93_42_EEC 中文版
MDD技术文档指导文件

发布地点,日期:
签名:
7. 技术文档语言 一个成员国家可能要求技术文档的 A 部分使用官方语言,如果能懂可以不翻译。在需要翻译的情 况,允许文件拥有者额外的时间递交第一部分的内容给检查机构。
而且,关于翻译不能增加更多条件,例如要求有授权的翻译,官方翻译或类似要求。
注:本指令的信息仅作为生产商的指南,生产商仍需仔细阅读并理解 MDD.
2. 技术文档的来历
根据 MDD 93/42/EEC,所有的医疗器械必须符合基本要求附录 I 才能贴 CE 标志。技术文档则提供产 品符合基本要求的文件证据。
然而,技术文档这个词对制造商而言将比较费解,因为在 MDD 中未提到这个词。技术文档是技术 性文件的一个通称,用于证明产品符合基本要求。MDD 的不同附录将会略微不同地描述技术文 档。接下来便介绍每一个 MDD 附录如何描述技术文档。
附录 A) 全部的制造和检验计划 风险分析(见附录 B) 临床数据(见附录 C) 标签,如产品标签,说明书,病人信息,广告材料 合格声明
B 部分包含所有的检测和验证报告,产品有关的质量体系信息,产品的详尽描述如设计图,产品数 据参数,制造过程描述等。如果 B 部分是分散的,应建立一个控制目录,列出每个相关文件。
III 类医疗器械: -附录 II,包括第 4 部分(常指 II.4) -附录 III+附录 IV -附录 III+附录 V
IIb 类医疗器械: -附录 II,不包括第 4 部分(常指 II.3) -附录 III+附录 IV -附录 III+附录 V -附录 III+附录 VI
IIa 类医疗器械: -附录 II,不包括第 4 部分(常指 II.3)
93-42-EEC中文版

欧洲共同体理事会关于医疗器械的93/42/EEC指令来源:日期:2005-4-28 10:44:371993年6月14日欧洲共同体理事会考虑到建立欧洲经济共同体的条约,特别是其第100a条;考虑到欧洲共同体委员会提交的议案;考虑到与欧洲议会合作;考虑到经济与社会委员会的意见;鉴于应在欧洲共同体内部市场范围内正式通过必要的措施;鉴于欧洲共同体内部市场是一个确保商品、人员、服务和资本自由流通的无内部边界的区域;鉴于各成员国有关医疗器械的安全、健康保护和工作特性的法律、法规和行政条款的内容和范围是不同的;鉴于各成员国之间对这类器械的认证和检验程序存在差异;鉴于这种差异在欧洲共同体内形成贸易壁垒;鉴于各国针对医疗器械的使用所制定的有关患者、使用者及其他人员的安全和健康保护的条款应予以协调,以保证此类器械在欧洲共同体内部市场自由流通;鉴于协调条款必须与成员国为管理直接或间接与这类器械有关的公共健康和医疗保险计划的资金筹措而采取的措施相区别;鉴于因而只要遵守欧洲共同体法律,这些条款并不影响各成员国实施上述措施的能力;鉴于医疗器械应向患者、使用者及第三方提供高水平的保护并达到制造商赋予其的性能水平;鉴于因此保持和提高各成员国已达到的保护水平是本指令的基本目的之一;鉴于在1965年1月26日欧洲共同体理事会关于使有关特许专卖药品的法律、法规或行政措施趋于一致的65/65/EEC指令中,某些医疗器械是用于施药的;鉴于在这种情况下,医疗器械投放市场通常由本指令管理,而药品投放市场由65/65/EEC指令管理;鉴于如果这种器械投放市场的方式使器械与药品构成一种规定只供组合使用且不能再次使用的整体,则这个单一整体产品应由65/65/EEC指令管理;鉴于必须将上述器械与包含某种物质,特别是当其单独使用时,按65/65/EEC指令可视为药物的医疗器械相区别;鉴于在这种情况下,若这种物质能配合医疗器械对人体产生辅助作用,则这类器械投放市场由本指令管理;鉴于这类物质的安全性、质量和有效性必须比照欧洲共同体理事会1975年5月20日关于使成员国有关分析标准、药物毒理学标准和临床标准及特许专卖药品试验协议的法律趋于一致的75/318/EEC指令规定的适当方法加以验证;鉴于本指令附录中的基本要求和其他要求,包括对“减小”或“降低”危险的任何引用,在解释和实施时必须考虑设计时的技术现状和实际做法及高水平的健康和安全相适应的技术与经济条件;鉴于按照1985年5月7日欧洲共同体理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和生产的规定必须限于满足基本要求所必须的条款;鉴于因为这些要求是基本的,因此它们应取代各国相应的条款;鉴于实施基本要求应审慎考虑设计时的技术水平和与高水平的健康及安全保护相适应的技术、经济条件;鉴于1990年6月20日欧洲共同体理事会关于使成员国有关有源植入式医疗器械的法律趋于一致的90/385/EEC指令是新方法指令在医疗器械领域中的首次应用案例;鉴于为了使欧洲共同体的统一规定适用于所有医疗器械,本指令基本上是以90/385/EEC指令的条款为依据的;鉴于为此必须修订90/385/EEC指令,以便放入本指令规定的一般性条款;鉴于电磁兼容性问题是医疗器械安全的一个组成部分;鉴于就1989年5月3日欧洲共同体理事会关于使成员国有关电磁兼容的法律趋于一致的89/336/EEC指令而言,应包含这方面的专门规定;鉴于本指令应包括有关发射电离辐射的器械在设计和制造方面的要求;鉴于本指令既不影响1980年7月15日欧洲共同体理事会80/836/Euratom指令对有关保护公众和工人免受电离辐射危险的基本安全标准的指令进行修订所要求的授权,也不影响欧洲共同体理事会1984年9月3日对接受医疗检查和治疗的人员的辐射防护规定了基本措施的84/466/Euratom指令的实施;鉴于欧洲共同体理事会1989年6月12日关于采取措施鼓励改善工人工作中的安全和健康的89/391/EEC指令以及有关这一问题的专门指令应继续予以实施;鉴于为了证实符合基本要求并使这种符合得到验证,需要制定欧洲协调标准来防止与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方机构制定的,应保持其非强制性的地位;鉴于为此欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)按照1984年11月13日欧洲共同体委员会与这两个机构之间签署的合作总指导原则,被认可为批准协调标准的主管机构;鉴于在本指令中,协调标准是受欧洲共同体委员会委托,由上述两机构之一,或两个机构共同根据欧洲共同体理事会1983年3月18日关于在技术标准和法规领域提供信息程序的83/189/EEC指令,依照上述总指导原则而批准的技术规范(欧洲标准或协调文件);鉴于对协调标准进行修订,欧洲共同体委员会应得到根据83/189/EEC 指令建立的常设委员会的帮助;鉴于应采取的措施必须按欧洲共同体理事会87/373/EEC决定中规定的程序Ⅰ而规定;鉴于在特定领域,欧洲药典专著这类现有的形式应包括在本指令的范围内;鉴于因此有几部欧洲药典专著可视为等同于上述协调标准。
普通医疗器械产品CE认证分类

根据 MDD 附录九 [93/42/EEC]规则 1 非插入式器械属于I类器械,但适用以下其他规则的除外.规则 2 用于输送和储存血液、体液或人体组织和其他液体和气体为人体吸收、服用或注入的非插入式样器械,属于IIa类:如果他们可以同IIa或更高类别的有源器械连接使用;或如果他们的预定功能属于储存和输送血液和其他体液、储存人体器官或人体组织.其他的都属于I类。
规则 3插入式器械用于改变血液、体液和其他注入人体的液体的生物和化学成分属于III 类器械。
但处理方法属于过滤、气体和热能的分离或交换的该类非插入式器械属于IIa.除此以外的其他情况属于I类。
规则 4同受伤皮肤接触的非插入式器械:如果用于形成机械屏障,阻止或吸收渗出液体,属于I类;如果主要用于辅助治疗已经伤及真皮的创伤,属于IIb类;其他情况属于IIa,包括主要用于处理创伤周围环境的器械.规则 5插入式器械,除非属于外科手术插入式器械或同有源器械连接使用,如果属于暂时性的使用方式,属于I类;如果属于短期性的使用方式,属于IIa类;但不包括在口腔中仅至咽部、在耳道中仅至耳鼓、在鼻腔中使用但不被粘膜吸收的器械;如果属于长期性的使用方式,属于IIb类;但在口腔中仅至咽部、在耳道中仅至耳鼓、在鼻腔中使用但不被粘膜吸收的器械属于IIa类;其他需要与IIa或更高类别的器械连接使用的插入式器械,属于IIa类;但不包括外科手术插入式器械。
规则 6暂时性使用方式的外科手术插入式器械属于IIa类,但以下情况除外: 如果为了诊断、监测或矫正心脏或主血管系统疾病,器械直接触及这些器官,在属于III类;可重复使用的外科器械属于I类;以电离辐射的方式提供活力(energy)的器械属于IIb类;对人体生理发生作用或为人体全部或大部分所吸收的器械属于IIb类;通过发送装置给人体施用药物,如果对人体具有某种危险,则属于IIb类.规则 7以短期方式使用的外科插入式器械属于IIa,但以下情况除外:如果为了诊断、监测或矫正心脏或主血管系统疾病,直接触及这些器官的器械,属于III类;直接触及中枢神经系统的专用器械属于III类;以电离辐射的方式提供活力(energy)的器械属于IIb类;对人体生理发生作用或为人体全部或大部所吸收的器械属于III类;在人体内促使人体发生某种化学变化,但不属于安装在牙齿上或为人体给药的器械,属于IIb类。
9342EEC 协调标准
信息 非侵入式血 压计.第 1 部 分:一般要求. 修改件 A1 非侵入式血 压表.第 2 部 分:机械血压 表的补充要 求 非侵入式血 压表.第 3 部 分:电机血压 表的补充要 求 非介入式血 压计.第 4 部 分:自动非介 入式血压计 整体系统精 确度测定的 试验过程 可移动式气 瓶.气瓶标识 ( 不 包 括 LPG).第 3 部 分:色码 麻醉气化装 置制剂特殊 添充系统.拒 形键控填充 系统 麻醉和呼吸 设备圆锥形 接头第 2 部 分:螺纹承重 连接头 气体切开插 管.第 2 部分: 儿科婴幼儿 用管 医用灭菌器. 环氧乙烷灭
EN 1639:2004
EN 1640:2004
EN 1640:1996
EN 1641:2004
EN 1641:1996
EN 1642:2004
EN 1642:1996
EN 1707:1996
Conical fittings with a 6 % (Luer) taper for syringes, needles and certain other medical equipment Lock fittings
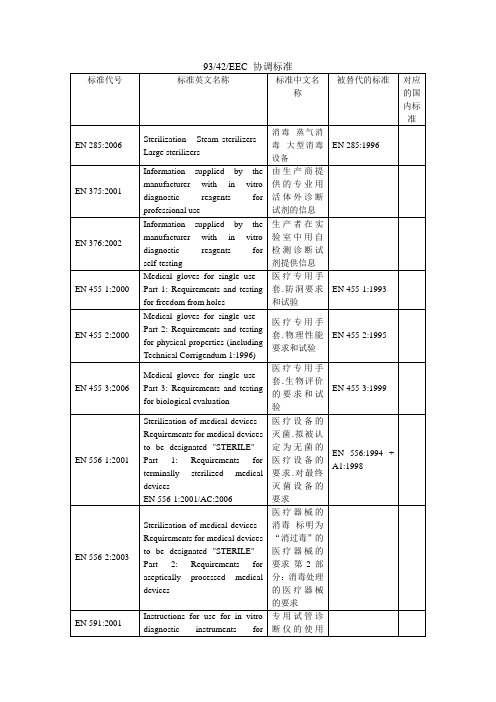
93/42/EEC 协调标准
标准代号 标准英文名称 标准中文名 称 被替代的标准 对应 的国 内标 准
EN 285:2006
Sterilization - Steam sterilizers Large sterilizers Information supplied by the manufacturer with in vitro diagnostic reagents for professional use Information supplied by the manufacturer with in vitro diagnostic reagents for self-testing Medical gloves for single use Part 1: Requirements and testing for freedom from holes Medical gloves for single use Part 2: Requirements and testing for physical properties (including Technical Corrigendum 1:1996) Medical gloves for single use Part 3: Requirements and testing for biological evaluation Sterilization of medical devices Requirements for medical devices to be designated "STERILE" Part 1: Requirements for terminally sterilized medical devices EN 556-1:2001/AC:2006 Sterilization of medical devices Requirements for medical devices to be designated "STERILE" Part 2: Requirements for aseptically processed medical devices Instructions for use for in vitro diagnostic instruments for
MDREU_2017_745欧盟医疗器械最新法规(中英对照版)

MDREU_2017_745欧盟医疗器械最新法规(中英对照版)公报中⽂版⽴法L117第60卷2017年5⽉5⽇内容I ⽴法法案法规★欧洲议会和理事会于2017年4⽉5⽇签发的关于医疗器械第2017/745号法规,修订了第2001/83/EC号指令,第178/2002号(EU)法规和第1223/2009号(EU)法规,并废除了理事会第90/385/EEC号和第93/42/EEC号指令(1) (1)★欧洲议会和理事会于2017年4⽉5⽇签发的关于体外诊断医疗器械第2017/746号(EU)法规并废除了第98/79/EC号指令和理事会第2010/227/EU号决议 (176)________________(1)EEA相关性⽂本。
以浅⾊字体打印标题的法案均为涉及农业⽇常管理的法案,⼀般在有限期内有效。
所有其他法案的标题均以粗体打印,并以星号开头。
I(⽴法法案)法规欧洲议会和理事会于2017年4⽉5⽇签发的关于医疗器械第2017/745号法规,修订了第2001/83/EC号指令,第178/2002号(EU)法规和第1223/2009号(EU)法规,并废除了理事会第90/385/EEC号和第93/42/EEC号指令(EEA相关性⽂本)欧洲议会和欧盟委员会,考虑到“欧盟运作条约”,特别是其中第114条和第168(4)(c)条规定,并考虑到欧盟委员会提案,于⽴法草案转交各国议会后,考虑到欧洲经济和社会委员会之意见(1),在咨询地区委员之后,根据⼀般⽴法程序运作(2),鉴于:(1)理事会第90/385/EEC号指令(3)和理事会第93/42/EEC号指令(4)构成有关医疗器械(不包括体外诊断医疗器械)的欧盟监管框架。
但需要对该指令进⾏⼤幅修订,以便建⽴稳健、透明、可预测和可持续的医疗器械监管框架,以确保⾼⽔平的安全和健康,同时为创新提供⽀持。
Council Directive 90/385/EEC (3) and Council Directive 93/42/EEC (4) constitute the Union regulatoryframework for medical devices, other than in vitro diagnostic medical devices. However, a fundamentalrevision of those Directives is needed to establish a robust, transparent, predictable andregulatory framework for medical devices which ensures a high level of safety and health whilst supporting innovation. (2)本法规旨在确保区域内医疗器械市场的平稳运作,在为患者和使⽤者提供⾼⽔平健康保护的基础上,同时考虑到活跃于本⾏业的中⼩型企业利益。
9342eec中文

20 鉴于按照一般规则,医疗器械应标示 CE 标志,表明它们符合本指令的条款,使其能在欧共体内自由流 通,并按其预定用途投入使用;
23 鉴于证实符合基本要求可能意味着必须由制造商负责完成临床试验;鉴于为了完成临床试验,必须确定 保护公众健康和公共秩序的适当方式;
24 鉴于在欧共体级别一体化的医疗器械警戒系统可以更有效地完成健康保护和相关的控制; 25 鉴于本指令覆盖了 1976 年 7 月 27 日理事会第 76/764/EEC 号关于使成员国有关临床用汞柱式温度计最高
15 鉴于为了证实符合基本要求并使该符合性得以验证,有必要协调欧洲标准以避免与医疗器械的设计、制 造和包装有关的危险;鉴于这类欧洲协调标准是由非官方立法机构制定的,应保持其非强制性的性质; 鉴于到目前为止,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(Cenelec),根据 1984 年 11 月 13 日执委会与这两个机构之间签署的合作通则,已被公认为是制定协调标准的职能机构;
Created by lisong Page 1 of 35
11 鉴于按照 1985 年 5 月 7 日理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计 和制造的规则必须限制在满足基本要求所必需的条款内;鉴于因为这些要求是最基本的,因此它们应取 代相应的国家规定;鉴于基本要求的实施应谨慎考虑设计当时的技术水平,并在符合健康和安全高度保 护的原则下考虑技术和经济因素;
17 鉴于在 1990 年 12 月 13 号决议关于用于技术协调指令不同合格评定程序的各阶段模式中,理事会制定了 协调合格评定程序;鉴于将这些模式用于医疗器械可使制造商和公告机构在合格评定中的责任根据有关 的器械型式予以确定;鉴于对这些模式的细节所作的补充,根据医疗器械必需验证的性质,证明是合理 的;
MDD9342EEC

医疗器械指令(Medical Device Directive)93/42/EEC欧共体医疗器械产品安全共同指令欧洲共同体公报,1993年7月12日,NO. L169/1(此法案对欧共体成员国而言,其公布与否属非强制性)1993年6月14日关于医疗器械的第93/42/EEC号理事会指令欧共体理事会,1考虑到建立欧洲经济共同体的(罗马)条约,特别是其第100a条,2考虑到执委会的提案,以及与欧洲议会的合作,3考虑到经济和社会委员会的意见,4鉴于应就内部市场的完成采取一些措施;鉴于内部市场是一无内部疆界的区域,区域内的货品、人员、服务和资金应可自由流通;5鉴于各成员国间现存有关医疗器械的安全,对健康的保护和功能特性方面的法律、法规和行政命令的内容与范围不尽相同;鉴于各成员国之间对此类器械的认证和检验程序也存在差异;鉴于前述的分歧将在共同体内部构成贸易壁垒;6鉴于为了保护患者、使用者以及必要时其他人员的安全与健康,有关医疗器械使用的国家规定应予以协调,以保证此类器械在内部市场能自由流通;7鉴于协调规定必须与各成员国为管理直接或间接与这类器械有关的公共健康和疾病保险计划的资金筹措所采取的措施相区别;鉴于共同体若与上述措施相符,则这些规定并不影响各成员国实施上述措施的能力;8鉴于医疗器械应向患者,使用者及第三方提供高度的保护并达到制造商赋予其的性能水准;鉴于,因此,维持和改进各成员国已达到的保护水平是本指令的基本目标之一;9鉴于在1965年1月26日的理事会第65/65/EEC号关于使有关根据特许专卖医药产品的法律、法规或管理行为所制定的实施规定趋于一致的指令中某些医疗器械是用于使用药品的;鉴于在这种情况下,医疗器械的市场投放通常受本指令管辖,而药品的市场投放则受第65/65/EEC号指令管辖;鉴于若有某种器械投放市场时器械与其它医疗产品构成一整体的组合单元,并以这种组合形式使用且不能二次使用,则该整体单元产品应受第65/65/EEC号指令管辖;鉴于必须将上述器械与和其它物质组合的医疗器械相区别,特别是若这些物质在单独使用时,按第65/65/EEC号指令可视为药物;鉴于在这种情况下,若这种物质是作为器械的辅助物作用于人体,则这类器械的市场投放受本指令管辖;鉴于,这类物质的安全,质量和效用必须由1975年5月20日理事会第75/318/EEC号关于使成员国有关分析标准,药物毒理学标准和临床标准及特许专卖药品检测协议的法律趋于一致的指令中规定的适当方法加以验证;10 鉴于本指令附录中的基本要求和其它要求包括任何涉及“最低”或“降低”危险的内容的阐述和实施必须考虑设计当时的技术与实际情况,并在符合健康和安全高度保护的原则下考虑技术和经济因素;11 鉴于按照1985年5月7日理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和制造的规则必须限制在满足基本要求所必需的条款内;鉴于因为这些要求是最基本的,因此它们应取代相应的国家规定;鉴于基本要求的实施应谨慎考虑设计当时的技术水平,并在符合健康和安全高度保护的原则下考虑技术和经济因素;12 鉴于1990年6月20日理事会第90/385/EEC号关于使成员国有关有源植入性医疗器械的法律趋于一致的指令是新方法指令在医疗器械领域中的首次应用;鉴于为了统一共同体的规则使之适用于所有医疗器械,本指令在很大程度上是以第90/385/EEC号指令的条款为依据;鉴于同样的原因第90/385/EEC号指令必须增加本指令制定的一般条款的部分;13 鉴于电磁兼容性问题已成为医疗器械安全不可缺少的组成部分;鉴于本指令应包含与1989年5月3日理事会第89/336/EEC号关于使成员国有关电磁兼容的法律趋于一致的指令中的内容有关的特别条款;14 鉴于本指令应包括对有关释放致电离辐射的器械的设计和制造的要求;鉴于本指令既不影响1980年7月15日理事会第80/836/Euratom指令对有关制定一般公众和工人避免离子辐射危险的健康保护的基本安全标准指令的修改中要求的授权,也不影响1984年9月3日理事会第84/466/Euratom号规定有关接受医疗检查和治疗的人员的辐射保护的基本措施的指令的适用;鉴于1989年6月12日理事会第89/391/EEC号关于鼓励改善工作场所中工人的安全和健康的措施介绍的指令和同样主题的其它特别指令应持续适用;15 鉴于为了证实符合基本要求并使该符合性得以验证,有必要协调欧洲标准以避免与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方立法机构制定的,应保持其非强制性的性质;鉴于到目前为止,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(Cenelec),根据1984年11月13日执委会与这两个机构之间签署的合作通则,已被公认为是制定协调标准的职能机构;16 鉴于本指令的协调标准是一种根据执委会的委托,由上述两机构之一,或两个机构共同根据1983年3月18日理事会第83/189/EEC号关于技术标准和法规领域信息传递程序的指令和上述通则的规定而制定的技术规范(欧洲标准或协调文件);鉴于由于协调标准有可能被修改,执委会应得到根据第83/189/EEC 号指令设立的常设委员会的协助;鉴于所采取的措施必须按理事会第87/373/EEC号决议制定的程序Ⅰ予以阐释;鉴于在特殊领域,如列入欧洲药典专著中的内容,应纳入本指令的框架内;鉴于因此,几部欧洲药典专著应视为等同于前述协调标准;17 鉴于在1990年12月13号决议关于用于技术协调指令不同合格评定程序的各阶段模式中,理事会制定了协调合格评定程序;鉴于将这些模式用于医疗器械可使制造商和公告机构在合格评定中的责任根据有关的器械型式予以确定;鉴于对这些模式的细节所作的补充,根据医疗器械必需验证的性质,证明是合理的;18 鉴于为进行合格评定程序,有必要将器械分为四个产品类别;鉴于分类原则是依据器械技术设计和制造中潜在的危险对人体的易损伤性;鉴于在一般情况下,第Ⅰ类器械具有较低的易损伤性,其合格评定程序可由制造商单独完成;鉴于对第Ⅱa类器械公告机构应在生产阶段强制性介入;鉴于第Ⅱb和第Ⅲ类器械具有较高的潜在危险,公告机构必须对器械的设计与制造阶段进行检验;鉴于第Ⅲ类器械属于最关键的器械,它们在投放市场前需预先就其符合性获得明确授权;19 鉴于在器械的符合性由制造商负责评定的情况下,主管当局必须能够,特别是在紧急情况下,联系到在欧共体内的负责器械市场投放的人员,无论是制造商还是其他在欧共体内经制造商授权的人员;20 鉴于按照一般规则,医疗器械应标示CE标志,表明它们符合本指令的条款,使其能在欧共体内自由流通,并按其预定用途投入使用;21 鉴于为对抗艾滋病和根据1989年5月16日通过的关于在欧共体级别的关于艾滋病预防和控制的进一步行动的理事会决议,用于预防HIV病毒感染的医疗器械必须提供高度的保护;鉴于这类产品的设计和制22 鉴于分类规则通常可对医疗器械进行恰当的分类;鉴于考虑到各种器械的不同性质及该领域内的技术进步,采取的步骤应包括授权执委会决定器械的适当分类或重新分类,必要时调整分类规则;鉴于这些问题与健康保护有密切关系,因此这些决议应按照第87/373/EEC号指令规定的第Ⅲa程序进行;23 鉴于证实符合基本要求可能意味着必须由制造商负责完成临床试验;鉴于为了完成临床试验,必须确定保护公众健康和公共秩序的适当方式;24 鉴于在欧共体级别一体化的医疗器械警戒系统可以更有效地完成健康保护和相关的控制;25 鉴于本指令覆盖了1976年7月27日理事会第76/764/EEC号关于使成员国有关临床用汞柱式温度计最高读数的法律趋于一致的指令中提到的医疗器械;鉴于上述指令应予撤销;鉴于同样原因,1984年9月17日理事会第84/539/EEC号关于使成员国有关用于人或兽医的电子医疗器械的法律趋于一致的指令必须修改。
欧洲医疗器械93-42-EEC

歐洲共同體理事會關於醫療器械的93/42/EEC指令1993年6月14日歐洲共同體理事會考慮到建立歐洲經濟共同體的條約,特別是其第100a條;考慮到歐洲共同體委員會提交的議案;考慮到與歐洲議會合作;考慮到經濟與社會委員會的意見;鑒於應在歐洲共同體內部市場範圍內正式通過必要的措施;鑒於歐洲共同體內部市場是一個確保商品、人員、服務和資本自由流通的無內部邊界的區域;鑒於各成員國有關醫療器械的安全、健康保護和工作特性的法律、法規和行政條款的內容和範圍是不同的;鑒於各成員國之間對這類器械的認證和檢驗程序存在差異;鑒於這種差異在歐洲共同體內形成貿易壁壘;鑒於各國針對醫療器械的使用所制定的有關患者、使用者及其他人員的安全和健康保護的條款應予以協調,以保證此類器械在歐洲共同體內部市場自由流通;鑒於協調條款必須與成員國為管理直接或間接與這類器械有關的公共健康和醫療保險計劃的資金籌措而採取的措施相區別;鑒於因而只要遵守歐洲共同體法律,這些條款並不影響各成員國實施上述措施的能力;鑒於醫療器械應向患者、使用者及第三方提供高水準的保護並達到製造商賦予其的性能水平;鑒於因此保持和提高各成員國已達到的保護水平是本指令的基本目的之一;鑒於在1965年1月26日歐洲共同體理事會關於使有關特許專賣藥品的法律、法規或行政措施趨於一致的65/65/EEC指令中,某些醫療器械是用於施藥的;鑒於在這種情況下,醫療器械投放市場通常由本指令管理,而藥品投放市場由65/65/EEC指令管理;鑒於如果這種器械投放市場的方式使器械與藥品構成一種規定只供組合使用且不能再次使用的整體,則這個單一整體產品應由65/65/EEC指令管理;鑒於必須將上述器械與包含某種物質,特別是當其單獨使用時,按65/65/EEC指令可視為藥物的醫療器械相區別;鑒於在這種情況下,若這種物質能配合醫療器械對人體產生輔助作用,則這類器械投放市場由本指令管理;鑒於這類物質的安全性、質量和有效性必須比照歐洲共同體理事會1975年5月20日關於使成員國有關分析標準、藥物毒理學標準和臨床標準及特許專賣藥品詴驗協定的法律趨於一致的75/318/EEC指令規定的適當方法加以驗證;鑒於本指令附錄中的基本要求和其他要求,包括對“減小”或“降低”危險的任何引用,在解釋和實施時必須考慮設計時的技術現狀和實際做法及高水準的健康和安全相適應的技術與經濟條件;鑒於按照1985年5月7日歐洲共同體理事會關於技術協調與標準化新方法的決議所規定的原則,有關醫療器械設計和生產的規定必須限於滿足基本要求所必須的條款;鑒於因為這些要求是基本的,因此它們應取代各國相應的條款;鑒於實施基本要求應審慎考慮設計時的技術水平和與高水準的健康及安全保護相適應的技術、經濟條件;鑒於1990年6月20日歐洲共同體理事會關於使成員國有關有源植入式醫療器械的法律趨於一致的90/385/EEC 指令是新方法指令在醫療器械領域中的首次應用案例;鑒於為了使歐洲共同體的統一規定適用於所有醫療器械,本指令基本上是以90/385/EEC指令的條款為依據的;鑒於為此必須修訂90/385/EEC指令,以便放入本指令規定的一般性條款;鑒於電磁相容性問題是醫療器械安全的一個組成部分;鑒於就1989年5月3日歐洲共同體理事會關於使成員國有關電磁相容的法律趨於一致的89/336/EEC指令而言,應包含這方面的專門規定;鑒於本指令應包括有關發射電離輻射的器械在設計和製造方面的要求;鑒於本指令既不影響1980年7月15日歐洲共同體理事會80/836/Euratom指令對有關保護公眾和工人免受電離輻射危險的基本安全標準的指令進行修訂所要求的授權,也不影響歐洲共同體理事會1984年9月3日對接受醫療檢查和治療的人員的輻射防護規定了基本措施的84/466/Euratom指令的實施;鑒於歐洲共同體理事會1989年6月12日關於採取措施鼓勵改善工人工作中的安全和健康的89/391/EEC指令以及有關這一問題的專門指令應繼續予以實施;鑒於為了證實符合基本要求並使這種符合得到驗證,需要制定歐洲協調標準來防止與醫療器械的設計、製造和包裝有關的危險;鑒於這類歐洲協調標準是由非官方機構制定的,應保持其非強制性的地位;鑒於為此歐洲標準化委員會(CEN)和歐洲電工標準化委員會(CENELEC)按照1984年11月13日歐洲共同體委員會與這兩個機構之間簽署的合作總指導原則,被認可為批准協調標準的主管機構;鑒於在本指令中,協調標準是受歐洲共同體委員會委託,由上述兩機構之一,或兩個機構共同根據歐洲共同體理事會1983年3月18日關於在技術標準和法規領域提供資訊程序的83/189/EEC指令,依照上述總指導原則而批准的技術規範(歐洲標準或協調文件);鑒於對協調標準進行修訂,歐洲共同體委員會應得到根據83/189/EEC指令建立的常設委員會的幫助;鑒於應採取的措施必須按歐洲共同體理事會87/373/EEC決定中規定的程序Ⅰ而規定;鑒於在特定領域,歐洲藥典專著這類現有的形式應包括在本指令的範圍內;鑒於因此有幾部歐洲藥典專著可視為等同於上述協調標準。
MDD 93-42-EEC简介

M用
MDD
简介
有源植入医疗器械指令 90/385/EEC(AIMDD) COUNCIL DIRECTIVE of 20 June 1990, on the approximation of the laws of the Member States relating to Active Implantable Medical Devices (90/385/EEC) 医疗器械指令93/42/EEC(MDD) COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993, concerning Medical Devices (Directive) 体外诊断医疗器械指令98/79/EC(IVMDD) DIRECTIVE 98/79/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 27 October 1998,on In Vitro Diagnostic medical devices
MDD 93/42/EEC
质量法规部 2014-07
目录
• CE Marking • MDD
CE Marking
简述
目的
适用范围
适用对象
获取方法
CE Marking
简述
① 1993年7月22日发表《欧盟产品指令Product Directive》,93/68/EEC,’CE’ 标识诞 生; ② ‘CE’,法语“Communate Europpene”的缩写,意思即“欧洲共同体”; ③ 具有’CE’标识的商品,意味着该商品满足欧盟相关法规及协调标准的要求,制造 商负有相应责任。
23项条款
条款号 Article 1 Article 2 Article 3 Article 4 Article 5 Article 6 Article 7 Article 8 Article 9 Article 10 Article 11 Article 12 Article 13 Article 14 Article 15 Article 16 Article 17 Article 18 Article 19 Article 20 Article 21 Article 22 Article 23 Definition,Scope Placing on the market and putting into service Essential Requirements Free movement, devices intended for special purposes Reference to standards Committee on standards and Technical regulations Committee on Medical device Safeguard clause Classfication Information on incidents occuring following placing of devices on the market Conformity assessment procedures Particular procedure for systems and procedure packs Decisions with regards to classfication, derogation clause Registration of persons responsible for placing devices on the market Clinical investigation Notified bodies CE marking Wrongly affixed CE Marking Decision in respect of refusal or restriction Confidentiality Repeal and amendment of Directives Implementation,transitional provisions This directive is addressed to the member states 内容 定义,范围 上市及使用 基本要求 自由流通及特殊目的的器械 标准的参考 标准及技术法规委员会 医疗器械委员会 保护条款 分类 器械上市后偶发事件的资讯 符合评鉴程序 成套医疗系统及程序的特殊程序 分类决定的排除条款 负责器械上市行销人员的登录 临床调查 公告机构 CE标示 误加的CE标示 有关拒绝或限制的决定 机密性 指令的撤销及修订 实施及过渡条款 本指令通告所有会员国
MDD 93-42-EEC简介

2014/07/01,欧盟网站共计公布25类需进行CE
Marking的商品大类
1. 有源植入医疗器械; 2. 燃气炉具;
3. 载人的索道装置;
4. 建筑产品;
5. 能源相关产品生态设计;
6. 电磁兼容;
7. 使用于具有爆炸性环境中的设备和保护系统;
8. 民用爆破器材;
9. 热水锅炉;
10. 体外诊断试剂;
② ‘CE’,法语“Communate Europpene”的缩写,意思即“欧洲 共同体”;
③ 具有’CE’标识的商品,意味着该商品满足欧盟相关法规及协 调标准的要求,制造商负有相应责任。
CE Marking
目的 消除商品流通壁垒,保护公众利益.
CE Marking
适用范围
Member states of EU 欧盟成员国(28个,24种官方语言)
基本要求 一般要求 有关设计及架构的要求
EC符合性声明(完全品质保证系统)
内容 Definition,Scope Placing on the market and putting into service Essential Requirements Free movement, devices intended for special purposes Reference to standards Committee on standards and Technical regulations Committee on Medical device Safeguard clause Classfication Information on incidents occuring following placing of devices on the market Conformity assessment procedures Particular procedure for systems and procedure packs Decisions with regards to classfication, derogation clause Registration of persons responsible for placing devices on the market Clinical investigation Notified bodies CE marking Wrongly affixed CE Marking Decision in respect of refusal or restriction Confidentiality Repeal and amendment of Directives Implementation,transitional provisions This directive is addressed to the member states
MDD_93_42_EEC(amended_by_2007_47_EC)中英对照版

1.1.1. Either a critical evaluation of the relevant scientific literature currently available relating to the safety,
performance, design characteristics and intended purpose of the device, where:
-
f4~1JJ37* ~fl:;fJ&l=5)[iWt ~ I¥J~~~~ In] , ~ ~ aJj ~;fflJGr{Ji'l;f§ ~ WI'IiWffr~
;fQ
;!jl;*~
0
0
8:fQ~
9=t m1;!J.iI¥J~ili&#;*~tffi 0
;1:11:, *3t1'fiJ(1J\o;"t, pM, t*J!il'l1J~:< 1l":nillJl&'IIJ:1o;J£. Wi!;, *3t1'fJl&tJ(~:'III'""pli" (geoeruooer@163. com), *~*.A.l'fjij",
JiJT:l2E I¥J iJ't 11' ffl :fQ jxl Jl#il 3't.@. H~ 1¥J"ilJ 3't tt I¥J i'f1i'i1!f~ m ~ [1{EPiC ~ *4 7J 1tf.ii X~·~ @~f.ii Et-JtSf11t , I2.L r :t~~$Z7J [1{EPiC tSf11t" , ~ m¥u*~~11J·i,fflj;fJFm, m~ a1, ~\~.mr~* ~ r37U~ 1t~:7-J ;!jl;;pllJ I¥J ~;tJE I¥J i'lJ'.!ll.I¥J 1J¥*"f:*¥J¥: 1.1.1. x'.!" ~1~1r1f m Et'~, l:ij -H:;fMS<.J ~-.i:'Ii" 'Ii f:m" lfstit4* F.1. :fQ tj)j:llJl ffl ~ if ~ I¥J H"f:)( mtitl:1T *U WItt Wffr, "!1Q
mdd9342eec简介

② “Directive 指令”是指设定所有欧盟成员国必须实现的目标的立法行为,但是 该设定目标的实现途径由各成员国自己决定。
注:http://europa.eu/eu-law/decision-making/legal-acts/index_en.htm
Turkey 土耳其
Kosovo科索沃
CE Marking
适用对象
面向欧盟成员国进行销售的规定商品。
① 对商品原产地无要求 ② 规定商品*
注:* 见下述网址 http://ec.europa.eu/enterprise/policies/single-marketgoods/cemarking/professionals/manufacturers/index_en.htm
2019/07/01,欧盟网站共计公布25类需进行CE Marking 的商品大类
1. 有源植入医疗器械; 2. 燃气炉具; 3. 载人的索道装置; 4. 建筑产品; 5. 能源相关产品生态设计; 6. 电磁兼容; 7. 使用于具有爆炸性环境中的设备和保护系统; 8. 民用爆破器材; 9. 热水锅炉; 10. 体外诊断试剂; 11. 升降机; 12. 低压器材; 13. 机械; 14. 测量仪器; 15. 医疗器械; 16. 环境噪声辐射; 17. 非自动称重仪器; 18. 个人防护设备; 19. 压力设备; 20. 烟火; 21. 无线电和电信终端设备; 22. 休闲用船只; 23. 电子电器设备中有害物质的限制; 24. 玩具安全; 25. 简单压力容器。
23项条款
条款号 Article 1 Article 2 Article 3 Article 4 Article 5 Article 6 Article 7 Article 8 Article 9 Article 10 Article 11 Article 12 Article 13 Article 14 Article 15 Article 16 Article 17 Article 18 Article 19 Article 20 Article 21 Article 22 Article 23
欧盟医疗器械指令 MDD 93-42-EEC(中英文)

Whereas the essential requirements and other requirements set out in the Annexes to this Directive, including any reference to 'minimizing' or 'reducing' risk must be interpreted and applied in such a way as to take account of technology and practice existing at the time of design and of technical and economical considerations compatible with a high level of protection of health and safety; 鉴于本指令附录所订的基本要求及其他要求, 包括[最低]或[降低]危险部分的应用, 应考虑设计当时的科技及实施情形, 并在符合健康和安全高度保护的原则下考虑技术及经济的因素;
医疗器材指令 93 42 eec 20141016

分級的原則一般是依照醫療器材的風險程度,有少部分特殊規 則(血袋或含藥醫材)的醫療器材採用例外的分及方法。 MDD指令附錄IX有18條分類規則作為分類指導。類別越高,意 味著風險級別越高,符合性評估的要求也就越高。
符合性評鑑 Conformity Assessment
歐盟的符合性評鑑程序乃依照歸類後的醫療器材分級而決定, 業者可以在醫療器材指令的規定程序內選擇對自己最便利的 符合性評鑑程序(Conformity Assessment Procedure)。 在歐盟的符合性評鑑證書其有效期限為3年,而其不同等級的 醫療器材會有不同的符合性評鑑程序路徑。 新版2007/47/EC指令與舊版93/42/EEC指令對所採用的符合性評 鑑方式是一致的, 在新版2007/47/EC指令中,部分醫療器材分級和舊版93/42/EEC 指令的分級不同,將會產生同樣醫療器材而變更成不同的符 合性評鑑程序。
需要注意的是以往歐盟的醫療器材指令(MDD)都 是遵循93/42/EEC指令,然而在2009年開始進行「歐 盟醫療器材指令93/42/EEC新方案」(New Approach EU Medical Device Directive 93/42/EEC)的修訂,並 於今年(2010年)3月21日起生效實施,新要求載錄 於修改的2007/47/EC指令中,並明確指出歐盟醫療器 材符合新指令的規定將無過渡期,故現今送審歐盟 的醫療器材則需遵循新指令,也就是2007/47/EC指令
選擇代施查核機構
符合性評鑑都需要代施查核機構(Notified Body) 的介入(除Annex Ⅶ程序例外),並且新版 2007/47/EC指令更強制規定申請CE mark時就必須要 有歐體代表。 代施查核機構的產生是由各國最高衛生主管機關 (Competent Authority)審查通過後,提供給歐盟 指委會,相關名單可以在歐盟指委會網頁上查詢。
MDD 9342EEC简介

2014/07/01,欧盟网站共计公布25类需进行CE Marking的商品大类
1. 有源植入医疗器械; 2. 燃气炉具; 3. 载人的索道装置; 4. 建筑产品; 5. 能源相关产品生态设计; 6. 电磁兼容; 7. 使用于具有爆炸性环境中的设备和保护系统; 8. 民用爆破器材; 9. 热水锅炉; 10. 体外诊断试剂; 11. 升降机; 12. 低压器材; 13. 机械; 14. 测量仪器; 15. 医疗器械; 16. 环境噪声辐射; 17. 非自动称重仪器; 18. 个人防护设备; 19. 压力设备; 20. 烟火; 21. 无线电和电信终端设备; 22. 休闲用船只; 23. 电子电器设备中有害物质的限制; 24. 玩具安全; 25. 简单压力容器。
Malta 马耳他
Czech Republic 捷克
Hungary 匈牙利
Netherlands 荷兰
Denmark 丹麦
Ireland 爱尔兰
Poland 波兰
Candidate countries 候选国家 (5个)
Iceland 冰岛
Montenegro 黑山
Serbia 塞尔维亚
The former Yugoslav Republic of Macedonia 马其顿
Turkey 土耳其
Kosovo科索沃
CE Marking
适用对象
面向欧盟成员国进行销售的规定商品。
① 对商品原产地无要求 ② 规定商品*
注:* 见下述网址 http://ec.europa.eu/enterprise/policies/single-marketgoods/cemarking/professionals/manufacturers/index_en.htm
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
医疗器械指令(Medical Device Directive)93/42/EEC欧共体医疗器械产品安全共同指令欧洲共同体公报,1993年7月12日,NO. L169/1(此法案对欧共体成员国而言,其公布与否属非强制性)1993年6月14日关于医疗器械的第93/42/EEC号理事会指令欧共体理事会,1考虑到建立欧洲经济共同体的(罗马)条约,特别是其第100a条,2考虑到执委会的提案,以及与欧洲议会的合作,3考虑到经济和社会委员会的意见,4鉴于应就内部市场的完成采取一些措施;鉴于内部市场是一无内部疆界的区域,区域内的货品、人员、服务和资金应可自由流通;5鉴于各成员国间现存有关医疗器械的安全,对健康的保护和功能特性方面的法律、法规和行政命令的内容与范围不尽相同;鉴于各成员国之间对此类器械的认证和检验程序也存在差异;鉴于前述的分歧将在共同体内部构成贸易壁垒;6鉴于为了保护患者、使用者以及必要时其他人员的安全与健康,有关医疗器械使用的国家规定应予以协调,以保证此类器械在内部市场能自由流通;7鉴于协调规定必须与各成员国为管理直接或间接与这类器械有关的公共健康和疾病保险计划的资金筹措所采取的措施相区别;鉴于共同体若与上述措施相符,则这些规定并不影响各成员国实施上述措施的能力;8鉴于医疗器械应向患者,使用者及第三方提供高度的保护并达到制造商赋予其的性能水准;鉴于,因此,维持和改进各成员国已达到的保护水平是本指令的基本目标之一;9鉴于在1965年1月26日的理事会第65/65/EEC号关于使有关根据特许专卖医药产品的法律、法规或管理行为所制定的实施规定趋于一致的指令中某些医疗器械是用于使用药品的;鉴于在这种情况下,医疗器械的市场投放通常受本指令管辖,而药品的市场投放则受第65/65/EEC号指令管辖;鉴于若有某种器械投放市场时器械与其它医疗产品构成一整体的组合单元,并以这种组合形式使用且不能二次使用,则该整体单元产品应受第65/65/EEC号指令管辖;鉴于必须将上述器械与和其它物质组合的医疗器械相区别,特别是若这些物质在单独使用时,按第65/65/EEC号指令可视为药物;鉴于在这种情况下,若这种物质是作为器械的辅助物作用于人体,则这类器械的市场投放受本指令管辖;鉴于,这类物质的安全,质量和效用必须由1975年5月20日理事会第75/318/EEC号关于使成员国有关分析标准,药物毒理学标准和临床标准及特许专卖药品检测协议的法律趋于一致的指令中规定的适当方法加以验证;10 鉴于本指令附录中的基本要求和其它要求包括任何涉及“最低”或“降低”危险的内容的阐述和实施必须考虑设计当时的技术与实际情况,并在符合健康和安全高度保护的原则下考虑技术和经济因素;11 鉴于按照1985年5月7日理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和制造的规则必须限制在满足基本要求所必需的条款内;鉴于因为这些要求是最基本的,因此它们应取代相应的国家规定;鉴于基本要求的实施应谨慎考虑设计当时的技术水平,并在符合健康和安全高度保护的原则下考虑技术和经济因素;12 鉴于1990年6月20日理事会第90/385/EEC号关于使成员国有关有源植入性医疗器械的法律趋于一致的指令是新方法指令在医疗器械领域中的首次应用;鉴于为了统一共同体的规则使之适用于所有医疗器械,本指令在很大程度上是以第90/385/EEC号指令的条款为依据;鉴于同样的原因第90/385/EEC号指令必须增加本指令制定的一般条款的部分;13 鉴于电磁兼容性问题已成为医疗器械安全不可缺少的组成部分;鉴于本指令应包含与1989年5月3日理事会第89/336/EEC号关于使成员国有关电磁兼容的法律趋于一致的指令中的内容有关的特别条款;14 鉴于本指令应包括对有关释放致电离辐射的器械的设计和制造的要求;鉴于本指令既不影响1980年7月15日理事会第80/836/Euratom指令对有关制定一般公众和工人避免离子辐射危险的健康保护的基本安全标准指令的修改中要求的授权,也不影响1984年9月3日理事会第84/466/Euratom号规定有关接受医疗检查和治疗的人员的辐射保护的基本措施的指令的适用;鉴于1989年6月12日理事会第89/391/EEC号关于鼓励改善工作场所中工人的安全和健康的措施介绍的指令和同样主题的其它特别指令应持续适用;15 鉴于为了证实符合基本要求并使该符合性得以验证,有必要协调欧洲标准以避免与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方立法机构制定的,应保持其非强制性的性质;鉴于到目前为止,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(Cenelec),根据1984年11月13日执委会与这两个机构之间签署的合作通则,已被公认为是制定协调标准的职能机构;16 鉴于本指令的协调标准是一种根据执委会的委托,由上述两机构之一,或两个机构共同根据1983年3月18日理事会第83/189/EEC号关于技术标准和法规领域信息传递程序的指令和上述通则的规定而制定的技术规范(欧洲标准或协调文件);鉴于由于协调标准有可能被修改,执委会应得到根据第83/189/EEC 号指令设立的常设委员会的协助;鉴于所采取的措施必须按理事会第87/373/EEC号决议制定的程序Ⅰ予以阐释;鉴于在特殊领域,如列入欧洲药典专著中的内容,应纳入本指令的框架内;鉴于因此,几部欧洲药典专著应视为等同于前述协调标准;17 鉴于在1990年12月13号决议关于用于技术协调指令不同合格评定程序的各阶段模式中,理事会制定了协调合格评定程序;鉴于将这些模式用于医疗器械可使制造商和公告机构在合格评定中的责任根据有关的器械型式予以确定;鉴于对这些模式的细节所作的补充,根据医疗器械必需验证的性质,证明是合理的;18 鉴于为进行合格评定程序,有必要将器械分为四个产品类别;鉴于分类原则是依据器械技术设计和制造中潜在的危险对人体的易损伤性;鉴于在一般情况下,第Ⅰ类器械具有较低的易损伤性,其合格评定程序可由制造商单独完成;鉴于对第Ⅱa类器械公告机构应在生产阶段强制性介入;鉴于第Ⅱb和第Ⅲ类器械具有较高的潜在危险,公告机构必须对器械的设计与制造阶段进行检验;鉴于第Ⅲ类器械属于最关键的器械,它们在投放市场前需预先就其符合性获得明确授权;19 鉴于在器械的符合性由制造商负责评定的情况下,主管当局必须能够,特别是在紧急情况下,联系到在欧共体内的负责器械市场投放的人员,无论是制造商还是其他在欧共体内经制造商授权的人员;20 鉴于按照一般规则,医疗器械应标示CE标志,表明它们符合本指令的条款,使其能在欧共体内自由流通,并按其预定用途投入使用;21 鉴于为对抗艾滋病和根据1989年5月16日通过的关于在欧共体级别的关于艾滋病预防和控制的进一步行动的理事会决议,用于预防HIV病毒感染的医疗器械必须提供高度的保护;鉴于这类产品的设计和制22 鉴于分类规则通常可对医疗器械进行恰当的分类;鉴于考虑到各种器械的不同性质及该领域内的技术进步,采取的步骤应包括授权执委会决定器械的适当分类或重新分类,必要时调整分类规则;鉴于这些问题与健康保护有密切关系,因此这些决议应按照第87/373/EEC号指令规定的第Ⅲa程序进行;23 鉴于证实符合基本要求可能意味着必须由制造商负责完成临床试验;鉴于为了完成临床试验,必须确定保护公众健康和公共秩序的适当方式;24 鉴于在欧共体级别一体化的医疗器械警戒系统可以更有效地完成健康保护和相关的控制;25 鉴于本指令覆盖了1976年7月27日理事会第76/764/EEC号关于使成员国有关临床用汞柱式温度计最高读数的法律趋于一致的指令中提到的医疗器械;鉴于上述指令应予撤销;鉴于同样原因,1984年9月17日理事会第84/539/EEC号关于使成员国有关用于人或兽医的电子医疗器械的法律趋于一致的指令必须修改。
兹通过本指令:第1条定义,范围1. 本指令适用于医疗器械及其附件。
在本指令中,附件应被视为医疗器械。
医疗器械和附件以下均称为器械。
2.在本指令中,适用以下定义:(a) “医疗器械”指制造商预定用于人体以下目的的任何仪器、器械、器具、材料或其它物品,无论它们是单独使用还是组合使用,包括为其正常使用所必需的软件:——疾病的诊断、预防、监测、治疗或减缓;——受伤或残障的诊断、监测、治疗、减缓或修补;——解剖学或生理学过程的探查,替换或变型;——妊娠的控制。
这些器材不可通过药理学,免疫学或代谢作用等方式在人体内外达到其预定的基本作用,但可用这些器材辅助其功能。
(b) “附件”本身不是器械,但由其制造商特别预定与器械一起使用,使器械能按照制造商为其预定的用途来使用的物件;(c) “用于体外诊断的器械”指用于对来自人体的样品进行体外检测的任何试剂、试剂产品、成套工具、仪器、装置或系统,无论它们单独使用或组合使用,其目的都是为了提供人体的生理状况、健康或疾病状况或先天性异常等信息;(d) “定制器械”指根据有资格的医生依据其职责给出特定设计特点的书面指示而制造的仅用于特别患者的器械。
上述指示也可以由具有专业资格经认可的其他人提供。
为满足执业医生或其它专业使用者特殊要求而改装且批量生产的器械不视为定制器械;(e) “临床试验器械”指有正式资格的执业医生在如附录X第2.1节中提到的适宜于人类的临床环境中进行试验所用的器械。
对于实施临床试验而言,任何凭借其专业资格被批准从事此项试验的人都可被承认相当于具有正式资格的执业医生;(f) “制造商”指在以其名义将器械投放市场前,负责器械的设计,制造,包装和标签的自然人或法人,无论这些工作是他自己完成的,还是由第三方代他完成的。
本指令规定制造商必须履行的义务也适用于负责对一件或多件制成品进行装配、包装、加工、重新处理和/或加贴标签和/或对其作为一件器械规定其预期用途,以便器械能以其名义投放市场的自然人或法人。
非属第一段制造商定义者,而是为个别患者组装或改装已上市的器械的人不适用于本段的规定;(g) “预定用途”指根据制造商在标签,说明书和/或宣传材料中提供的资料对器械预定的用途;(h) “投放市场”指首次使一种器械不是用于临床试验,而是以付款交易或免费赠送的方式在欧共体市场销售或使用,不论其是新的还是经过重新处理的;(i) “投入使用”指器械在欧共体市场上第一次按预定用途预备使用的阶段;3. 如果器械是用于控制第65/65/EEC号指令第一条覆盖的药品,则该器械受本指令管辖,但不得违反第65/65/EEC号指令中有关药品的条款。
然而,假如器械与药品组合成一整体产品,只可以给定的组合形式使用且不能二次使用时,则该完整产品受第65/65/EEC号指令管辖。