美国药典重金属指导原则
2020版《中国药典》重金属检验操作规程(USP)

一、目的:制订详尽的工作程序,规范检验操作,保证检验数据的准确性。
二、范围:本标准适用于参考美国药典标准检验品种重金属的测定。
三、职责:1、检验员:严格按操作规程操作,认真、及时、准确地填写检验记录;2、化验室负责人:监督检查检验员执行本操作规程。
四、内容:1、特殊试剂:1.1硝酸铅原液:将159.8毫克的硝酸铅溶于100毫升水中,加入1毫升硝酸,然后用水稀释至1000毫升。
制备此溶液并将其储存在无可溶性铅盐的玻璃容器中。
1.2标准铅溶液:临用新制,用水稀释10.0毫升硝酸铅原液至100.0毫升。
每毫升标准铅溶液含有相当于10微克的铅。
以每克被测物质100微升标准铅溶液为基础制备的对比溶液包含相当于每百万份被测物质1部分的铅。
2、方法一:2.1 pH3.5乙酸盐缓冲液:溶解25克醋酸铵在25毫升水中,加入6mol/l盐酸38毫升。
如果需要调节,可用6mol/l氢氧化铵或6mol/l盐酸调节pH值为3.5,用水稀释至100毫升,并混合。
2.2标准制备:将标准铅溶液(20微克铅)2毫升放入50毫升比色管中,用水稀释至25毫升。
使用pH计或短程pH指示纸作为外部指示剂,用1mol/l乙酸或6mol/l氢氧化铵调节到3.0到4.0之间的pH,用水稀释至40毫升,混匀。
2.3供试品制备:按照各专著的指示,将试验准备的溶液放入50mL比色管中,或使用各专著中指定体积的酸,溶于水中,用水稀释至25mL,单位为按公式计算的待测物质:2.0/(1000L)其中L是重金属限度,占百分数。
使用pH计或短程pH指示剂纸作为外部指示剂,用1mol/l 乙酸或6 mol/l氢氧化铵调节pH值在3-4之间,用水稀释至40毫升,并混合。
2.4 监测制备:在第三根50mL比色管中,放入按供试品制备指示制备的溶液25mL,并加入2.0mL标准铅溶液。
使用pH计或短程pH指示剂纸作为外部指示剂,用1mol/l乙酸或6mol/l氢氧化铵调节pH值在3-4之间,用水稀释至40毫升,并混合。
美国药典标准

猪八戒照镜子歇后语
猪八戒照镜子歇后语
小编相信一提起猪八戒,没有人是不知道的,这是《西游记》贡献给我国文化长廊中的一个经典角色。
虽说那么的好吃懒做。
但也憨厚惹人爱。
民间就流传着很多关于猪八戒的歇后语,可见他的魅力哦:
猪八戒照镜子——里外不是人
【关于猪八戒的歇后语集锦】
猪八戒照镜子——里外不是人
猪八戒的后脊梁——无能之辈(悟能之背)
猪八戒戴花——自美
和尚打伞——无法无天
猪八戒读书——竟冲识字的
八戒保媒把把成功——猪连必合(珠联壁合)
猪八戒进女儿国——看花了眼
猪八戒娶媳妇——背着走
猪八戒背媳妇——舍得花力气
猪八戒不成仙——坏在嘴上
猪八戒吃黄连/猪八戒吃人参果——苦了大嘴的
猪八戒吃猪啼——自残骨肉
猪八戒充英雄——只是嘴皮子拱得欢
猪八戒戴耳环——自以为美
猪八戒戴花——越多越丑
猪八戒的武艺/猪八戒过火焰山/猪八戒耍把式——倒打一耙
猪八戒的嘴巴——自我欣赏
猪八戒掉进万花筒——丑态百出
猪八戒发眸气——又丑又恶
猪八戒拱帘子——嘴先进
猪八戒进了女儿国——看花了眼
猪八戒进屠场——自己贡献自己
猪八戒啃地梨——什么仙人吃什么果
猪八戒了天拜佛——掸心不稳
猪八戒买猪肝——难得心肠
猪八戒卖炒肝——这是哪道肺
猪八戒卖凉粉——样数不多,滋味不少
猪八戒三十六变——没有一副好嘴脸
猪八戒摔镜子——怕露丑
猪八戒西天取经——三心二意
猪八戒相亲——怕露嘴脸
猪八戒想娶媳妇——一厢情愿
猪八戒背媳妇——心甘情愿
猪八戒招亲——黑灯黑人
猪八戒照镜子/猪八戒照像——自找难堪(看)。
2020版《中国药典》重金属检验操作规程(USP)
一、目的:制订详尽的工作程序,规范检验操作,保证检验数据的准确性。
二、范围:本标准适用于参考美国药典标准检验品种重金属的测定。
三、职责:1、检验员:严格按操作规程操作,认真、及时、准确地填写检验记录;2、化验室负责人:监督检查检验员执行本操作规程。
四、内容:1、特殊试剂:1.1硝酸铅原液:将159.8毫克的硝酸铅溶于100毫升水中,加入1毫升硝酸,然后用水稀释至1000毫升。
制备此溶液并将其储存在无可溶性铅盐的玻璃容器中。
1.2标准铅溶液:临用新制,用水稀释10.0毫升硝酸铅原液至100.0毫升。
每毫升标准铅溶液含有相当于10微克的铅。
以每克被测物质100微升标准铅溶液为基础制备的对比溶液包含相当于每百万份被测物质1部分的铅。
2、方法一:2.1 pH3.5乙酸盐缓冲液:溶解25克醋酸铵在25毫升水中,加入6mol/l盐酸38毫升。
如果需要调节,可用6mol/l氢氧化铵或6mol/l盐酸调节pH值为3.5,用水稀释至100毫升,并混合。
2.2标准制备:将标准铅溶液(20微克铅)2毫升放入50毫升比色管中,用水稀释至25毫升。
使用pH计或短程pH指示纸作为外部指示剂,用1mol/l乙酸或6mol/l氢氧化铵调节到3.0到4.0之间的pH,用水稀释至40毫升,混匀。
2.3供试品制备:按照各专著的指示,将试验准备的溶液放入50mL比色管中,或使用各专著中指定体积的酸,溶于水中,用水稀释至25mL,单位为按公式计算的待测物质:2.0/(1000L)其中L是重金属限度,占百分数。
使用pH计或短程pH指示剂纸作为外部指示剂,用1mol/l 乙酸或6 mol/l氢氧化铵调节pH值在3-4之间,用水稀释至40毫升,并混合。
2.4 监测制备:在第三根50mL比色管中,放入按供试品制备指示制备的溶液25mL,并加入2.0mL标准铅溶液。
使用pH计或短程pH指示剂纸作为外部指示剂,用1mol/l乙酸或6mol/l氢氧化铵调节pH值在3-4之间,用水稀释至40毫升,并混合。
美国药典溶出度试验方法的建立与验证指导原则的解读-yh20170630.pptx

美国药典溶出度试验⽅法的建⽴与验证指导原则的解读-yh20170630.pptx美国药典溶出度试验⽅法的建⽴与验证指导原则的解读2017.6.30涵盖内容总体评价溶出介质溶出仪器实验设计测定⽅法与验证总体评价总体评价-限度限度范围应考虑的问题1、多批次的考量2、具有代表性3、具有针对性(针对重点药品)4、考虑样品的稳定性总体评价-区分⼒区分可能影响体内活性的改变处⽅组成、API的晶型关键辅料的品规⽣产⼯艺关键⼯艺参数等等总体评价-稳定性稳定性(重点是影响因素)总体评价-变异范围变异区间的考量总体评价-变异范围药物本⾝的原因举例总体评价-变异范围试验设计的原因举例溶出介质溶出介质选择的基本原则应考察API及制剂的理化性质和介质对API的影响:溶解性稳定性尽可能模拟药物在体内释药过程的环境溶出介质漏槽条件(SINKCONDITION)在静态循环溶出装置中随着溶出的进⾏杂质中药物浓度↑浓度梯度(Cs-C)↓扩散动⼒↓溶出速度↓☆漏槽条件:溶出介质量应超过药物全溶时所需量,⾄少3倍以上。
☆如果溶出介质的区分⼒⾜够,且经过验证,不满⾜漏槽条件也是允许的。
溶出介质释药介质的基本种类1、⽔(适合⾮pH依赖释药)2、pH1.2-6.8的溶液(速释制剂)3、pH1.2-7.5的溶液(缓释制剂)4、⼈⼯胃液2h+⼈⼯肠液5、⽔+表⾯活性剂(SDS或SLS、胆酸盐等)6、缓冲盐⽔+表⾯活性剂(SDS或SLS、胆酸盐等)7、有机溶媒(不推荐)8、其他如:结肠、⼝腔、眼内释药等特殊介质溶出介质关于表⾯活性剂和有机溶剂有机溶剂:在进⾏⼯艺摸索溶出⽐对时可以使⽤,在拟定质量标准时不建议使⽤。
表⾯活性剂:1)必要时可添加。
2)所使⽤浓度应在多⽔平进⾏考核。
3)应通过考核耐⽤性,确定是否规定表⾯活性剂的纯度。
所⽤缓冲盐与酸的浓度如影响溶出效果时,也应予以考察。
溶出介质其他因素溶出介质体积溶出介质脱⽓处理溶出介质酶允许添加酶的原因:因明胶表⾯会产⽣交联,影响溶出度测定结果,但在体内由于酶的作⽤,不会影响药物的体内释放。
美国药典重金属检测方法-中文

铑
10
40
0.0001
钌
10
40
0.0002
铬
25
100
0.002
钼
25
100
0.0002
镍
25
100
0.002
钒
25
100
0.005
铜
250
1000
0.002
镁
250
1000
0.001
样品制备
范围广泛的各种样品都可以用 USP<232>/<233> 进行分 析,所以提供适合所有样品类型的详细样品处理方法并不现 实。有些药物样品可以直接分析(不用溶解),而其他样品 可以用水性溶剂(如水或稀酸)或适当的有机溶剂(如 2-丁 氧乙醇 : 水(25 : 75)[3],DMSO 或 DGME)简单稀释或 溶解进行制备。用水性或有机溶剂进行简单稀释或溶解的 方法必须考虑样品的化学稳定性,并且对于有机溶剂溶解, 还要考虑样品中组分化合物的不同挥发性。对许多 API 来 说,用有机溶剂稀释是首选方法,这种情况下有必要采取有 助于稳定分析物的方法,以避免因较高或较低挥发性(与校 正标准品相比)成分的存在而造成的回收率波动 [7]。
USP<232> 包括一个涉及元素形态的章节,指出 As 和 Hg 的某些形态值得关注,因为其毒性比其它形态要大得 多。As 的 PDE 是指无机 As,如果总 As 浓度超出限度, 必须用一种能够对不同 As 形态进行分离和定量的方法对样 品进行重新分析。这样做的原因是无机 As 比常见的有机形 式(如,砷甜菜碱)毒性大得多,因此形态分析必须能够分 离其不同化学形态,确定无机 As(亚砷酸盐(三价 As)和 砷酸盐(四价 As))的总量低于限量。同样,Hg 限量也是 指无机 Hg(Hg2+),虽然甲基汞(MeHg)是毒性更大的 形态,但通常认为药物中不可能存在 MeHg。但如果样品来 自于可能含有相当量甲基汞的原料(如,鱼组织),也必须 对其进行特别的分离和测定。
USP标准物质指导原则

确认产品特性
o o
测定纯度
光谱分析(FTIR、NMR、MS 、UV/VIS ) 色谱分析(TLC 、HPLC、GC )
o
直接纯度测试
o
色谱纯度 非有机杂质 溶剂(水、残留溶液)
间接纯度测试(示例)
比旋度 元素分析 官能团滴定
提供附加信息,以支持质量平衡指定以及评测各批次的持续性(即根据另一种规范标准进行试验) 对其他特性进行评测
7. 包装
每种标准物质的内部审核和批准过程可分为 3 个阶段。 只有在第 3 阶段中获得内部科学审查小组的一致同意,标准物质才能提交到各论或附录专家委员会进行投票。 只要科学审查小组成员中有一人投反对票,该标准物质就必须返回进行额外测试和/或校正。 此外,专家委员会志愿者成员也可以请求进行额外测试和/或校正,并最终决定标准物质的适用性。
USP 标准物质建立:从雏形到目录
1. 确定是否需要开发新的标准物质
确立 USP 标准物质首先要解决对散装材料的需求,首先从制定新的各论或现有各论或标准的新应用开始。
2. 采购待用材料
USP 与 USP 文件标准倡议者合作,必要时也联合其他产品制造商,共同获取待用材料。同时还可以向供应商请求提供诸 如稳定性数据、包装、储藏以及处理情况之类的重要信息。USP 将积极听取制造厂商的建议,这是因为他们都是制药、辅 料、食物添加剂、食品化学以及生物材料方面的专家。下载美国药典 (USP) 标准物质材料供应商指导原则 (仅提供英文版)。 为标准物质采购待用材料之前,需要首先以药典用途作为依据确定采购说明。我们要求待用材料
8. 包装后质量控制 (QC) 和质量保证 (QA) 审核
USP 符合 ISO 9001:2000 质量管理体系认证和 cGMP 质量管理规范。每批标准物质在包装和贴完标签后要进行清场。每 批标准物质在发往仓库之前要执行包装后质量控制测试、标签使用前检查以及最终质量保证批次记录审查。
各国重金属和农残限量和标准

锻石膏
重金属:含重金属不得过百万分之十。
白矶
重金属:含重金属不得过百万分之二十。
玄明粉
重金属:含重金属不得过百万分之二十。
含石申量不得过百万分之二十0
地龙
重金属:含重金属不得过百万分之三十。
芒硝
重金属:含重金属不得过百万分之十。 含砷量不得过百万分之十。
氯氰菊酯<1.0mg/kg
九、德国:
重金属限量:
铅(Pb)<5mg/kg。
汞(Hg)<0.1mg/kg。
镉(Cd)<0.2mg/kg。
十、法国:
重金属限量:
铅(Pb)<5mg/kg。
汞(Hg)<0.1mg/kg。
镉(Cd)<0.2mg/kg。
十一、英国:
重金属及砷盐限量:
砷(As)食品总量 <1mg/kg,草药w5mg/kg。
18)三泰芬(Triadimefo n)
芍药0.01
19)赛福宁(Triforine)
芍药0.1
20)赛福唑(Triflumizole)
黄芪0.1芍药1.0
21)芬瑞莫(Fenarimol)
黄芪0.5
22)二甲戊乐灵(Pendimethalin)当归0.2麦门冬0.2柴胡0.2芍药0.2红花0.1
23)芬普宁(Fenpropathrin)
1、中药材:(生药农药残留量的行业标准)
适用范围:黄芪、远志、甘草、桂皮、细辛、山茱萸、苏叶、大枣、陈皮、
枇杷叶、牡丹皮
BHC总量<0.2mg/kg
DDT总量<0.2mg/kg
2、中药制剂:(汉方及生药制剂农药残留量的行业标准)
ICH指导原则-Q3D_Final_Business_Plan-中文版

最终商业计划Q3D:杂质:元素杂质指南2009年7月17日指导委员会于2009年10月29日批准问题及耗费目前并没有统一的指南对药物制剂及辅料中的金属杂质作出合理的控制。
ICH Q3D指南将杂质分类为有机杂质、无机杂质及残留溶剂。
然而ICH指南中只对有机杂质及残留溶剂作了恰当限定,对无机杂质未作限定。
在ICH Q3C中仅有以下关于无机杂质的信息描述:“通常采用药典或其他恰当的方法对无机杂质进行常规检测及定量。
在药物研发过程中应对残留在新原料药中的催化剂进行评估。
应对新原料药质量标准中关于无机杂质的引入或去除进行必要的讨论。
可接受标准应基于药典标准中已知的安全性数据制定得出。
”从以下的限度指南中有明显的问题存在:“1. 现行药典中对于无机杂质的控制方法包括:重金属检验、炽灼残渣/硫酸盐灰分法及其他湿化学检验。
这些检验方法在某些情况下适用于无机杂质的检验,但是也存在一些缺陷,如不属于特征性检验、对现代化合成工艺过程中可能引入的低水平的残留金属催化剂及试剂难以检测到等。
在目前不具备基于风险因素的金属控制方法的情况下,其中,重金属检验往往被用于对潜在金属杂质的初筛。
2. 药典标准是参照旧版制定,而不必基于恰当的安全性数据。
采用药典常用方法,尤其是重金属检验,可检测出金属的数量并加以限制,但并未从最大毒性角度来对其中大部分金属进行考虑。
”目前,药典标准对无机杂质的控制仍存在其他的问题。
药典中对无机杂质的分析方法及标准见于通则或在辅料、原料药及药物制剂的特定章节中。
通则及辅料章节目前在PDG(药典讨论组)的讨论范畴内,但不包括原料药及药物制剂章节。
无机杂质中的某些章节在PDG 范畴内,不包括其他章节。
即使对于PDG中所考虑的通则,不同药典中的基本方法也并不是完全一致的,而使得统一的协调指南难以制定出来。
总的来说,与通常的分析方法及药物制剂、辅料中金属杂质的限度控制标准不一致,未对与病人安全最为相关的金属进行充分控制。
USP35(231)重金属检查第二法中英对照

Method II方法ⅡpH 3.5 Acetate Buffer— Prepare as directed under Method I.取醋酸铵25.0g,加水25ml溶解后,加38.0ml6N盐酸,如有必要用6N氨水或6N 盐酸调PH至3.5 ,用水稀释至100ml,摇匀。
Standard Preparation— Prepare as directed under Method I.标准溶液的制备取50ml比色管,用移液管移取2.0ml标准铅溶液(20 gPb),用水稀释到25ml,用1N醋酸或6N氨水溶液调PH至3.0~4.0之间,用窄范围的精密pH试纸作指示,然后加水稀释至40ml,摇匀。
Test Preparation— Use a quantity, in g, of the substance to be tested as calculated by the formula:2.0 / (1000L),供试品溶液的制备称取一定量的样品(以g计算) ,按公式2.0/1000L计算,in which L is the Heavy metals limit, in percentage. Transfer the weighed quantity of the substance to a suitable crucible, add sufficient sulfuric acid to wet the substance, and carefully ignite at a low temperature until thoroughly charred. (The crucible may be loosely covered with a suitable lid during the charring.)其中L为重金属的限度(%),置适宜的坩埚中,加适量的硫酸预以湿润,在低温小心炽灼,直至全部炭化,(炭化过程坩埚盖可不盖严),Add to the carbonized mass 2 mL of nitric acid and 5 drops of sulfuric acid, and heat cautiously until white fumes no longer are evolved. Ignite, preferably in a muffle furnace, at 500 to 600, until the carbon is completely burned off.炭化物中加2ml硝酸和5滴硫酸,小心加热,直至不再挥发白色烟雾,于500~600℃炽灼, 直至炭完全灰化。
fda药物重金属含量标准

fda药物重金属含量标准FDA(美国食品和药物管理局)是负责监管和管理药物安全的重要机构之一。
在药物质量控制中,重金属含量是一个非常重要的标准。
本文将介绍FDA对药物重金属含量的标准以及其重要性。
一、背景介绍重金属是指密度高、刚性大的金属元素,如铅、汞、砷等。
它们具有一定的毒性,在人体内长期积累可能导致严重的健康问题。
因此,在药物生产中,控制药物中重金属的含量非常重要。
二、FDA的药物重金属含量标准根据FDA的规定,药物中重金属的含量必须符合一定的标准。
具体标准如下:1. 铅(Lead):每日剂量不得超过0.5微克。
2. 汞(Mercury):每日剂量不得超过0.3微克。
3. 砷(Arsenic):每日剂量不得超过0.5微克。
这些标准是为了保证药物的安全性和质量,确保人们使用药物时不会受到重金属的危害。
三、重金属含量标准的重要性药物的主要作用是治疗疾病和保护人们的健康。
然而,如果药物中含有过多的重金属,就会对人体产生负面影响,并可能导致中毒。
1. 铅的危害:铅是一种常见的重金属,长期摄入过多的铅可能导致神经系统受损、肾功能异常等问题。
2. 汞的危害:汞对人体的中枢神经系统和免疫系统有毒性影响,长期摄入过多的汞可能导致脑损伤和免疫系统紊乱。
3. 砷的危害:砷是一种高度有毒的重金属,长期摄入过多的砷可能引起多种癌症和其他疾病。
因此,严格控制药物中重金属的含量是保障患者用药安全的重要手段之一。
四、重金属检测方法为了确保药物的质量,需要使用精确、可靠的方法来检测重金属的含量。
常见的重金属检测方法包括原子吸收光谱法、电感耦合等离子体质谱法和感应耦合等离子体质谱法等。
这些方法能够快速、准确地测定药物中的重金属含量,确保药物的质量符合标准要求。
五、国际标准与合规要求除了FDA的标准外,国际上也有许多其他机构和组织对药物中的重金属含量提出了要求。
例如,欧洲药典委员会(European Pharmacopoeia, EP)也制定了类似于FDA标准的重金属含量限制。
美国药典USP32-重金属测试

<231> 重金属本试验系在规定的试验条件下,金属离子与硫化物离子反应显色,通过制备的标准铅溶液目视比较测定,以确证供试品中重金属杂质含量不超过各论项下规定的限度(以供试品中铅的百分比表示,以重量计)。
(见分光光度法和光散射项下测定法目视比较法<851>)[ 注意:对本试验有响应的典型物质有铅、汞、铋、砷、锑、锡、镉、银、铜和钼等]。
除各论另有规定外,按第一法测定重金属。
第一法适用于在规定试验条件下,能产生澄清、无色溶液的物质。
第二法适用于在第一法规定试验条件下不能产生澄清、无色溶液的物质,或者适用于由于性质复杂,易干扰硫化物离子与金属离子形成沉淀的物质,或者是不易挥发的和易挥发的油类物质。
第三法为湿消化法,仅用于第一法、第二法都不适合的情况。
特殊试剂特殊试剂特殊试剂特殊试剂硝酸铅贮备液—取硝酸铅159.8mg,溶于100ml水中,加1ml硝酸,用水稀释至1000ml。
制备和贮存本溶液的玻璃容器应不含可溶性铅。
标准铅溶液—使用当天,取硝酸铅贮备液10.0ml,用水稀释至100.0ml。
每1ml的标准铅溶液含相当于10µg的铅。
按每克供试品取100µl标准铅溶液制备的对照溶液,相当于供试品含百万分之一的铅。
方法方法方法方法IIII pH3.5醋酸盐缓冲液—取醋酸铵25.0g溶于25ml水中,加6N盐酸液38.0ml,必要时,用6N氢氧化铵液或6N盐酸液调节pH至3.5,用水稀释至100ml,混匀。
标准溶液准备—精密量取标准铅溶液2ml,(相当于20µg的Pb),置50ml比色管中,加水稀释至25ml,以精密pH试纸作为外指示剂,用1N醋酸液或6N 氢氧化铵液调节pH至3.0~4.0,用水稀释至40ml,混匀。
供试品溶液制备—取各论项下规定的供试品溶液25ml,置50ml比色管中,或用各论项下规定用量的酸溶解样品,再用水稀释至25ml,供试品以g计,按下式计算: 2.0/(1000L)式中L是重金属限度(%)。
美国药典重金属指导原则

General Chapter on Inorganic Impurities: Heavy MetalsUSP Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals andUSP Staff*The following Stimuli article is provided to interested parties in advance of publishing in the September-October Pharmacopeial Forum 34(5). You may send comments on this article to Kahkashan Zaidi, PhD, Senior Scientist, Documentary Standards Division, US Pharmacopeia, 12601 Twinbrook Parkway, Rockville, MD 20852-1790; tel. 301.816.8269; e-mail kxz@. The deadline for comments is December 15, 2008.ABSTRACT In the ICH Q3A Impurities in Drug Substances guidance, impurities are classified as organic, inorganic, and residual solvents. Within the inorganic impurities classification, the metals listed in Table 1 are important to control in food, dietary supplements, and drug articles. Many toxic metal impurities found in pharmaceutical articles have been controlled for years by application of the Heavy Metals test described in USP–NF General Chapter Heavy Metals<231>. However, the procedures and the methods contained in <231> lack the sensitivity, specificity, and recovery to monitor properly the levels of these metals. A number of additional chapters for the control of specific metals and other inorganic impurities are contained in USP–NF. This Stimuli article proposes a new USP General Chapter for the control of inorganic impurities in drug and dietary supplement articles intended for use in humans. For the purposes of this article, inorganic impurity, metal, and element all refer to those elements listed in Table 1. The proposed new General Chapter recommends procedures that rely on modern analytical technology and includes limits that are based on toxicity and exposure levels for the selected metals. The new General Chapter also introduces a performance-based approach for the selection of the appropriate technology. This chapter is proposed to replace <231> and may impact other General Chapters that control metals.INTRODUCTIONAmong the category of inorganic impurities, metal impurities have long been monitored in food and drug articles intended for consumption by humans and other animals. For purposes of this General Chapter, drug articles include: drug substances and products (including natural-source and rDNA biologics) and excipients. Dietary supplements and their ingredients are also included, but other foods and food ingredients will not be addressed. Some metals may pose no significant health hazard at sufficiently low exposure levels, when present as certain complexes, at certain oxidation states, or in organic combinations. This chapter should be considered a screening method to identify the presence of potentially hazardous elements. Where speciation of an element is important, further testing is necessary. In these cases, the monograph will include specific instructions for appropriate identification and control. The topic of speciation will not be covered further in this article.Some inorganic impurities are toxic at low levels, and these impurities should be monitored to ensure safety. Sources of inorganic impurities include those that are deliberately added to the process (e.g., catalysts), those that are carried through a process that is conducted according to good manufacturing practices (e.g., undetected contaminants from starting materials or reagents), those coming from the process (e.g., leaching from pipes and other equipment), and those thatcontrol of these impurities may be certified by a vendor, but purchasers also must corroborate the absence of impurities before using these materials in a manufactured article.The General Chapters Expert Committee of the USP Council of Experts formed an Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals to assist the Expert Committee in revision of General Chapter Heavy Metals <231>. As drafted by this Ad Hoc Advisory Panel and revised by the Expert Committee, the proposed revision specifies that the level of each inorganic impurity should not exceed the limit defined in Table 1 or otherwise specified in the individual monograph. This level is determined by concomitant comparison with a monitor solution and USP Reference Standard solution(s).The selection of an instrumental technique and a procedure for the evaluation of the inorganic impurities specified in Table 1 requires the evaluation of a large number of variables including, among others, sensitivity, precision, accuracy, compatibility, time, and cost. The method selected may include plasma spectrochemistry, atomic absorption spectroscopy, or any other method that displays requisite accuracy (trueness and uncertainty) and established sensitivity and specificity. Meeting this requirement must be demonstrated experimentally using the USP Reference Standard(s). Any procedure that provides measurement values within ± 20% of the certified concentration for each element in the appropriate USP Reference Standard(s) is considered to be an acceptable procedure to demonstrate compliance. A guide for the selection of a procedure is presented in Figure 1. When a manufacturer does not have a preferred procedure, or when the preferred procedure does not meet criteria for performance described above, proceed as directed in the remainder of this General Chapter.Procedure—Determine the levels of individual inorganic impurities by the test, unless the individual monograph specifies otherwise.Reagents—All reagents used for the preparation of sample and standard solutions should be free of inorganic impurities in accordance with Plasma Spectrochemistry <730>. Commercial, National Institute of Standards and Technology–traceable elemental stock standards, either single element or multi-element, containing Al, Sb, As, Be, B, Cd, Cr, Co, Cu, In, Ir, Fe, Pb, Li, Mg, Mn, Hg, Mo, Ni, Os, Pd, Pt, Rh, Rb, Ru, Se, Sr, Tl, Sn, W, or Zn at a recommended concentration of 100 µg/mL or greater also are used as reagents.Performance-based USP Reference Standards—USP Inorganic Impurities Class 1 Reference Standard for test articles soluble in aqueous solutions.USP Inorganic Impurities Class 2 Reference Standard for test articles soluble in organic solvents.USP Inorganic Impurities Class 3 Reference Standard for closed-vessel microwave digestions. Equipment—One of the following plasma spectrometers is required for an analyst to perform this multi-element analysis:1. Inductively coupled plasma–atomic (optical) emission spectrometer.2. Inductively coupled plasma–mass spectrometer.In addition, a closed-vessel microwave digestion system may be required for the preparation of test materials (see Figure 1).METHODSample PreparationDetermine the means of sample preparation using the flow chart in Figure 1. The sample preparation scheme should provide sufficient sample loading to allow quantification of each element at the specified limit stated in the corresponding monograph or as stated in Table 1. For closed-vessel microwave digestions follow the manufacturer’s recommended procedures to ensure safe usage. Use utmost caution if concentrated hydrofluoric acid (HF) is used for the preparation of test articles, and review or establish local procedures for safe handling, safe disposal, and HF-tolerant instrumental configurations. [NOTE: The specific details of the Sample preparation have not been included in this Stimuli article but have been developed by the Ad Hoc Advisory Panel. The decision to exclude the specific method details from the Stimuli article is based on the desire of the Ad Hoc Advisory Panel to receive feedback on the concepts proposed herein rather than on the specific method. Based on the feedback received, these details may be included in future chapter development.]System Suitability Criteria—Method reporting limitThe method reporting limit (MRL) is defined as the lowest element concentration of a solution prepared in the working calibration standard matrix that can be experimentally determined to within ± 30% of the prepared concentration. The sensitivity criterion for the method is that the MRL is 0.5 × the USP limit for each applicable element.RecoveryThe suitability of the sample preparation scheme must be demonstrated by preparation and analysis of a suitable USP Reference Standard and by spike recovery measurements of the specific test article according to <730>. The spiked test article solution will be referred to as a Monitor solution. The experimental concentration results shall be ± 20% of the certified concentration for each required element in the analysis. The spike recovery results for the Monitor solution must be ± 20% of the spike concentration for each element. Analysis of a suitable USP Reference Standard shall be included with the analyses of test articles and must be within ± 20% of the certified concentration for each required element for the results to be considered acceptable.CalibrationPrepare calibration standards in the same solution as used for preparation of the test articles. Analysts are encouraged to use internal standards according to <730> for preparation of test article and calibration standard solutions. Prepare 4 working standards plus a blank at element concentrations encompassing the required USP limits for the test article, the USP Reference Standard, and the Monitor solution. Standard curve acceptance criteria must be met according to <730>. If the concentration of an element in the test article solution is determined to be greater than 110% of the highest calibration standard concentration, the test article solution should be appropriately diluted within the range of the standard curve and then reanalyzed.DriftTo monitor instrument drift, analyze a working standard solution at an intermediate concentration of each element immediately following standardization, following the final test solution, and during the analysis at a frequency of one working standard solution analysis per not more than 10 sample analyses during the analytical run. The check standard results should agreetest article solutions that are not bracketed with results within the tolerance for the check standard.Analysis [NOTE: The specific details of the methods have not been included in this Stimuli article but have been developed by the Ad Hoc Advisory Panel. The decision to exclude the specific method details from the Stimuli article is based on the desire of the Ad Hoc Advisory Panel to receive feedback on the concepts proposed herein rather than on the specific method. Based on the feedback received, these details may be included in future chapter development.] Calculations and Reporting—Upon completion of the analysis, calculate the final concentration of a given element in the test article in µg/g from the solution element concentration in μg/mL as follows:C = [(A×V1)/W] × (V2/V3)where:C = concentration of analyte in μg/g,A = instrument reading in μg/mL,V1 = volume of initial test article preparation,W = weight of test article preparation in g,V2 = total volume of any dilution performed in mL, andV3 = aliquot of initial test article preparation used in any dilution performed in mL. Similarly, calculate the final concentration of a given element in the test article in µg/g from the solution element concentration in ng/mL as follows:C = [(A × V1)/W] × (1 μg/1000 ng)(V2/V3)where:C = concentration of analyte in μg/g,A = instrument reading in ng/mL,V1 = volume of initial test article preparation,W = weight of test article preparation in g,V2 = total volume of any dilution performed in mL, andV3 = aliquot of initial test article preparation used in any dilution performed in mL. Calculate the results for each analyte, and compare the values obtained for the test article to those provided in Table 1. The results should not exceed the values in the table.CONCLUSIONThe USP Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals invites comments on the recommendations regarding the use of appropriate analytical instrumentation with limits that are based on toxicity and exposure levels for the metals and the new approach for the determination of an appropriate analytical procedure by the application of USP Reference Standards described in this Stimuli article. Please send detailed comments to: Kahkashan Zaidi, PhD, Senior Scientist, Documentary Standards Division, US Pharmacopeia, 12601 Twinbrook Parkway, Rockville, MD 20852-1790; tel. 301.816.8269; e-mail kxz@.Figure 1. Inorganic impurity decision tree.Table 1. Element limits for oral and parenteral materials. [NOTE: The contents of this table represent a first approximation by members of the Ad Hoc Advisory Panel and are under active discussion internationally.]aUSP Oral Limit, µg/g USP Parenteral Limit, µg/gPermittedElement OralDaily Exposurefor DosageForms, µg/dayAluminum (Al) 50,000 5000 500Antimony (Sb) 20 2 0.2Arsenic (As) 15 1.5 0.15Beryllium (Be) 100 10 1Boron (B) 10,000 1000 100Cadmium (Cd) 25 2.5 0.25Chromium (Cr) 150 15 1.5Cobalt (Co) 1000 100 10Copper (Cu) 500 50 5Indium (In) 100 10 1Iridium (Ir) 100 10 1Iron (Fe) 15,000 1500 150Lead (Pb) 10b 1 0.1Lithium (Li) 600 60 6Magnesium (Mg) c c cManganese (Mn) 7000 700 70Mercury (Hg) 15 1.5 0.15250 25 2.5 Molybdenum(Mo)Nickel (Ni) 1000 100 10Osmium (Os) 100 10 1Palladium (Pd) 100 10 1Platinum (Pt) 100 10 1Rhodium (Rh) 100 10 1Rubidium (Rb) c c cRuthenium (Ru) 100 10 1Selenium (Se) 250 25 2.5Strontium (Sr) 30,000 3000 300Thallium (Tl) 4 0.4 0.04Tin (Sn) 30,000 3000 300Tungsten (W) 375 37.5 3.8Zinc (Zn) 15,000 1500 150a Some of the limits in this table were calculated using the criteria given in the EMEA Guideline on the Specification Limits for Residues of Metal Catalysts, available at: http://www.emea.europa.eu/pdfs/human/swp/444600.pdf, accessed 25 March 2008.b Limit for lead calculated from the FDA limit for bottled drinking water: 5 µg/L assuming consumption of 2 L/day.c Under deliberation.。
美国FDA铅镉溶出量限量标准
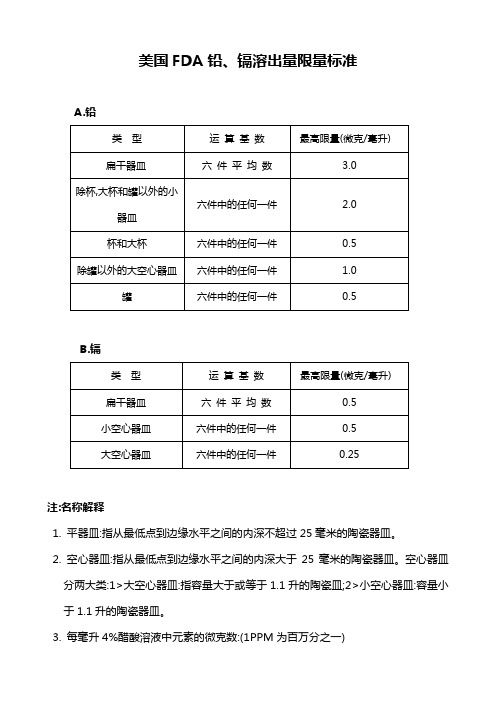
六件中的任何一件
0.5
大空心器皿
六件中的任何一件
0.25
注:名称解释
1.平器皿:指从最低点到边缘水平之间的内深不超过25毫米的陶瓷器皿。
2.空心器皿:指从最低点到边缘水平之间的内深大于25毫米的陶瓷器皿。空心器皿分两大类:1>大空心器皿:指容量大于或等于1.1升的陶瓷皿;2>小空心器皿:容量小于1.1升的陶瓷器皿。
美国FDA铅、镉溶出量限量标准
A.铅
类型
运算基数件平均数
3.0
除杯,大杯和罐以外的小器皿
六件中的任何一件
2.0
杯和大杯
六件中的任何一件
0.5
除罐以外的大空心器皿
六件中的任何一件
1.0
罐
六件中的任何一件
0.5
B.镉
类型
运算基数
最高限量(微克/毫升)
扁干器皿
六件平均数
0.5
3.每毫升4%醋酸溶液中元素的微克数:(1PPM为百万分之一)
美国药典对原料药和制剂中杂质 的控制要求

新原料药中的杂质 新药品中的杂质 杂质:残留溶剂指导原则 杂质:元素杂质指导原则
Q4 Q5 Q6 Q7 Q8 Q9 Q10 Q11新原料药和药品稳定性测试” Q1B – “新原料药和药品光学稳定性测试” Q2A – “分析方法验证” Q3A – “原料药中的杂质” Q3B – “制剂中的杂质” Q3C – “残留溶剂” Q3D – “元素杂质” Q6A – “质量标准:新原料药和制剂的检测方法和可接受标准; 化学物质”
其他关于杂质的通则
干燥失重<731> 检测未知挥发物 眼膏中金属微粒 <751> 检测分散的金属颗粒数目和大小 熔程或熔点<741> 纯度和鉴别 示差<831> 注射剂中颗粒检查<788> 溶液完全性和澄清度 — 注射用氮烯唑胺当按照标签指示溶解,溶液 呈澄清,微黄或黄色 可氧化物质(高锰酸盐溶液呈持久粉红色)各论: 丙酮,羊毛脂等 氮元素检测<431> - 例如:糊精 –蛋白质NMT1.0%
27
内容
介绍 • 历史 • 召回 • 定义 • 杂质分类/来源 • 杂质调查 • 杂质限度设定 注册方面 • ICH • 药典 • FDA 非药物相关性杂质 • 提取物和浸出物 • 人为添加杂质 • 水和辅料中的杂质
28
可提取物和浸出物
可提取物(潜在的可浸出物)在实验室实验条件下,可强行自容器密封系统材料 和组成中转移出的化合物 浸出物 是由药物容器密封系统成分中转移到药物制剂中的化学物质,包括有机物 和无机物 能由玻璃(金属氧化物)、橡胶塞、塑料容器中转出 来自供应商的信息通常是不完整的
“生产设备不应存在任何对该药品的危害。与药品相接触的生产设备部分
原料药中有机杂质研究与控制

原料药中有机杂质研究与控制——综述1.1 前言随着公众和媒体对药物安全性的日益关注,控制药物中的杂质已成为药品质量控制中的重要问题。
人用药品注册技术要求国际协调会(ICH)已经制定了与杂质控制相关的切实可行的指导原则[1],其基本理念已经逐步被国际社会接受。
目前,英国药典(BP)[2]、欧洲药典(EP)[3]和美国药典(USP)[4]均在附录中设有专门的杂质检查通则;中国药典(ChP)[5]二部从ChP(2005)开始,附录中开始设有“药物杂质研究指导原则”;2005年国家食品药品监督管理局(SFDA)发布了《化学药物杂质研究的技术指导原则》[6];之后,2007年又颁布了《药品注册管理办法》,在药品注册审评过程中,对药品中的杂质评价都予以高度重视,极大地促进了药品质量的提高。
1.2 原料药中杂质控制理念的变迁原料药(化学药物)中所含有的杂质,按照《化学药物杂质研究技术指导原则》定义是指任何影响药物纯度的物质。
其杂质一般分为三类:有机杂质、无机杂质和残留溶剂,是包括工艺中引入的杂质和降解产物。
无机杂质是指在原料药及制剂生产或传递过程中产生的杂质,包括:反应试剂、配位体、催化剂、重金属、无机盐等。
残留溶剂是指在原料药及制剂生产过程中使用的有机溶剂。
对于无机杂质和残留溶剂,各国药典都收载了经典、简便、有效的检测方法,故可采用药典的相关方法对其进行控制。
而有机杂质因其化学结构一般与活性成分类似或具渊源关系,故又称为有关物质。
由于有机杂质其产生的途径与工艺流程密切相关,且可能产生毒副作用,从而给原料药本身在药物使用的安全性和有效性方面带来诸多影响,所以各国药品监督部门在对原料药的质量标准制定上,均对有机杂质的控制予以高度重视,并随着新测试仪器的开发利用、分析技术水平的提高,不断地对有机杂质的分析与控制提出更高的要求。
下文中所提“杂质控制”均指有机杂质的控制。
追溯人们对药品中杂质控制理念的变迁,可概括为三个主要阶段:主成分纯度控制阶段、非确定性杂质限度控制阶段和确定性杂质定量限度控制阶段。
美国药典USP32-重金属测试
<231> 重金属本试验系在规定的试验条件下,金属离子与硫化物离子反应显色,通过制备的标准铅溶液目视比较测定,以确证供试品中重金属杂质含量不超过各论项下规定的限度(以供试品中铅的百分比表示,以重量计)。
(见分光光度法和光散射项下测定法目视比较法<851>)[ 注意:对本试验有响应的典型物质有铅、汞、铋、砷、锑、锡、镉、银、铜和钼等]。
除各论另有规定外,按第一法测定重金属。
第一法适用于在规定试验条件下,能产生澄清、无色溶液的物质。
第二法适用于在第一法规定试验条件下不能产生澄清、无色溶液的物质,或者适用于由于性质复杂,易干扰硫化物离子与金属离子形成沉淀的物质,或者是不易挥发的和易挥发的油类物质。
第三法为湿消化法,仅用于第一法、第二法都不适合的情况。
特殊试剂特殊试剂特殊试剂特殊试剂硝酸铅贮备液—取硝酸铅159.8mg,溶于100ml水中,加1ml硝酸,用水稀释至1000ml。
制备和贮存本溶液的玻璃容器应不含可溶性铅。
标准铅溶液—使用当天,取硝酸铅贮备液10.0ml,用水稀释至100.0ml。
每1ml的标准铅溶液含相当于10µg的铅。
按每克供试品取100µl标准铅溶液制备的对照溶液,相当于供试品含百万分之一的铅。
方法方法方法方法IIII pH3.5醋酸盐缓冲液—取醋酸铵25.0g溶于25ml水中,加6N盐酸液38.0ml,必要时,用6N氢氧化铵液或6N盐酸液调节pH至3.5,用水稀释至100ml,混匀。
标准溶液准备—精密量取标准铅溶液2ml,(相当于20µg的Pb),置50ml比色管中,加水稀释至25ml,以精密pH试纸作为外指示剂,用1N醋酸液或6N 氢氧化铵液调节pH至3.0~4.0,用水稀释至40ml,混匀。
供试品溶液制备—取各论项下规定的供试品溶液25ml,置50ml比色管中,或用各论项下规定用量的酸溶解样品,再用水稀释至25ml,供试品以g计,按下式计算: 2.0/(1000L)式中L是重金属限度(%)。
美国药典对正文修订的杂质检查新要求

美国药典对正文修订的杂质检查新要求最近,美国药典对申报正文修订时的杂质检查有新的要求,非复杂有效成分、生物制品和生物技术制品、辅料、疫苗的正文修订申报指导原则(USP guideline for submitting requests for revision to the USP-NF)正在征求意见。
现将其中非复杂有效成分正文修订中的杂质部分简述如下:正文有关内容修订所用术语可参考文件ICH Q3A(R)。
其中指定杂质包括已知杂质(identified impurity)和未知杂质(unidentified impurity)。
原料药中的杂质原料药中的杂质检查,对指定杂质(specified impurities)应规定限量,并且对所有非指定杂质(unspecified impurities)的限量定为0.10%( The impurity test of a drug substance monograph is intended to limit all specified impurities, with a further limit of 0.10% for all unspecified impurities)。
新的正文要求用表1中的命名。
表1杂质检查杂质类型Q3A杂质分类USP传统检查新的USP检查有机起始原料普通杂质色谱纯度有关物质——的限度指定杂质副产物中间体降解产物指定和未指定杂质试剂,配位体,催化剂指定杂质无机试剂,配位体,催化剂无指定杂质重金属和其他残留金属重金属——的限度重金属无机盐炽灼残渣炽灼残渣残留溶剂有机挥发性杂质——的限度有机挥发性杂质USP正文原料药仅检查实际存在的杂质,不检查理论存在的。
如果采用不同的合成工艺,由此产生不同的杂质,则需要用不同的杂质检查方法,并在包装中标明可行的检查方法。
如果杂质的毒性已经FDA评价过,则在修订报告中应包括毒性数据。
有机杂质常用液相和气相法检查,对杂质鉴别和定量,最好用外标法,而不用内标法,用内标法可能混入其他杂质。
美药典关于重金属

Revision BulletinOfficial April 1, 2015〈231〉 Heavy Metals1quantity, in g, of the substance to be tested, as calculated Delete the following:by the formula:2.0/(1000L)v〈231〉 HEAVY METALSin which L is the Heavy metals limit, as a percentage. Usinga pH meter or short-range pH indicator paper as externalindicator, adjust with 1N acetic acid or 6N ammonium This test is provided to demonstrate that the content of hydroxide to a pH between 3.0 and 4.0, dilute with water metallic impurities that are colored by sulfide ion, under to 40mL, and mix.the specified test conditions, does not exceed the HeavyMonitor Preparation—Into a third 50-mL color-compar-metals limit specified in the individual monograph in per-ison tube place 25mL of a solution prepared as directed for centage (by weight) of lead in the test substance, as deter-Test Preparation, and add 2.0mL of Standard Lead Solution. mined by concomitant visual comparison (see Visual Com-Using a pH meter or short-range pH indicator paper as ex-parison in the section Procedure under Spectrophotometryternal indicator, adjust with 1N acetic acid or 6N ammo-and Light-Scattering 〈851〉) with a control prepared from anium hydroxide to a pH between 3.0 and 4.0, dilute with Standard Lead Solution. [N OTE—Substances that typically willwater to 40mL, and mix.respond to this test are lead, mercury, bismuth, arsenic,Procedure—To each of the three tubes containing the antimony, tin, cadmium, silver, copper, and molybdenum.]Standard Preparation, the Test Preparation, and the Monitor Determine the amount of heavy metals by Method I, un-Preparation, add 2mL of pH 3.5 Acetate Buffer, then add less otherwise specified in the individual monograph.1.2mL of thioacetamide–glycerin base TS, dilute with water Method I is used for substances that yield clear, colorlessto 50mL, mix, allow to stand for 2minutes, and view preparations under the specified test conditions. Method IIdownward over a white surface*: the color of the solution is used for substances that do not yield clear, colorlessfrom the Test Preparation is not darker than that of the preparations under the test conditions specified for Methodsolution from the Standard Preparation, and the color of the I, or for substances that, by virtue of their complex nature,solution from the Monitor Preparation is equal to or darker interfere with the precipitation of metals by sulfide ion, orthan that of the solution from the Standard Preparation.for fixed and volatile oils. Method III, a wet-digestion[N OTE—If the color of the Monitor Preparation is lighter than method, is used only in those cases where neither Method Ithat of the Standard Preparation, use Method II instead of nor Method II can be used.Method I for the substance being tested.]Special ReagentsMethod II Lead Nitrate Stock Solution—Dissolve 159.8mg ofN OTE—This method does not recover mercury.lead nitrate in 100mL of water to which has been added1mL of nitric acid, then dilute with water to 1000mL. Pre-pH 3.5 Acetate Buffer—Prepare as directed underpare and store this solution in glass containers free from Method I.soluble lead salts.Standard Preparation—Prepare as directed under Standard Lead Solution—On the day of use, dilute Method I.10.0mL of Lead Nitrate Stock Solution with water to Test Preparation—Use a quantity, in g, of the substance 100.0mL. Each mL of Standard Lead Solution contains the to be tested as calculated by the formula:equivalent of 10µg of lead. A comparison solution pre-pared on the basis of 100µL of Standard Lead Solution per 2.0/(1000L)g of substance being tested contains the equivalent of1part of lead per million parts of substance being tested.in which L is the Heavy metals limit, in percentage. Transferthe weighed quantity of the substance to a suitable cruci-ble, add sufficient sulfuric acid to wet the substance, and Method I carefully ignite at a low temperature until thoroughly char-red. (The crucible may be loosely covered with a suitable pH 3.5 Acetate Buffer—Dissolve 25.0g of ammonium lid during the charring.) Add to the carbonized mass 2mL acetate in 25mL of water, and add 38.0mL of 6N hydro-of nitric acid and 5drops of sulfuric acid, and heat cau-chloric acid. Adjust, if necessary, with 6N ammonium hy-tiously until white fumes no longer are evolved. Ignite, droxide or 6N hydrochloric acid to a pH of 3.5, dilute with preferably in a muffle furnace, at 500° to 600°, until the water to 100mL, and mix.carbon is completely burned off. Cool, add 4mL of 6N Standard Preparation—Into a 50-mL color-comparison hydrochloric acid, cover, digest on a steam bath fortube pipet 2mL of Standard Lead Solution (20µg of Pb),15minutes, uncover, and slowly evaporate on a steam bath and dilute with water to 25mL. Using a pH meter or short-to dryness. Moisten the residue with 1 drop of hydrochloric range pH indicator paper as external indicator, adjust with acid, add 10mL of hot water, and digest for 2minutes.1N acetic acid or 6N ammonium hydroxide to a pH be-Add 6N ammonium hydroxide dropwise until the solution tween 3.0 and 4.0, dilute with water to 40mL, and mix.is just alkaline to litmus paper, dilute with water to 25mL,and adjust with 1N acetic acid to a pH between 3.0 and Test Preparation—Into a 50-mL color-comparison tube4.0, using short-range pH indicator paper as an external place 25mL of the solution prepared for the test as di-indicator. Filter if necessary, rinse the crucible and the filter rected in the individual monograph; or, using the desig-with 10mL of water, combine the filtrate and rinsing in a nated volume of acid where specified in the individual50-mL color-comparison tube, dilute with water to 40mL, monograph, dissolve in and dilute with water to 25mL theand mix.Procedure—To each of the tubes containing the Stan-dard Preparation and the Test Preparation, add 2mL of pH*In those countries or jurisdictions where thioacetamide cannot be used,add 10mL of freshly prepared hydrogen sulfide TS to each of the tubes,mix, allow to stand for 5minutes, and view downward over a white surface.Revision Bulletin 2〈231〉 Heavy Metals Official April 1, 2015 3.5 Acetate Buffer, then add 1.2mL of until no further darkening occurs, then heat strongly to the thioacetamide–glycerin base TS, dilute with water to 50mL,production of dense, white fumes. Cool, cautiously add mix, allow to stand for 2minutes, and view downward over5mL of water, boil gently to the production of dense,a white surface*: the color of the solution from the Test white fumes, and continue heating until the volume is re-Preparation is not darker than that of the solution from the duced to a few mL. Cool, cautiously add 5mL of water, Standard Preparation.and examine the color of the solution. If the color is yellow,cautiously add 1mL of 30percent hydrogen peroxide, andagain evaporate to the production of dense, white fumes Method III and a volume of 2 to 3mL. If the solution is still yellow,repeat the addition of 5mL of water and the peroxide pH 3.5 Acetate Buffer—Prepare as directed under treatment. Cool, dilute cautiously with a few mL of water, Method I.and rinse into a 50-mL color-comparison tube, taking carethat the combined volume does not exceed 25mL.Standard Preparation—Transfer a mixture of 8mL ofsulfuric acid and 10mL of nitric acid to a clean, dry,If the substance is a liquid—Transfer the weighed quan-100-mL Kjeldahl flask, and add a further volume of nitric tity of the test substance to a clean, dry, 100-mL Kjeldahl acid equal to the incremental volume of nitric acid added flask. [N OTE—A 300-mL flask may be used if the reactionto the Test Preparation. Heat the solution to the production foams excessively.] Clamp the flask at an angle of 45°, and of dense, white fumes; cool; cautiously add 10mL of water;cautiously add a few mL of a mixture of 8mL of sulfuric and, if hydrogen peroxide was used in treating the Test acid and 10mL of nitric acid. Warm gently until the reac-Preparation, add a volume of 30percent hydrogen peroxide tion commences, allow the reaction to subside, and pro-equal to that used for the substance being tested. Boil gen-ceed as directed for If the substance is a solid, beginningtly to the production of dense, white fumes. Again cool,with “add portions of the same acid mixture.”cautiously add 5mL of water, mix, and boil gently to the Monitor Preparation—Proceed with the digestion, us-production of dense, white fumes and to a volume of 2 to ing the same amount of sample and the same procedure as 3mL. Cool, dilute cautiously with a few mL of water, add directed in the subsection If the substance is a solid in the 2.0mL of Standard Lead Solution (20µg of Pb), and mix.section Test Preparation, until the step “Cool, dilute cau-Transfer to a 50-mL color-comparison tube, rinse the flask tiously with a few mL of water.” Add 2.0mL of Lead Stan-with water, adding the rinsing to the tube until the volume dard Solution (20µg of lead), and mix. Transfer to a 50-mL is 25mL, and mix.color comparison tube, rinse the flask with water, adding Test Preparation—Unless otherwise indicated in the in-the rinsing to the tube until the volume is 25mL, and mix. dividual monograph, use a quantity, in g, of the substance Procedure—Treat the Test Preparation, the Standardto be tested as calculated by the formula:Preparation, and the Monitor Preparation as follows. Using apH meter or short-range pH indicator paper as external in-2.0/(1000L)dicator, adjust the solution to a pH between3.0 and4.0with ammonium hydroxide (a dilute ammonia solution may in which L is the Heavy metals limit, as a percentage.be used, if desired, as the specified range is approached), If the substance is a solid—Transfer the weighed quantity dilute with water to 40mL, and mix.of the test substance to a clean, dry, 100-mL Kjeldahl flask.To each tube add 2mL of pH 3.5 Acetate Buffer, then add [N OTE—A 300-mL flask may be used if the reaction foams 1.2mL of thioacetamide–glycerin base TS, dilute with water excessively.] Clamp the flask at an angle of 45°, and add a to 50mL, mix, allow to stand for 2minutes, and view sufficient quantity of a mixture of 8mL of sulfuric acid and downward over a white surface*: the color of the Test Prep-10mL of nitric acid to moisten the substance thoroughly.aration is not darker than that of the Standard Preparation, Warm gently until the reaction commences, allow the reac-and the color of the Monitor Preparation is equal to ortion to subside, and add portions of the same acid mixture,darker than that of the Standard Preparation.heating after each addition, until a total of 18mL of the Official: December 1, 2015 acid mixture has been added. Increase the amount of heat,•Official:January 1, 2018•(RB 1-Apr-2015) and boil gently until the solution darkens. Cool, add 2mLv USP38of nitric acid, and heat again until the solution darkens.Continue the heating, followed by addition of nitric acid。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
General Chapter on Inorganic Impurities: Heavy MetalsUSP Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals andUSP Staff*The following Stimuli article is provided to interested parties in advance of publishing in the September-October Pharmacopeial Forum 34(5). You may send comments on this article to Kahkashan Zaidi, PhD, Senior Scientist, Documentary Standards Division, US Pharmacopeia, 12601 Twinbrook Parkway, Rockville, MD 20852-1790; tel. 301.816.8269; e-mail kxz@. The deadline for comments is December 15, 2008.ABSTRACT In the ICH Q3A Impurities in Drug Substances guidance, impurities are classified as organic, inorganic, and residual solvents. Within the inorganic impurities classification, the metals listed in Table 1 are important to control in food, dietary supplements, and drug articles. Many toxic metal impurities found in pharmaceutical articles have been controlled for years by application of the Heavy Metals test described in USP–NF General Chapter Heavy Metals<231>. However, the procedures and the methods contained in <231> lack the sensitivity, specificity, and recovery to monitor properly the levels of these metals. A number of additional chapters for the control of specific metals and other inorganic impurities are contained in USP–NF. This Stimuli article proposes a new USP General Chapter for the control of inorganic impurities in drug and dietary supplement articles intended for use in humans. For the purposes of this article, inorganic impurity, metal, and element all refer to those elements listed in Table 1. The proposed new General Chapter recommends procedures that rely on modern analytical technology and includes limits that are based on toxicity and exposure levels for the selected metals. The new General Chapter also introduces a performance-based approach for the selection of the appropriate technology. This chapter is proposed to replace <231> and may impact other General Chapters that control metals.INTRODUCTIONAmong the category of inorganic impurities, metal impurities have long been monitored in food and drug articles intended for consumption by humans and other animals. For purposes of this General Chapter, drug articles include: drug substances and products (including natural-source and rDNA biologics) and excipients. Dietary supplements and their ingredients are also included, but other foods and food ingredients will not be addressed. Some metals may pose no significant health hazard at sufficiently low exposure levels, when present as certain complexes, at certain oxidation states, or in organic combinations. This chapter should be considered a screening method to identify the presence of potentially hazardous elements. Where speciation of an element is important, further testing is necessary. In these cases, the monograph will include specific instructions for appropriate identification and control. The topic of speciation will not be covered further in this article.Some inorganic impurities are toxic at low levels, and these impurities should be monitored to ensure safety. Sources of inorganic impurities include those that are deliberately added to the process (e.g., catalysts), those that are carried through a process that is conducted according to good manufacturing practices (e.g., undetected contaminants from starting materials or reagents), those coming from the process (e.g., leaching from pipes and other equipment), and those thatcontrol of these impurities may be certified by a vendor, but purchasers also must corroborate the absence of impurities before using these materials in a manufactured article.The General Chapters Expert Committee of the USP Council of Experts formed an Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals to assist the Expert Committee in revision of General Chapter Heavy Metals <231>. As drafted by this Ad Hoc Advisory Panel and revised by the Expert Committee, the proposed revision specifies that the level of each inorganic impurity should not exceed the limit defined in Table 1 or otherwise specified in the individual monograph. This level is determined by concomitant comparison with a monitor solution and USP Reference Standard solution(s).The selection of an instrumental technique and a procedure for the evaluation of the inorganic impurities specified in Table 1 requires the evaluation of a large number of variables including, among others, sensitivity, precision, accuracy, compatibility, time, and cost. The method selected may include plasma spectrochemistry, atomic absorption spectroscopy, or any other method that displays requisite accuracy (trueness and uncertainty) and established sensitivity and specificity. Meeting this requirement must be demonstrated experimentally using the USP Reference Standard(s). Any procedure that provides measurement values within ± 20% of the certified concentration for each element in the appropriate USP Reference Standard(s) is considered to be an acceptable procedure to demonstrate compliance. A guide for the selection of a procedure is presented in Figure 1. When a manufacturer does not have a preferred procedure, or when the preferred procedure does not meet criteria for performance described above, proceed as directed in the remainder of this General Chapter.Procedure—Determine the levels of individual inorganic impurities by the test, unless the individual monograph specifies otherwise.Reagents—All reagents used for the preparation of sample and standard solutions should be free of inorganic impurities in accordance with Plasma Spectrochemistry <730>. Commercial, National Institute of Standards and Technology–traceable elemental stock standards, either single element or multi-element, containing Al, Sb, As, Be, B, Cd, Cr, Co, Cu, In, Ir, Fe, Pb, Li, Mg, Mn, Hg, Mo, Ni, Os, Pd, Pt, Rh, Rb, Ru, Se, Sr, Tl, Sn, W, or Zn at a recommended concentration of 100 µg/mL or greater also are used as reagents.Performance-based USP Reference Standards—USP Inorganic Impurities Class 1 Reference Standard for test articles soluble in aqueous solutions.USP Inorganic Impurities Class 2 Reference Standard for test articles soluble in organic solvents.USP Inorganic Impurities Class 3 Reference Standard for closed-vessel microwave digestions. Equipment—One of the following plasma spectrometers is required for an analyst to perform this multi-element analysis:1. Inductively coupled plasma–atomic (optical) emission spectrometer.2. Inductively coupled plasma–mass spectrometer.In addition, a closed-vessel microwave digestion system may be required for the preparation of test materials (see Figure 1).METHODSample PreparationDetermine the means of sample preparation using the flow chart in Figure 1. The sample preparation scheme should provide sufficient sample loading to allow quantification of each element at the specified limit stated in the corresponding monograph or as stated in Table 1. For closed-vessel microwave digestions follow the manufacturer’s recommended procedures to ensure safe usage. Use utmost caution if concentrated hydrofluoric acid (HF) is used for the preparation of test articles, and review or establish local procedures for safe handling, safe disposal, and HF-tolerant instrumental configurations. [NOTE: The specific details of the Sample preparation have not been included in this Stimuli article but have been developed by the Ad Hoc Advisory Panel. The decision to exclude the specific method details from the Stimuli article is based on the desire of the Ad Hoc Advisory Panel to receive feedback on the concepts proposed herein rather than on the specific method. Based on the feedback received, these details may be included in future chapter development.]System Suitability Criteria—Method reporting limitThe method reporting limit (MRL) is defined as the lowest element concentration of a solution prepared in the working calibration standard matrix that can be experimentally determined to within ± 30% of the prepared concentration. The sensitivity criterion for the method is that the MRL is 0.5 × the USP limit for each applicable element.RecoveryThe suitability of the sample preparation scheme must be demonstrated by preparation and analysis of a suitable USP Reference Standard and by spike recovery measurements of the specific test article according to <730>. The spiked test article solution will be referred to as a Monitor solution. The experimental concentration results shall be ± 20% of the certified concentration for each required element in the analysis. The spike recovery results for the Monitor solution must be ± 20% of the spike concentration for each element. Analysis of a suitable USP Reference Standard shall be included with the analyses of test articles and must be within ± 20% of the certified concentration for each required element for the results to be considered acceptable.CalibrationPrepare calibration standards in the same solution as used for preparation of the test articles. Analysts are encouraged to use internal standards according to <730> for preparation of test article and calibration standard solutions. Prepare 4 working standards plus a blank at element concentrations encompassing the required USP limits for the test article, the USP Reference Standard, and the Monitor solution. Standard curve acceptance criteria must be met according to <730>. If the concentration of an element in the test article solution is determined to be greater than 110% of the highest calibration standard concentration, the test article solution should be appropriately diluted within the range of the standard curve and then reanalyzed.DriftTo monitor instrument drift, analyze a working standard solution at an intermediate concentration of each element immediately following standardization, following the final test solution, and during the analysis at a frequency of one working standard solution analysis per not more than 10 sample analyses during the analytical run. The check standard results should agreetest article solutions that are not bracketed with results within the tolerance for the check standard.Analysis [NOTE: The specific details of the methods have not been included in this Stimuli article but have been developed by the Ad Hoc Advisory Panel. The decision to exclude the specific method details from the Stimuli article is based on the desire of the Ad Hoc Advisory Panel to receive feedback on the concepts proposed herein rather than on the specific method. Based on the feedback received, these details may be included in future chapter development.] Calculations and Reporting—Upon completion of the analysis, calculate the final concentration of a given element in the test article in µg/g from the solution element concentration in μg/mL as follows:C = [(A×V1)/W] × (V2/V3)where:C = concentration of analyte in μg/g,A = instrument reading in μg/mL,V1 = volume of initial test article preparation,W = weight of test article preparation in g,V2 = total volume of any dilution performed in mL, andV3 = aliquot of initial test article preparation used in any dilution performed in mL. Similarly, calculate the final concentration of a given element in the test article in µg/g from the solution element concentration in ng/mL as follows:C = [(A × V1)/W] × (1 μg/1000 ng)(V2/V3)where:C = concentration of analyte in μg/g,A = instrument reading in ng/mL,V1 = volume of initial test article preparation,W = weight of test article preparation in g,V2 = total volume of any dilution performed in mL, andV3 = aliquot of initial test article preparation used in any dilution performed in mL. Calculate the results for each analyte, and compare the values obtained for the test article to those provided in Table 1. The results should not exceed the values in the table.CONCLUSIONThe USP Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals invites comments on the recommendations regarding the use of appropriate analytical instrumentation with limits that are based on toxicity and exposure levels for the metals and the new approach for the determination of an appropriate analytical procedure by the application of USP Reference Standards described in this Stimuli article. Please send detailed comments to: Kahkashan Zaidi, PhD, Senior Scientist, Documentary Standards Division, US Pharmacopeia, 12601 Twinbrook Parkway, Rockville, MD 20852-1790; tel. 301.816.8269; e-mail kxz@.Figure 1. Inorganic impurity decision tree.Table 1. Element limits for oral and parenteral materials. [NOTE: The contents of this table represent a first approximation by members of the Ad Hoc Advisory Panel and are under active discussion internationally.]aUSP Oral Limit, µg/g USP Parenteral Limit, µg/gPermittedElement OralDaily Exposurefor DosageForms, µg/dayAluminum (Al) 50,000 5000 500Antimony (Sb) 20 2 0.2Arsenic (As) 15 1.5 0.15Beryllium (Be) 100 10 1Boron (B) 10,000 1000 100Cadmium (Cd) 25 2.5 0.25Chromium (Cr) 150 15 1.5Cobalt (Co) 1000 100 10Copper (Cu) 500 50 5Indium (In) 100 10 1Iridium (Ir) 100 10 1Iron (Fe) 15,000 1500 150Lead (Pb) 10b 1 0.1Lithium (Li) 600 60 6Magnesium (Mg) c c cManganese (Mn) 7000 700 70Mercury (Hg) 15 1.5 0.15250 25 2.5 Molybdenum(Mo)Nickel (Ni) 1000 100 10Osmium (Os) 100 10 1Palladium (Pd) 100 10 1Platinum (Pt) 100 10 1Rhodium (Rh) 100 10 1Rubidium (Rb) c c cRuthenium (Ru) 100 10 1Selenium (Se) 250 25 2.5Strontium (Sr) 30,000 3000 300Thallium (Tl) 4 0.4 0.04Tin (Sn) 30,000 3000 300Tungsten (W) 375 37.5 3.8Zinc (Zn) 15,000 1500 150a Some of the limits in this table were calculated using the criteria given in the EMEA Guideline on the Specification Limits for Residues of Metal Catalysts, available at: http://www.emea.europa.eu/pdfs/human/swp/444600.pdf, accessed 25 March 2008.b Limit for lead calculated from the FDA limit for bottled drinking water: 5 µg/L assuming consumption of 2 L/day.c Under deliberation.。