寡核苷酸介导的定点诱变ppt课件
第七章定点诱变

第七章定点诱变第七章定点诱变[本章摘要]突变有重复、缺失、倒位、易位四种类型。
定点诱变主要是关于:简单的插入或缺失,单个碱基的置换以及系统的缺失、插入或成串碱基的置换。
寡核苷酸介导的定点诱变主要是对单个或少数几个碱基的操作;外切核酸酶Ⅲ、BAL31、DNase Ⅰ介导嵌套缺失,可以得到系列缺失体。
第一节:寡核苷酸介导的诱变70年代初j×174 DNA (ss DNA 5386bp),当用带琥珀突变的ssDNA 与变性的野生型DNA片段一起转染细菌时,观察到“标记获救”现象。
即产生带野生型基因组的噬菌体,原因是野生型DNA 片段与突变体的相应序列退火,形成错配的异源双链体,后者可被宿主编码的错配修复系统转变为全长的野生型基因组。
由此可利用突变的DNA 片段将突变引入到野生型DNA 中。
一、Kunkel法或称“U” 法1.背景介绍dut-dUTP 酶缺陷,细胞不能把dUTP 转化为dUMP ,因此细胞内dUTP 的含量大为增加,其中一些dUTP 可掺入DNA 中正常情况下由胸苷嘧啶占据的位置。
ung -UDG 酶缺陷,UDG 酶[尿嘧啶(uracil)-N-糖基化酶(glycosylase)]可除去掺入DNA 中的尿嘧啶残基。
2.原理Kunkel 法原理如图7-1 所示,过程包括:①当目标DNA 插入phagemid 载体并进入E.coli CJ236 后。
②由于该菌体ung- dut-突变,合成的DNA 上含有少量尿嘧啶(取代胸腺嘧啶)。
③M13K07 辅助感染下,产生带U 的单链DNA 。
④与引入诱变的Oligo 复性。
⑤在T7DNA polymerase 和ligase 作用下,以带U 的单链DNA 为模板合成一条新链,形成杂合双链。
⑥感染dut+ung+ E.coli MV1190 后,在细胞分裂过程中,只有新合成的DNA 链才能起模板作用合成新的并引入了诱变位点的子代双链DNA 。
《定点诱变技术》课件

本课程将介绍定点诱变技术的原理、实验步骤、应用案例和未来展望,以及 它对人类社会的影响和挑战。
概述
什么是定点诱变技术?
定点诱变技术是一种精准编辑基因术?
传统的基因编辑技术往往是非特异性的,不能达到精准编辑的效果,而定点诱变技术可以 避免对非目标区域的影响。
实验步骤
1
实验前的准备工作
首先需要构建引导RNA分子、购买
细胞培养与转染
2
Cas9核酸酶,以及筛选细胞线等。这 些都需要提前准备。
将引物与Cas9,利用转染技术导入到
目标细胞中,完成复合物的构建。
3
PCR扩增和测序
利用PCR扩增技术检测目标基因组位
置是否发生了修饰,并对其进行测序,
数据分析和结果解读
定点诱变技术的应用领域
定点诱变技术可以应用于基础科研、农业生产、医疗诊断等领域,为人类社会带来了诸多 益处。
基本原理
基于CRISPR-Cas9技术实 现的定点诱变
CRISPR-Cas9技术可以在基因 组中精准识别目标区域,并将 核酸酶Cas9导入到目标位置, 再通过引导RNA分子的作用, 完成对基因组上的目标位点的 修饰。
定点诱变技术可能会带来一系 列的伦理、法律和社会问题, 例如导致人们撕裂不和,社会 不公等问题。
定点诱变技术未来发展 的瓶颈和解决方案
目前,定点诱变技术在某些肿 瘤细胞和人类胚胎细胞中并没 有得到广泛应用,需要进一步 研究,探索更为高效、精准、 安全的定点诱变技术。
4
验证修饰深度和准确性。
对PCR扩增和测序的数据进行分析, 解读实验结果,得出结论,为下一步
的研究提供指导。
应用案例
定点诱变技术在基因 修复中的应用
基因定点诱变技术与DNA与蛋白质互作及定点突变介绍

30
酵母单杂交的基本 原理示意图
从 拟 南 芥 cDNA 文 库 中 筛选与顺式元 件DRE结 合的 转录因子示意图。
31
三)凝胶阻滞实验 (electrophoretic mobility shift assay, EMSA)
六)体内足迹试验
• 用适量DMS处理完整的游离细胞,使染色 质中的G残基甲基化,提取DNA并加入六氢 吡啶切割DNA链。与对照的裸露DNA经甲基 化处理后形成42
43
七)染色质免疫沉淀实验 Chromatin Immunoprecipitation(ChIP) Assay
33
34
• EMSA • 还被
用于 研究 与蛋 白质 相结 合的 DNA • 序列 的特
异型。
35
四)DNaseI足迹试验 (DNase I foot printing)
• 主要步骤: ① 用32P标记DNA双链末端,并用RE切去一端; ② 加入细胞特定周期蛋白质提取物,温育; ③ 加入适量DNaseI或硫酸二甲酯-六氢吡啶,使 DNA链发生断裂。 这一反应中,DNaseI或硫酸二甲酯的用量非常关键, 要保证一条链只发生一次断裂! ④沉淀DNA(包括与DNA相结合的蛋白质); ⑤进行DNA凝胶分析。
真核生物DNA序列(非编码序列)和被 转录的结构基因距离较近,和转录调控有关。
A 启动子(启动子上游近侧序列) B 增强子 C 沉默子
29
启动子(promtor)在转录起始点上游约100-200bp以内, 每个元件长度约为7-20bp,决定RNA聚合酶Ⅱ转录起始 点和转录频率的关键元件。
定点诱变技术

Family gene shuffling library of chimeras
Generating chimeras with crossovers of large blocks of sequences
How DNA shuffling works ?
一、单基因和基因家族的重组装
Fragment with DNAseI
第三章 DNA突变技术
基因突变包括单个碱基或片断的替换,基
因片断的插入与删除等。
根据其特点可将基因突变技术分两大类:
1.位点特异性突变
定点突变
2.随机突变 表型筛选
随机突变
易错PCR法(Error-prone PCR)
降低一种dNTP的量(降至5%-10%) 加入dITP来代替被减少的dNTP 缓冲液中另加0.5mmol/L Mn2+
适用于插入片段长度在 1.0kb以下的多点突变。
Steps of Multipoints Mutagenesis Kit (TaKaRa)
X
XXX
X
Reassemble fragments
XXX
X
Select best recombinants
XXX
XX
X
XX
X
XX
X
XXX X
XXX X
XXX X
XX XXX X X X X X
XX X
X
XX X
X
XX X
X
XX X
XXXXXX NhomakorabeaXX
X
Repeat for multiple cycles
枯草杆菌蛋白酶E热稳定性的分子进化 (Huimin Zhao等,1999)
8 DNA诱变

易错PCR原理图
盒式诱变
盒式取代
通过限制性内切酶切除特定的双链DNA片段,再与含有突变 的单一同源DNA片段连接。
混合核苷酸诱变
取代的是含有随机突变的混合双链寡核苷酸,可在限定区域 内引入大量的随机突变。 由于核苷酸一般由化学法合成,因此可变区域受到限制。
简单的盒式取代诱变
混合寡核苷 酸盒式诱变
增变菌株的诱变作用
增变菌株的诱变作用是在活体细胞中进行的,但是诱变 的目的基因不是增变菌株的基因,而是克隆化的外源片 段DNA。 常用的大肠杆菌增变菌株为XL1-Red (mutD mutS mutT)。
mutD的突变造成DNA聚合酶3’-5’外切核酸酶活性缺陷; mutS的突变使DNA错配修复系统失去功能; mutT的突变不能水解dGTP的氧化产物8-oxo-dGTP,使得DNA发 生突变。
突变频率一般为10-3/bp/代。
DNA洗牌
DNA洗牌法(DNA shuffling)是1994年Stemmer实验 室开发的用于改造单基因的体外诱变方法,针对有一 定同源性的基因家族进行基因家族洗牌。 步骤
选择一组具有一系列形状的基因序列作为重组的亲本,亲本 之间有序列同源性(>60%); 超声波或酶处理,消化DNA片段为50-100 bp大小的片段; 无引物PCR; 有引物PCR; 克隆产物。
2.
•
重叠延伸PCR诱变
需要1对内部引物和1对侧翼引物,经过3个PCR反应,效率 非常高。
3.
•
双向PCR快速定点诱变
需要1对引物,经过1次PCR反应,但2条引物5’端所对应的 序列是连续的。
大引物PCR诱变
重叠延伸PCR诱变
第十章 基因工程的应用[可修改版ppt]
![第十章 基因工程的应用[可修改版ppt]](https://img.taocdn.com/s3/m/514014e8376baf1ffd4fad58.png)
诱变型
⑨分离提取M13双链DNA,再转化大肠杆菌,用寡聚核 苷酸探针挑选突变型载体克隆。
⑩切下突变基因,接到表达载体中表达诱变的蛋白质。
(3)缺点: 通常只有1%-5%的噬菌斑含有突变的基因。
(4)改进: 目的基因插入双链形式的M13噬菌体后,转入特 殊的受体菌株中。
2. 寡核苷酸介导的PCR诱变 (1)方法 ①将目的基因克隆到质粒上。
⑥转化修复缺陷型大肠杆菌。
⑦模板链由于已经成为线性分子,且又不能被修 复,所以不能在菌体内稳定存在,只剩下新生的 突变链在菌体内复制。 4. 重叠延伸诱变 ①目的基因克隆到载体上(不一定是M13)。
②设计两对引物:
引物1和引物2是目的基因两端的载体上的通用引物; 引物3和引物4是与突变位点对应的反向互补序列。
粘性末端1 ATGAAຫໍສະໝຸດ GCATGCGTACG 4
ATGC TACGTACTTAC
粘性末端
⑤环化成有两个切口的开环质粒。用连接酶 连接或直接转入细菌(细菌会修复切口)。
3.改进型双引物诱变 方法(是目前较常用的方法之一):
①目的基因克隆到M13噬菌体载体中。 ②分离提取M13单链DNA作模板。
③设计两个不同区段的PCR引物,延伸方向一致, 其中一个引物的5’端磷酸化。
单一酶切位点
④用DNA聚合酶延伸后用T4DNA连接酶连 接成杂交双链。
TACTTACGTACG ATGCATGCATGCATGC
EcoRI酶切口
CTGGAATTCATGCGAC GACCTTAAATACG
⑤用引物2对应的单一限制性内切酶切杂交双链,会 得到模板链被切开一个口但新生的链完整的杂交双 链环。
重链上有3个(CH1、CH2、CH3),轻链上有 1个(CL)。 不同的抗体分子的恒定区的差异只有一个或两 个氨基酸。
第十章_DNA诱变

第一节 随机诱变
体外随机诱变:随机地在克隆化DNA中引入碱基臵
换突变。 特点:不需要有序列针对性的合理设计,引入突变 的位臵及其性质是随机的;在目的DNA片段中引入 大量的序列多样性,得到的突变体可能是单点突变 ,也可能是多点突变。 成功的关键有二:1、选择合适的突变率;2、有效 的定向选择或筛选方法。 方法:错误掺入诱变、盒式诱变、增变菌株诱变、 化学诱变。
二、盒式诱变
盒式诱变(cassette mutagenesis):是一种定点
突变技术,将靶基因的一段DNA删掉,并用人工化 学合成所具有的突变核苷酸的双链寡核苷酸片段取 代。 包括简单的盒式取代诱变和混合寡核苷酸诱变两种 方式。 简单的盒式取代诱变是通过限制性内切酶切除特定 的双链DNA片段,再与含有突变的单一序列双链寡 核苷酸连接,得到取代突变,是一种定点诱变。 若用于取代的是含有随机突变的混合双链寡核苷酸 ,则可在限定区域内引入大量的随机突变。
2、重叠延伸PCR诱变
使用带突变碱基的互补引物对两段目的基因序列分别进行第一轮PCR
扩增;将扩增产物进行重叠退火延伸,获得带突变碱基的完整序列 ;使用该完整序列的两端引物进行第二轮PCR扩增。
应用:重叠延伸剪接术(SOE)
可用于将两个 DNA 片段在所期望的位点进行连接。
包括两个步骤:
一、DNA洗牌法
DNA洗牌法(DNA Shuffling):体外同源重组技术
。将来源不同但功能相同的一组同源基因,用核酸 酶Ⅰ消化成随机片段,由这些随机片段组成一个文 库,使之互为引物和模板进行PCR扩增,当一个基因 拷贝片段作为另一基因拷贝的引物时,引起模板互 换,重组因而发生,导入体内后,选择正突变体作 为新一轮的体外重组。
基因工程第七章DNA定点诱变

• 3’端所形成的杂交体足以引导DNA合成。如果
错配核苷酸太靠近3’端,3’端将不能形成稳定的 杂交体,易被外切活性降解。所以3’端需有79bp完全配对;
• 为便于筛选,应选用可形成稳定杂交体而长
度最短的诱变寡核苷酸。一般17-19bp, 错配在 中央,使得完全配对的杂交体与错配杂交体 之间的热稳定性差异足够大。
位点选择定点诱变法
三、Transformer SiteDirected mutagenesis
转化子诱变法
Synthesize second strand
Digest DNA (primary digestion)
Transform E.coli mutS to propagate plasmids
ATG ATG
ATG ATG
BamHI
ATG
BamHI
PCR ATG
ATG
EcoRI
PCR
ATG
EcoRI
重叠延伸
BamHI
ATG
PCR EcoRI
第二节 嵌套缺失
第二节 嵌套缺失
1. 外切核酸酶III的消化
Exonuclease III 5‘ 3‘
5‘ 3‘
5‘ 3‘
2. BAL 31的消化
DNA ligase
U
U
U
U
U
2.原理
Insert target DNA
转化 E.coli CJ236
U
U
U U
U
U
Isolate phagemid ss DNA 与突变寡核苷酸退火
转化E.coli MV1190 (dut+/ung+)
UU
基因定点诱变
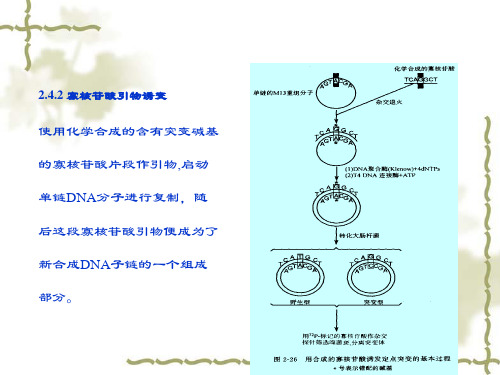
使用化学合成的含有突变碱基 的寡核苷酸片段作引物,启动 单链DNA分子进行复制,随 后这段寡核苷酸引物便成为了 新合成DNA子链的一个组成 部分。
寡核苷酸引物诱变过程
正链DNA合成 突变引物合成 异源双链DNA分子制备 闭环异源双链DNA分子富集 转化 突变体筛选
寡核苷酸引物诱变法的局限性
异源双链DNA分子并非真正异源 突变体子代中突变碱基被错配修 复体系修复
提高寡核苷酸引物诱变突变效率的方法
(1) Kunkel 定点诱变法: 1985年由
(2) 硫代磷酸诱变法: 已知有些限制酶不能切割 硫代磷酸DNA分子.在异源双链DNA分子 制备时,加入硫代核苷酸,使之掺入突变 链中.然后用前述酶切割,并用外切酶局 部消化后,进行聚合反应,从而产生具有 定点突变的异源双链DNA
2.4.3 PCR诱变
(1) 重组PCR定点诱变:克服寡 核苷酸引物诱变能力仅限于5’ 端.
特点:经3轮PCR反应,一对互补 的带有突变碱基的内侧引物和 2个外侧引物
(2) 大引物诱变:核心是第一 轮PCR产物为第二轮PCR引 物
返回目录
返回第二章
定点突变技术

操作:设计引物,分别PCR,前两次PCR 反应产物经琼脂糖凝胶电泳鉴定后无需 纯化,直接将胶条切下置于EP管中, 80℃冷冻10min,然后将胶条离心后分 别取上清液作为模板进行第三次PCR,以 获得全长的突变目的基因
PCR介导的定点突变法其优点是操作较简 单,突变的成功率可达100%。但它亦 有两个缺点:①后续工作较复杂,PCR扩 增产物通常需要连接到载体分子上,然
寡核有酸引物介导的定点突变 法其优点是保真度比PCR突变法 高,经过改进后使该方法突变少 成功率大大提高,缺点是操作过 程环节复杂制。
盒式突变法具有简单易行、突变效率高 等优点,还可以在一对限制酶切位点内 一次突变多个位点。缺点是合成多条引 物的成本较高。另外,在一般情况下, 在靶DNA片段的两侧往往难以满足存在 一对限制性酶切位点的要求,限 制了该 方法的广泛应用。然而一旦具备了这样 的条件该方法则为首选。
定点突变技术
刘微
基因的定点突变技术
点突变的技术有很多种,常见的有: ㈠寡核苷酸介导的定点突变技术
(M13噬菌体法) ㈡ 盒式诱变 ㈢PCR点突变技术
1.引物PCR定点诱变法 2.重组PCR定点诱变法 3.重叠延伸PCR技术
寡核苷酸引物诱变技术 (M13噬菌体)
噬菌体M13的生活周期有二个阶段,在噬菌体 粒子中其基因组为单链,侵入宿主细胞以后, 通过复制以双链形式存在。将待研究的基因 插入载体M13,制得单链模板,人工合成一 段寡核苷酸(其中含一个或几个非配对碱基) 作为引物,合成相应的互补链,用T4连接酶 连接成闭环双链分子。经转染大肠杆菌,双 链分子在胞内分别复制,因此就得到两种类 型的噬菌斑,含错配碱基的就为突变型。
重组PCR定点诱变2
操作:设计引物,分别PCR, 从两个PCR反 应管中各取出3μl PCR反应产物,混匀后用 CaCl2转化法转化至感受态大肠杆菌中。涂 平板后,从转化的细菌菌落中随机挑选若干,筛 选。
基因工程8-DNA诱变ppt课件

易错PCR采用的方法:
• 增加MgCl2浓度到7mM,稳定非互补的碱基配对; • 加入0.5mM的MnCl2,Mn 2+能降低聚合酶对模板的特异性; • 增加聚合酶量到5U,促使在错配碱基处继续延伸反应; • 限定4种碱基中的一种,通常为正常浓度的1-10%,在缺乏正 确核苷酸时,DNA聚合酶经短暂停顿后,会插入另外3种可用 核苷酸的一种; • 3种为正常浓度的正常碱基,第四种为次黄嘌呤dITP(可与C、 T、A配对); • 增加dCTP和dTTP的浓度到1mM,促进错误掺入; • 使用突变DNA聚合酶,如Mutazyme。
在体外用诱变引物合成的杂合双链DNA在导入正常ung+ dut+菌 株后,只有带有突变位点的新合成链和作模板进一步复制,而 野生型不能复制。这样能够生长的细胞就带有突变位点。
(二)位点选择诱变
(altered sites in vitro mutagenesis)
位点选择诱变法使用了2个寡核苷酸引物,一个是用来引入突变 的引物,另一个是用来选择用的。选择性引物可用来恢复有缺陷 的抗生素抗性基因,同时可用来选择引入突变的DNA链。
特点:可正向选择突变链,使用高保真的T4DNA聚合酶合成 DNA,可以使用双良DNA作模板,可进行多轮筛选。但需特定载 体,即含有一个有缺陷的抗生素抗性基因。
(三)转化子诱变
(transformer site-directed mutagenesis)
寡核苷酸介导的定点诱变

利用一段含有突变序列的 寡核苷酸片段作为引物,经 DNA复制过程合成靶DNA片段, 使其子代链发生突变,称为引 物诱变。 引物诱变的基本操作步骤: ①获得单链目的基因;②人工 合成带突变序列的引物;③制 备异源双链DNA;④转染宿主 细胞;⑤筛选重组体;⑥突变 基因的鉴定和回收
寡核苷酸介导的定点诱变
• 盒式诱变(Cassette Mutagenesis) • 利用一段人工合成 的含有突变序列的寡核 苷酸片段,取代野生型 基因中的相应序列,从 而达到定点突变的目的, 称为盒式诱变。实施盒 式诱变时,要求靶DNA 插入点两侧有合适的限 制酶单一切点(图 4~5)。
盒式诱变的例子
快速引物诱变
(QuikChange Primer Mutagenesis) 这是由Stratagene公 司开发的一种引物定点诱 变法,该法的操作步骤如 下:①将带目的基因的重 组体在dam+菌株中扩增, 使DNA序列被dam甲基化 酶所修饰;②使用带突变 碱基的寡核苷酸引物,在 较高温度下复制子代链, 形成双链突变体;③用限 制酶DpnⅠ将带甲基化标记 的亲代双链水解;④将突 变体转化宿主细胞
在dut+的E. coli中
dUTP酶
dUTP
dUMP
在dUTP 酶缺失体中(dut- )
dUTP
dUMP
dut:dUTPase突变,可导致E.colidUTP 的量增加,当DNA复制时,可在很多应 该加dTTP的位置加入dUTP。
•
在正常情况下,尿嘧啶-N-糖 基化酶(ung+)可以去除掺入DNA中 的尿嘧啶残基。但在ung-的菌株 中,此酶失活。
•
限制酶位点消除引物诱变(Primer Mutagenesis with Restriction Enzyme Site Elimination)
第二章 基因工程主要技术原理-基因定点诱变 ppt课件
ppt课件
6
(2) 硫代磷酸诱变法: 已知有些限制 酶不能切割硫代磷酸DNA分 子.在异源双链DNA分子制备 时,加入硫代核苷酸,使之掺 入突变链中.然后用前述酶切 割,并用外切酶局部消化后, 进行聚合反应,从而产生具有 定点突变的异源双链DNA
ppt课件
7
2.4.3 PCR诱变
(1) 重组PCR定点诱 变:克服寡核苷酸引 物诱变能力仅限于5’ 端.
特点:经3轮PCR反 应,一对互补的带有 突变碱基的内侧引 物和2个外侧引物
ppt课件
8
(2) 大引物诱变:核心 是第一轮PCR产物为 第二轮PCR引物
ppt课件
9
返回目录
返回第二章
ppt课件
10
ppt课件
2
精品资料
基因工程
第二章 基因工程技术原理
• 你怎么称呼老师? • 如果老师最后没有总结一节课的重点的难点,你
是否会认为老师的教学方法需要改进? • 你所经历的课堂,是讲座式还是讨论式? • 教师的教鞭 • “不怕太阳晒,也不怕那风雨狂,只怕先生骂我
笨,没有学问无颜见爹娘 ……” • “太阳当空照,花儿对我笑,小鸟说早早早……”
ቤተ መጻሕፍቲ ባይዱ返回目录
第四节 基因定点诱变
2.4.1 盒式诱变
利用一段人工合成 的具有突变序列的 寡核苷酸片段取代 野生型基因中的相 应序列.
返回第二章
ppt课件
1
2.4.2 寡核苷酸引物诱变
使用化学合成的含有突变碱基 的寡核苷酸片段作引物,启动 单链DNA分子进行复制,随 后这段寡核苷酸引物便成为了 新合成DNA子链的一个组成 部分。
寡核苷酸引物诱变过程
正链DNA合成 突变引物合成 异源双链DNA分子制备 闭环异源双链DNA分子富集 转化 突变体筛选
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
引物诱变的基本操作步骤: ①获得单链目的基因;②人工 合成带突变序列的引物;③制 备异源双链DNA;④转染宿主 细胞;⑤筛选重组体;⑥突变 基因的鉴定和回收
Kunkel诱变法(The
+
限制酶位点消除法的操作步骤是:①将带突变碱基的寡核苷
酸引物与重组体单链模板退火后,加入硫代磷酸核苷酸衍生物进行复
制,产生具有硫代磷酸核苷酸的异源双链DNA;②代模板链由于无硫代磷酸核苷酸而
被切开,而新合成的子代链则无法切割;③使用核酸外切酶将亲代模
板链全部水解去除,然后再以含硫代磷酸核苷酸的子代单链为模板,
+
在大肠杆菌dut- ung-菌株
中生长的M13噬菌体的单链基因组
DNA中将含有20-30个尿嘧啶残基。
用这些噬菌体感染ung+菌株,尿嘧
啶被迅速去除,DNA链遭到破坏,
感染力下降约5个数量级。
+
此法的成功关键,是要得到好
的含U单链模板DNA。
+
这种方法得到的突变率太低,约为1-5%。主要原因是含突变位
+ 限制酶位点消除引物诱变(Primer Mutagenesis with Restriction Enzyme Site Elimination)
+
限制酶位点消除引物诱变技术是一种利用某些限制酶不能切割由
硫代磷酸核苷酸衍生物取代正常碱基所形成的DNA片段的特性,去除
不含突变碱基的亲代模板链,以提高定点突变效率的技术方法。
点的双链DNA,转入E.coli后,被其修复系统修复了。
快速引物诱变
(QuikChange Primer Mutagenesis)
这是由Stratagene公 司开发的一种引物定点诱 变法,该法的操作步骤如 下:①将带目的基因的重 组体在dam+菌株中扩增, 使DNA序列被dam甲基化 酶所修饰;②使用带突变 碱基的寡核苷酸引物,在 较高温度下复制子代链, 形成双链突变体;③用限 制酶DpnⅠ将带甲基化标记 的亲代双链水解;④将突 变体转化宿主细胞
Kunkel Mutagenesis Method)
这是1985年由Kunkel等人建 立的一种寡核苷酸引物诱变法, 该法的操作步骤如下:①将复制 型的M13噬菌体转染到脱氧尿苷 三磷酸酶和尿嘧啶脱糖苷酶双缺 陷的E. coli(dut-,ung-)菌株中 生长,使U取代T掺入到DNA链中 (一般每个重组体20~30个);② 以此种带U的DNA链为模板,用带 突变碱基的寡核苷酸引物进行复 制,产生异源双链;③将此异源 双链转化到ung+菌株中生长,含 U的模板链被破坏,从而使子代 DNA链大部分(~80%)含突变碱 基序列
经二次复制合成双链
+ 盒式诱变(Cassette Mutagenesis)
+ 利用一段人工合 成的含有突变序列的 寡核苷酸片段,取代 野生型基因中的相应 序列,从而达到定点 突变的目的,称为盒 式诱变。实施盒式诱 变时,要求靶DNA插 入点两侧有合适的限 制酶单一切点(图 4~5)。
盒式诱变的例子
引物诱变(Primer Mutagenesis)
在dut+的E. coli中
dUTP酶
dUTP
dUMP
在dUTP 酶缺失体中(dut- )
dUMP dUTP
dut:dUTPase突变,可导致E.colidUTP 的量增加,当DNA复制时,可在很多应 该加dTTP的位置加入dUTP。
+ 在正常情况下,尿嘧啶-N-糖基 化酶(ung+)可以去除掺入DNA中的 尿嘧啶残基。但在ung-的菌株中, 此酶失活。