、大小以及与其他峰的关系。
例如,峰的位置可以帮助我们确定组分的类型,而峰的形状和大小则可以提供关于组分纯度和浓度的信息。
此外,通过比较不同峰之间的相对位置和面积,我们还可以推断出组分之间的相互作用和可能存在的化学关系。
在分析过程中,我们还需要注意一些可能影响结果的因素,如色谱柱的选择、流动相的成分和流速、样品的预处理等。
这些因素都可能对色谱峰的位置、形状和大小产生影响,因此需要仔细控制和优化。
除了基本的峰分析外,我们还可以利用一些高级技术来进一步挖掘色谱峰中的信息。
例如,质谱联用技术可以将色谱峰与质谱数据相结合,从而提供关于组分结构的详细信息。
此外,通过多维色谱技术,我们还可以在同一张色谱图上获得更多关于样品组成和性质的信息。
最后,我们需要对色谱峰分析的结果进行解释和总结。
通过对比已知标准品色谱峰和未知样品色谱峰的信息,我们可以鉴定出样品中的各个组分,并推测它们可能的来源和性质。
同时,我们还需要评估分析的准确性和可靠性,以便为后续的科研或工业应用提供有力的支持。
总之,色谱峰分析是一项复杂而精细的工作,需要综合运用化学、物理和计算机科学等多个领域的知识和技术。
通过对20个色谱峰的深入探索和分析,我们不仅可以了解样品的组成和性质,还可以为相关领域的研究和应用提供有价值的参考信息。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
色谱分析中各种图谱现象的判断
色谱分析中各种图谱现象的判断
可能产生的原因及处理办法
一.基线噪音
1 流动池脏,用极性试剂清洗。
当有填料进入,拆开流通池。
2 检测器灯有问题,如能量偏低,更换氘灯。
3 周期性的波动,则起源于泵的脉冲,检修泵或更换垫片等。
4 温度对检测器的影响,控制温度。
5 气泡经过检测器,用大流量冲洗。
6 可能难出峰的样品连续不断出来,用强极性流动相冲柱。
7 流动相本底高,如水的纯度不够,换超纯水。
或试剂纯度不够,换色谱纯的试剂。
二.基线漂移(上漂和下漂)
1 柱中的流动相没有平衡,延长平衡时间,尤其在流动相中添加了有紫外吸收的添加剂。
2 在梯度洗脱中,基线上漂是正常的,在空白梯度中有可能是柱子中有杂质洗出。
其次是流动相中有干扰物,换流动相。
3 温度不稳定(示差检测器),控温。
4 在等度分析中,样品缓慢洗出,改变淋洗液强度或用梯度分析。
5 样品进入检测器,吸附在池中,可能每进样一次,本底一次比一次高,很少见。
三.倒峰的产生和消除
1.柱切换的脉冲效应,一般不是很明显,必要时考虑换阀。
2.在低波长分析时,流动相本底比较高时,而样品用本底低的流动相溶解,肯定出现倒峰,其程度同进样量和本底差有关。
解决办法,用流动相溶解样品,减少进样量,消除倒峰的影响。
高波长时,影响比较小。
3.如果倒峰不影响峰的分离,对外标法定量不影响。
但影响面积归一化法。
4.样品中有比流动相本底低的物质存在,如无机盐等,将出倒峰。
这种情况下,倒峰的位置不一定在死体积位置出现(大多数在死体积位置出现)。
四.鬼峰的产生和消除
1.样品分析时峰没出完,在下一针或下下一针出现,判断办法,延长分析时间,计算可能出现的保留时间。
然后调整流动相。
2.连续进样,在某个位置出现忽高忽低的峰,最可能是进样针污染,清洗进样针,注意污垢的干扰,有些样品易残留在针管里。
可重新取样分析。
3.定量管污染,处理方法同上。
4.在死体积位置出现的小峰,可能是柱切换造成的。
5.流动相与样品溶剂不一致,也会出现鬼峰,尤其在低波长时,出现位置在死体积的地方。
6.气泡,如果有小气泡通过流通池,也出现随机的假峰,大气泡存在,其出现的峰往往直上直下,脱气解决。
7.样品发生变化反应,重新取样快速分析。
五.峰前移或退后的判断
1.有规律的变化一般可能是流动相没平衡,如低浓度的缓冲液或离子对试剂,样品对pH变化极敏感。
2.温度变化,当天气变化快,分析时间长,比较明显,用柱温箱,空调解决此类问题。
3.流动相挥发,尤其用挥发性的酸时,时间长了酸度会发生变化,流动相新鲜配置。
4.仪器尤其泵的运行时间长后,重现性不好,仪器老化,在梯度分析时尤其明显,也可能流速不稳,或有气泡。
5.在分析某样品后,柱子可能发生改性,再次分析重现性变差,清洗或再生柱子。
6.进样浓度不一,溶剂不一,也会造成保留时间不一致。
六.前沿峰和拖尾峰的判断和处理
前沿峰的产生和处理(对称性小于0.9)
1.色谱柱漏穿,柱效明显偏低,柱压偏低,如所有峰都出现,换柱。
2.样品溶剂极性远大于流动相,局部改变了流动相平衡体系,办法是减少进样量,更换样品溶剂。
3.分离不完全,有大量峰重叠,(如系列物),办法,改变流动相,采用梯度洗脱。
拖尾峰的判断和处理(对称性大于1.2)
1.色谱柱柱头塌陷,有死体积,柱效明显下降,所有峰都如此,换柱。
2.色谱柱填料流失,例如C18在碱性流动相中,一般是使用较长时间后造成的,换柱。
3.用C18柱分析碱性化合物,可用BDS柱或未端封尾柱,或在流动相中添加减尾剂(三乙胺等)。
4.过载或超载,减少样品浓度和进样量。
5.保留性太强,出峰太迟,办法,增加洗脱强度。
6.管路有死体积,或连接管路太长,或在管路中有保留。
7.在分离过程中,可能发生结构变化,或存在结构互变,办法,用其它分析体系。
8.分离不完全,后面带有小峰,采用梯度或改变流动相。
9.样品溶解体系同流动相不匹配,样品在柱子中析出。
七.色谱双峰的判断
1.柱头或筛板堵塞,污染,处理办法,清洗筛板或对柱头填料修补。
2.溶剂极性太强或在二相中差异太大,换溶剂,或减少进样量。
3.过载,减少进样量,或进样体积。
4.样品的溶解度,样品难溶于流动相时,会出现双峰。
5.存在互变异构现象,双峰基本齐平。
6.样品存在动态反应,如酸与盐(间苯甲酸与间苯甲酸钠),在特定pH条件下会出双峰。
7.确实是二个组分混合在一起,改变流动相体系。
八.色谱峰变胖的判断和处理
1.死体积增加,如管路和连接管,可能用粗的代替细的,解决办法,缩短连接管路,用细管。
2.柱效降低,色谱柱受损,换柱。
3.流动相不合理,单一如只用甲醇水体系,没有添加改性剂,pH调节剂,也可能柱子选择有问题。
一般除了非极性化合物外,流动相中建议都加盐等化合物,可以提高化合物的分离柱效。
4.溶解与分离体系不一致,如用乙醇溶解样品在甲醇体系中进样,会造成峰位移和峰变宽。
5.大体积进样,在分析柱中进样超过100ul。
6.柱和仪器匹配不合理,如2.1mm内径的色谱柱(包括一体化的柱子,检测器的死体积问题),其管路要求比较细,4.6mm做的好的,2.1的就不行。