实沸点AET计算
LED结温热阻计算方法详解

LED结温热阻计算方法详解.Ta: 环境温度Rsa:铝基散热装置的热阻、散热器与环境间的热阻Ts: 散热装置的温度. Rms:铝基板到铝散热装置的热阻Tm: 铝基板的温度. Rcm:引脚到铝基板的热阻Tc: 引脚的温度. Rjc:PN结到引脚的热阻、结壳间的热阻Rja:PN结点到环境的热阻 Tj:晶体管的结温、芯片PN结最大能承受之温度( 100-130℃)P表示功耗 Rcs表示晶体管外壳与散热器间的热阻,L50: LED光源亮度降至50%的寿命L70: LED光源亮度降至70%的寿命结温计算的过程:1.热阻与温度、功耗之间的关系为: Ta=Tj-*P(Rjc+Rcs+Rsa)=Tj-P*Rja,2.当功率晶体管的散热片足够大而且接触足够良好时,壳温Tc=Ta晶体管外壳与环境间的热阻Rca=Rcs+Rsa=0。
此时Ta=Tj-*P(Rjc+Rcs+Rsa)演化成公式Ta=Tc=Tj-P*Rjc。
厂家规格书一般会给出,最大允许功耗Pcm、Rjc及(或) Rja等参数。
一般Pcm 是指在Tc=25℃或Ta=25℃时的最大允许功耗。
当使用温度大于25℃时,会有一个降额指标。
3.以ON公司的为例三级管2N5551举个实例:1)2N5551规格书中给出壳温Tc=25℃时的最大允许功耗是1.5W,Rjc是83.3度/W。
2)代入公式Tc=Tj- P*Rjc有:25=Tj-1.5*83.3可以从中推出最大允许结温Tj为150度。
一般芯片最大允许结温是确定的。
所以,2N5551的允许壳温与允许功耗之间的关系为:Tc=150-P*83.3。
3)比如,假设管子的功耗为1W,那么,允许的壳温Tc=150-1*83.3=66.7度。
4)注意,此管子Tc =25℃时的最大允许功耗是1.5W,如果壳温高于25℃,功率就要降额使用。
规格书中给出的降额为12mW/度(0.012W/度)。
5)我们可以用公式来验证这个结论。
假设壳温为Tc,那么,功率降额为0.012*(Tc-25)。
熔点及沸点测定

熔点及沸点测定熔点、沸点测定1、熔点的测定化合物的熔点就是所指在常压下用物质的遏―液两二者达至均衡时的温度。
但通常把晶体物质熔化后由固态转变为液态时的温度做为该化合物的熔点。
清澈的液态有机化合物通常都存有紧固的熔点。
在一定的外压下,固液两态之间的变化就是非常灵敏的,自初熔至全系列熔(称作熔程)温度不少于0.5-1℃。
若混有杂质则熔点存有明晰变化,不但熔点距不断扩大,而且熔点也往往上升。
因此,熔点就是晶体化合物纯度的关键指标。
有机化合物熔点通常不少于350℃,更易测量,故需借测量熔点去辨别未明有机物和推论有机物的纯度。
在鉴定某未知物时,如测得其熔点和某已知物的熔点相同或相近时,不能认为它们为同一物质。
还需把它们混合,测该混合物的熔点,若熔点仍不变,才能认为它们为同一物质。
若混合物熔点降低,熔程增大,则说明它们属于不同的物质。
故此种混合熔点试验,是检验两种熔点相同或相近的有机物是否为同一物质的最简便方法。
熔点装置图:沸点装置图:2、沸点的测定2、沸点测量液体的分子由于分子运动有从表面逸出的倾向,这种倾向随着温度的升高而增大,进而在液面上部形成蒸气。
当分子由液体逸出的速度与分子由蒸气中回到液体中的速度相等,液面上的蒸气达到饱和,称为饱和蒸气。
它对液面所施加的压力称为饱和蒸气压。
实验证明,液体的蒸气压只与温度有关。
即液体在一定温度下具有一定的蒸气压。
当液体的蒸气甩减小至与外界其行液面的总压力(通常就是大气压力)成正比时,就存有大量气泡从液体内部逸出,即为液体融化。
这时的温度称作液体的沸点。
通常所说的沸点是指在101.3kpa下液体沸腾时的温度。
在一定外压下,纯液体有机化合物都有一定的沸点,而且沸点距也很小(0.5-1℃)。
所以测定沸点是鉴定有机化合物和判断物质纯度的依据之一。
测定沸点常用的方法有常量法(蒸馏法)和微量法(沸点管法)两种。
五、实验步骤1、熔点的测量毛细管法:o①准备工作熔点管:将毛细管截成6~8cm长,将一端用酒精灯外焰封口(与外焰成40角旋转冷却)。
高中化学 有机化合物的标准热力学数据
CH3SH
l
-46.4
-7.7
169.2
90.5
g
-22.3
-9.3
255.2
50.3
26
甲 胺
CH5N
l
-47.3
35.7
150.2
102.1
g
-22.5
32.7
242.9
50.1
27
甲 肼
CH6N2
l
54.0
180.0
165.9
134.9
g
94.3
187.0
278.8
71.1
28
三氯乙腈
C2Cl3N
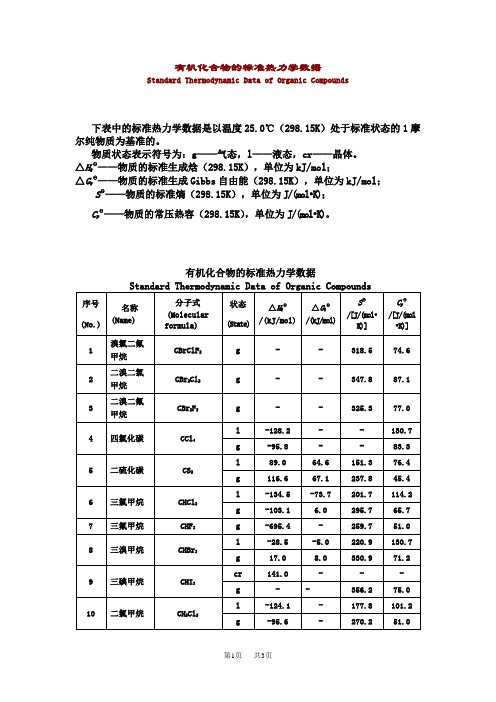
有机化合物的标准热力学数据
Standard Thermodynamic Data of Organic Compounds
下表中的标准热力学数据是以温度25.0℃(298.15K)处于标准状态的1摩尔纯物质为基准的。
物质状态表示符号为:g——气态,l——液态,cr——晶体。
△HfΘ——物质的标准生成焓(298.15K),单位为kJ/mol;
序号
(No.)
名称
(Name)
分子式
(Molecular formula)
状态
(State)
△HfΘ
/(kJ/mol)
△GfΘ
/(kJ/mol)
SΘ
/[J/(mol•K)]
CpΘ
/[J/(l•K)]
1
溴氯二氟甲烷
CBrClF2
g
-
-
318.5
74.6
2
二溴二氯甲烷
CBr2Cl2
g
-
-
347.8
87.1
按照爱因斯塔模型,求出单原子晶体的晶格热容量,并求出高低温极限情况下的表达式

按照爱因斯塔模型,求出单原子晶体的晶格热容量,并求出
高低温极限情况下的表达式
根据爱因斯塔模型,单原子晶体的晶格热容量可以用以下公式表示:
Cv = 3R
其中,Cv为晶格热容量,R为气体常数,其值约为8.314 J/(mol·K)。
在高温极限情况下,可以使用经典统计物理学中的杜隆-珀蒂定律来求解晶格热容量。
根据该定律,高温下的晶格热容量与晶格振动频率有关,表达式如下:
Cv = 3NR
其中,N为晶格中的原子数。
在低温极限情况下,由于晶格振动的量子性质开始显现,需要考虑量子力学效应。
在低温下,晶格热容量可由爱因斯坦固体模型来描述,其表达式如下:
Cv = 3NR[ (θE/T)^2 * exp(θE/T) / (exp(θE/T) - 1)^2 ]
其中,θE为爱因斯坦温度,T为绝对温度。
需要注意的是,以上表达式仅适用于单原子晶体,并且在实际应用中可能会存在一定的误差。
碳材料达恩公式

碳材料达恩公式
碳材料的达恩公式表示的是在恒定温度下,碳材料的热导率与频率和温度的关系。
其公式为:
k=∑(εi/λi)Gi
其中,k表示热导率,εi表示第i个组分的体积分数,λi表示第i个组分的热导率,Gi 表示第i个组分的热容。
需要注意的是,达恩公式只适用于恒定温度下的碳材料,且组分的热导率和热容也需要已知。
同时,该公式假设碳材料的热导率是由各个组分的热导率加权平均得到的,忽略了碳材料中可能存在的其他效应,如声子散射等。
因此,在实际应用中,需要结合具体的情况进行分析和修正。
实验指导书(一)-第一性原理方法计算

《计算材料学导论》实验指导书实验一:第一性原理方法计算模拟化合物的晶体结构和电子结构实验目的:1)近十年来,随着计算机技术和材料科学的发展,基于密度函数理论的第一性原理方法计算在材料科学中的应用十分普遍和活跃,发展异常迅速。
其应用领域涉及材料晶体结构优化,态密度和能带结构等电子结构,掺杂效应,相变热力学、光、电磁学性质的计算和设计。
量子化学计算软件包较多,如免费软件包ABINIT(详见教材), 商业化软件包V ASP, CASTEP,GAUSSIAN。
本实验运用VASP4.6软件包,计算AB型的ZnS或相似结构的晶体结构和电子结构。
实验要求:2)首先完成下列基础知识的问答填空,然后运用运用V ASP4.6软件包,计算AB型的ZnS或相似结构的晶体结构和电子结构,并画出图形。
实验内容:(一) 基础填空1) 简述第一性原理方法(或从头算)的基本概念。
()2)简述第一性原理方法在材料科学中有哪些具体应用?()3) 什么叫多粒子体系的总能?()4) 什么叫能带结构?它是如何形成的?()(二)第一性原理方法计算模拟AB型化合物(如ZnS)的晶体结构和电子结构。
1.ZnS具有多种晶形,如闪锌矿结构(The Zincblende (B3) Structure)和纤锌矿结构(The Wurtzite (B4)Structure),与之结构相同的化合物还有很多,不少化合物具有独特的光电特性。
请根据计算指南和模板,计算ZnS或者ZnO, SiC, AlN, CdSe,AgI, AlAs, AlP, AlSb, BAs, BN, BP, BeS, BeSe, BeTe, CdS,CdSe,CdTe, CuBr, CuCl, CuF, CuI, GaAs, GaP, GaSb, HgS, HgSe, HgTe, INAs, InP, MnS, MnSe, SiC, ZnSe, ZnTe)的晶体结构(含晶胞参数a,b,c,V,原子位置的可变内部参数),电子结构(含态密度(含总态密度,分态密度)和能带结构。
熔、沸点的测定-实验一bp、mp的测定.pdf

熔、沸点的测定一、实验目的1、了解熔、沸点测定的意义。
2、掌握用b 形管测定熔、沸点的原理和方法。
二、实验原理为什么测熔、沸点?1、作为特定物理常数,在化合物的初步鉴定、分离和纯化过程中具有重要的意义。
2、可用于初步判断化合物的纯度。
熔点:在标准大气压下,物质的固态和液态处于平衡时的温度,即为该物质的熔点。
通常只有纯净的物质才有固定的熔点,并且熔化范围极短,约为0.5~1℃;而不纯的物质由于杂质的存在,而使熔点降低,熔化温度范围变宽;通常把物质从开始熔化到完全熔化的温度范围称为熔程。
物质的杂质越多,其熔程就越大,不同的化合物有不同的熔程。
因此,通过熔程的测定,可用于化合物的初步鉴定,同时也可根据熔程的长短来定性的判断有机化合物的纯度。
常用毛细管法、显微熔点仪来测定。
沸点:当化合物受热时,其蒸气压升高,当蒸汽压达到与外界压力(通常为1个大气压,0.1Mpa ,760mmHg )相等时,液体开始沸腾的温度,就是该物质的沸点。
由于物质的沸点与外界大气压的有关,因此,在讨论或报道一个化合物的沸点时,一定要注明测定时的外界大气压,如果没注明,就是默认的一个大气压。
纯液态有机化合物在蒸馏过程中沸点范围很小(0.5~1℃),常用微量法(毛细管法)和常量法(蒸馏法)来测量。
当用毛细管法测定时,先加热到内管有连续气泡快速逸出后,停止加热,使温度自行下降,气泡逸出速度逐渐减慢,当最后一个气泡刚要缩进内管而还没有缩进,即与内管管口平行时,这时待测液体的蒸气压就正好等于外界大气压,这时的温度就是待测液体的沸点。
三、药品及物理常数待测物质分子量(mol wt)比重 (d 420) 熔点 (℃)沸点 (℃)性状 折光率nD 水溶解度(g/100g )二苯胺 169.10 1.160 52.5-53.5302 无色晶体稍溶 丙酮 58.05 0.7898 -94.6 56.5 无色液体1.359 易溶 未知液体未知固体四、仪器装置图熔点、沸点测定装置b形管温度计固体样品液体样品温度计沸点外管沸点内管温度计五、实验流程1、熔点测定2、沸点测定六、注意事项:1、制作毛细管时,封端头不能带尖,不能弯曲,应整体一样粗细,端头熔成一个小玻璃球。
测定熔点的方法

测定熔点的方法熔点是一种重要的物理性质,对于确定化学物质的纯度,确认其身份以及研究其结构具有重要意义。
测定熔点的方法有许多种,其中最常用的方法是热差法、热量仪法和显微镜法。
一、热差法热差法是一种常见的测定固体熔点的方法。
该方法基于热量传导的原理,通过记录样品加热以及融化的温度差来测定样品的熔点。
具体操作如下:1. 准备样品:取适量的样品,将其在研钵中加热至液态状态,然后迅速倒入冷却的研钵中,至少重复此步骤三次,使样品充分均匀。
2. 放置样品:准备热差装置,将预备好的样品放置在装置的样品缸中。
3. 开始加热:开启加热源,向样品缸中加热。
使用专业的温度计进行温度监测。
4. 记录温度:当样品开始融化时,记录它的温度。
随着加热的继续,继续记录样品温度,直到样品完全融化。
5. 差值计算:计算样品融化前后的温度差,这个差值即为该样品的熔点。
二、热量仪法热量仪法是一种测定固体或液体熔点的方法,它基于样品吸收或放出热量的原理,通过测量样品的温度变化和吸放热量的大小来测定样品的熔点。
具体操作如下:1. 准备样品:取适量的样品,称重,放入样品室中。
注意样品应该足够纯净。
2. 开始实验:打开热量仪系统并将其预热。
启动系统并选择适当的程序,以便得到准确的热量曲线。
3. 加热样品:使用电热加热系统加热样品,加热速率应该足够慢,使得温度每次提高1-2℃。
4. 记录数据:当样品开始融化时,热量仪系统会显示一个明显的热峰。
记录该峰的时间和温度,以及热量的值。
5. 数据分析:通过分析热量曲线,并计算样品吸放热量的大小,可以精确确定样品的熔点。
三、显微镜法显微镜法是一种测定熔点的标准方法。
该方法利用显微镜来观察样品的熔化过程,并测定其熔点。
具体操作如下:1. 准备样品:取适量的样品,将其以均匀的方式放置在熔点计的平台上。
确保样品呈现连续的片状。
可以使用特定的工具将样品压片,以使其具有符合要求的形态。
2. 启动实验:打开显微镜,并调整它的焦距,以便清楚观察样品的反射性质。
空穴化合物的电子结构计算

空穴化合物的电子结构计算电子结构计算方法是研究材料性质的重要手段之一。
其中,对于空穴化合物的电子结构计算具有特殊的意义和挑战。
空穴化合物指的是材料中的空位、夹杂物或缺陷,这些空缺位能引入价带中的空穴,从而在材料中形成正电荷。
本文将介绍一些常见的空穴化合物电子结构计算的方法和应用。
一、常见的空穴化合物电子结构计算方法1. 密度泛函理论(DFT)密度泛函理论是目前最常用的电子结构计算方法之一。
其基本思想是通过求解电子密度的泛函来获取材料的基态性质。
对于空穴化合物的计算,DFT通常利用近似的交换-相关泛函来描述电子间的相互作用,考虑到材料中的正电荷,并通过使用合适的赝势方法来处理相对论效应。
2. GW方法GW方法是一种自洽的非扰动计算方法,包括Green函数理论和空间相互作用的Hartree-Fock近似。
GW方法在描述电子相关性和激发态性质方面具有很高的精度和预测能力,因此在研究空穴化合物的电子结构计算中被广泛应用。
3. 杂化函数方法杂化函数方法结合了精确交换泛函和DFT的惯例泛函,通过引入一部分HF交换使得泛函更加适合描述材料中的空穴和正电荷。
常用的杂化函数方法包括B3LYP、PBE0等。
这些方法在描述能隙、电子态和光吸收等性质时表现出了较好的预测能力。
二、空穴化合物电子结构计算的应用1. 光电催化空穴化合物在光催化过程中起着重要作用。
通过电子结构计算,可以研究其能带结构和电子态密度,进而揭示光电催化机理和电子传输行为。
这对于设计和优化光催化材料具有指导意义。
2. 半导体材料空穴化合物在半导体材料中起到掺杂剂或夹杂物的作用,对材料的电子结构和导电性能具有重要影响。
通过电子结构计算,可以预测材料的能隙大小、载流子迁移率等关键参数,为半导体器件的设计和性能优化提供指导。
3. 电池材料在锂离子电池等储能设备中,空穴化合物的电子结构和离子扩散性能对储能性能具有重要影响。
电子结构计算可以帮助研究材料的电荷传输和离子扩散机制,为设计高性能的电池材料提供理论依据。
原油蒸馏标准试验方法 ASTM D

原油蒸馏标准试验方法ASTM D2892-98a(15层理论塔盘塔)该标准固定编号为D2892;紧跟在编号后的数字是首次采用或最后一次修订的年份。
括号内的数字是上次重新批准的年份。
小写希腊字母ε表示自上次修订或重新批准以来的编辑变化。
1.适用范围1.1本试验方法说明了稳定原油蒸馏到最终切割温度400℃AET(相当于大气压下的温度)的程序。
本试验方法采用14-18层理论塔盘的分馏塔,回流比为5:1。
指定了必须设备的性能要求。
以简单表格形式列出了一些典型的可用仪器。
本试验方法是一个效率和时间平衡的折衷方法,便于实验室之间蒸馏数据的比较。
1.2本试验方法详细讲述了生产液化气、馏份油和标准质量的渣油的程序以及测定上述馏份的质量和体积收率。
用上述数据画出质量百分数相对于温度的曲线。
蒸馏曲线列出实验条件:(15层理论塔盘塔,回流比5:1)或实沸点。
1.3除了液化石油气、非常轻的石脑油和初馏点高于400℃的馏份外,本试验方法也适用于其它石油混合馏份。
1.4本试验方法包括下列附录:1.4.1附录A1-蒸馏塔效率测定方法。
1.4.2附录A2-蒸馏塔动态藏量测定法1.4.3附录A3-蒸馏塔热损失测定法(静态)1.4.4附录A4-确定测温计位置的方法1.4.5附录A5-温度响应时间测定法1.4.6附录A6-检测器标定规程1.4.7附录A7-回流分配阀确定法1.4.8附录A8-含水原油试样脱水方法1.4.9附录A9-观测到的气温转变成大气压下的气温方法1.4.10附录A10-性能考核规程1.5以国际单位制表示的值是标准值,括号内值只用于提供信息。
1.6本标准并没有讲到所有安全注意事项,如果有,也只是与使用有关。
在使用前建立适当的安全和健康规程并确定规程的适用性是标准使用者的责任。
具体使用注意事项见第9节。
2.可引用的标准2.1 ASTM标准D941用LipkinBicapillary比重瓶测定液体密度和相对密度的方法D1217用Bingham比重瓶测定液体密度和相对密度的方法D1298用密度计测定原油和油品的密度和相对密度的方法D2427用气相色谱仪测定汽油中的C2-C5烃D4052用数字密度计测定液体的密度和相对密度D4057石油和油品采样法D4177石油和油品自动采样规程3.专有名词3.1定义3.1.1绝热-在塔的整个长度内没有大的热量增加或损失的状态3.1.1.1讨论-当蒸馏混合物时,例如原油,沿着塔向下通常回流比增加。
原油—实沸点蒸馏—常压蒸馏法

FNYSHYY00205 原油 实沸点蒸馏 常压蒸馏法F-NY-SH-YY-00205原油—实沸点蒸馏—常压蒸馏法1 范围本方法适用于原油及除液化石油气、很轻的石脑油和初馏点高于400℃以上石油馏分以外的石油混合物。
2 主题内容本方法规定了使用实沸点蒸馏仪(具有14~18块理论板,回流比为5∶1),对稳定原油进行蒸馏到相当于常压温度400℃的蒸馏方法。
3 相关技术术语3.1 绝热性 adiabaticity整个蒸馏柱没有明显的热量增加或热量损失的状态。
当蒸馏柱发生热损失时,其内回流会异常,大于柱头回流量,而当蒸馏柱加热套加热过量时,则其情况相反。
3.2 蒸出速率 boil up rate单位时间内蒸气进入蒸馏柱的量。
在给定的蒸馏柱中以cm 3/h 表示,或以每小时单位横截面的cm 3数表示(cm 3/h·cm 2)。
可参考关于正庚烷-甲基环己烷评定蒸馏柱效率部分(见附录A),测定在蒸馏柱的底部进行。
正庚烷-甲基环己烷试验的最大蒸出速率是在稳定无液泛的状态下测定的。
常规的绝热操作,蒸出速率可由馏出率乘以回流比加1估算。
3.3 原油的脱丁烷 debutanization of crude petroleum原油脱去包括丁烷及C 4以下的轻烃,保留较重的烃类。
实际上,原油脱丁烷就是:收集在冷阱中的轻烃,C 2~C 4的烃类为其存在于初始试样中的95%以上,而C 5烃类为其存在于初始试样中的5%以下。
3.4 蒸馏压力 distillation prcssure压力的测量点尽可能靠近蒸气温度测量点,一般在冷凝点的顶端。
3.5 蒸馏温度 distillation temperature在蒸馏柱头部测定的饱和蒸气温度。
此温度即是柱头温度或称气相温度。
3.6 动滞馏量 dynamic hold-up在正常操作条件下,蒸馏柱中滞馏液体的量。
对填料柱以填充的体积百分率表示,该数据能反映出各种填料间的差异。
materials_studio内聚能密度_溶度参数计算_概述说明

materials studio内聚能密度溶度参数计算概述说明1. 引言1.1 概述本文旨在对Materials Studio内聚能密度计算和溶度参数计算进行概述与说明。
Materials Studio是一种用于材料模拟和计算的软件平台,其内聚能密度计算方法可以用于研究材料的结构和稳定性,而溶度参数计算则可以帮助我们了解溶剂与溶质之间的相容性。
本文将介绍这两种计算方法的基本原理、计算方法以及应用领域。
1.2 文章结构文章共分为五个部分。
首先,引言部分将给出文章的概述,并解释文章的目的。
接下来,第二部分将详细介绍Materials Studio内聚能密度计算的基本原理、计算方法以及其应用领域。
第三部分将对溶度参数计算进行概述,包括定义与应用、计算流程以及实例分析。
然后,在第四部分中,我们将讨论并分析内聚能密度计算和溶度参数计算的结果。
最后,在第五部分中进行总结,并提出一些展望。
1.3 目的本文旨在向读者介绍Materials Studio内聚能密度计算和溶度参数计算这两种重要的材料模拟方法,并说明它们在材料研究中的应用价值。
通过了解和掌握这些计算方法,我们可以更好地理解材料的结构与稳定性,以及溶解与相容性等关键属性。
同时,本文还将提供一些实例和结果分析,以帮助读者进一步理解和应用这些计算方法。
通过阅读本文,读者将对Materials Studio内聚能密度计算和溶度参数计算有一个清晰的概念,并为进一步研究和应用提供指导和参考。
2. Materials Studio内聚能密度计算:2.1 基本原理:内聚能密度(Cohesive Energy Density, CED)是材料科学中一种重要的计算参数,用于描述物质中分子间相互作用的强度。
在Materials Studio软件中,可以通过密度泛函理论(Density Functional Theory, DFT)方法计算得到材料的内聚能密度。
2.2 计算方法:在Materials Studio中进行内聚能密度的计算主要分为以下几个步骤:- 准备模型:首先需要根据所研究的材料的晶体结构,利用Materials Studio 提供的建模工具构建起所需模型。
第六讲第一原理计算方法简介及MaterialsStudio中Castep使用案例

密度泛函理论
Hohenberg-Kohn第一定理指出体了以基态密度为变量,将体系能 量最小化之后就得到了基态能量。 根据以上两定理,将薛定谔方程转变为Kohn-Sham 方程
密度函数
电子与原子核间的库仑势
电子间的库仑势
交换关联势 (未知)
密度泛函理论 LDA和GGA近似 Kohn-Sham方程原则是精确的,但遗憾 的是交换关联势是未知的。要进行具体计 算,就必须使用近似方法求出交换关联势。 常用的近似方法有局域密度近似(Local Density Approximation)和广义梯度近 似(Generalized Gradient Approximation),在某些情况下,广义梯 度近似改善了局域密度近似的计算结果, 但它并不总是优于局域密度近似。
提示: CASTAP计算所需时间随原子数平方的增加而增加。 因此,建议是用最小的初晶胞来描述体系,可使用 Build\Symmetry\Primitive Cell菜单选项来转换成初晶
CASTEP的任务
计算设置:合适的3D模型文件一旦确定,必须选择计算类型 和相关参数,例如,对于动力学计算必须确定系综和参数, 包括温度,时间步长和步数。选择运行计算的磁盘并开始 CASTEP作业。 结果分析:计算完成后,相关的CASTEP作业的文档返回用户, 在项目面板适当位置显示。这些文档进一步处理能获得所需 的观察量如光学性质。 CASTAP中选择一项任务 1 从模块面板(Module Explorer)选择CASTAP\Calculation 2 选择设置表 3 从任务列表中选择所要求的任务
密度泛函理论 基组(basis set) 求解Kohn-Sham方程,选取适当的基组, 将波函数对其展开,将方程求解转化为线 性代数问题。 一般选用如下基组展开:
辽宁石油化工大学化工石油加工专业课程设计—常压塔的选型设计

课程设计说明书题目:常压塔的工艺设计课程名称:专业课程设计学号:班级:专业:化学工程与工艺(石油加工)姓名:日期: 2014年12月14日化学化工与环境学部目录1 前言 (3)2 设计说明书 (3)3工艺计算及说明 (5)3.1产品有关性质参数计算 (5)3.1.1 产品的实沸点蒸馏温度计算 (5)3.1.2产品的体积平均沸点计算 (8)3.1.3恩氏蒸馏10%~90%馏分的曲线斜率 (8)3.1.4立方平均沸点 (8)3.1.5中平均沸点 (8)3.1.6 d420与d15.615.6的换算 (9)3.1.7产品的分子量M、比重指数API°、特性因数K (9)3.1.8平衡汽化温度 (9)3.1.9临界温度、临界压力、焦点温度、焦点压力的求法 (10)3.2 物料衡算 (11)3.2.1切割点和产品收率的确定 (11)3.2.2汽提蒸汽用量 (12)3.2.3塔板型式和塔板数 (12)3.2.4操作压力 (12)3.2.5汽化段温度 (13)3.2.6tF的校核 (14)3.2.7塔底温度 (15)3.2.8塔顶及侧线温度的假设与回流热的分配 (15)3.2.9侧线及塔顶温度的校核 (16)3.2.10常压塔的计算草图和重柴油抽出板以下塔段热平衡图 (18)3.3全塔气、液相负荷分布 (18)4 塔的工艺参数计算 (19)设计评述 (20)附录气、液相负荷计算 (21)1 前言本次设计主要是针对年处理量685万吨原油的常压设计。
原油常压蒸馏作为原油的一次加工工艺,在原油加工总流程中占有重要作用,在炼厂具有举足轻重的地位,其运行的好坏直接影响到后续的加工过程。
其中重要的分离设备—常压塔的设计,是能否获得高收率、高质量油的关键。
近年来常减压蒸馏技术和管理经验不断创新,装置节能消耗显著,产品质量提高。
但与国外先进水平相比,仍存在较大的差距。
为了更好地提高原油的生产能力,本着投资少,能耗低,效益高的思想对原油进行常压蒸馏设计。
晶体结构的计算方法

晶体结构的计算方法晶体结构的计算方法是通过计算机模拟和各种实验技术来确定晶体的原子排列方式和结构特征。
通过计算方法可以预测晶体的力学性质、电学性质、光学性质和热学性质等。
这些预测以及对晶体结构的理解有助于设计新材料、优化材料性能和解释实验结果。
下面将介绍常见的晶体结构计算方法。
1. 密度泛函理论(Density Functional Theory,DFT)密度泛函理论是现代材料计算中最常用的方法之一、该理论基于电子结构的泛函理论,通过求解系统的电子密度函数来计算晶体的能量、结构和性质。
DFT的基本思想是将体系的总能量表示为电子的密度的函数。
通过求解Kohn-Sham方程,可以得到体系中的电荷密度分布和电子能级结构。
DFT方法可以模拟大多数晶体和材料的结构和性质,并且具有较高的计算效率。
2. 分子动力学模拟(Molecular Dynamics,MD)分子动力学模拟是一种基于牛顿运动定律的方法,它模拟原子或分子在经典力场作用下的运动轨迹,从而获得晶体的结构和动力学性质。
通过冷却、加热、压缩、拉伸等操作,可以模拟实验中无法实现的条件,并研究晶体的变形、相变、热膨胀和热导等特性。
MD方法可以提供分子尺度上晶体的变形和热运动信息,并揭示材料的物理机制。
3. 第一性原理计算方法(First-Principles Calculation)4. 蒙特卡罗模拟(Monte Carlo Simulation)蒙特卡罗模拟是一种统计模拟方法,通过随机抽样和概率统计的方法模拟系统的行为。
在晶体结构计算中,蒙特卡罗模拟可以模拟晶体的随机行为、相变和热力学等过程。
通过引入不同的物理模型和相互作用势能,可以模拟不同条件下的晶体结构和性质。
蒙特卡罗模拟方法可以有效地研究相变、精细结构和相互作用动力学等问题。
除了这些方法,还有许多其他的计算方法被应用于晶体结构计算,例如微扰理论、格林函数方法、电子迁移路径分析等。
不同的计算方法具有不同的适用范围和计算复杂度,根据具体问题的需求选择不同的方法进行晶体结构的计算和模拟。
高分子平衡熔点的统计热力学及相关分子能量参数的估算

( 6)
其次考虑链间局部平行排列相互作用 Ep 。在链单元和溶剂分子的混合过程中 ,伴随着构象的变化 , 高分子链在基态具有的平行有序堆砌也将被破坏 ,由此带来高分子间相互作用势能的变化。每一对平行 排列的键变成非平行排列时将带来的势能升高为 Ep 。体系中共有 ( r - 1) n2 个键 ,每个键周围有 q - 2 个 最近邻平行键位可以被其它键占据 ,这里同样忽略链端基的特殊性 , 并扣除了沿链前后两个连接键的贡 献 。由于格子空间中总共有 qnΠ 2 个键位可以被 ( r - 1) n2 个键所占据 ,由均匀混合假定可知 ,这样一个平
3
© 1994-2010 China Academic Journal Electronic Publishing House. All rights reserved.
・2 ・
高 分 子 通 报
2009 年 7 月
宏观体系通常具有数目庞大的微观状态 ,不可能穷尽这些微观状态来计算精确的配分函数 。广泛采用的 对策是引入平均场假定 ,就是采用一个平均化的场来表示体系中某一分子所受到的均匀分布的其它分子 的作用 ,这样就可以将微观状态能级数及其简并度分别加以简化处理计算 。平均场假定将大大简化配分 函数的计算 ,且较准确地反映了分子所处环境的微观物理图像 ,在相变研究中已有广泛的应用 ,例如 1873 年范德瓦耳斯状态方程 、 1907 年外斯分子场理论 、 1934 年布喇格 - 威廉姆斯合金有序化理论和 1937 年朗 道二类相变 “普遍” 理论等 ,都运用了平均场假定 。 Flory 和 Huggins 等于 20 世纪 40 年代藉助于格子模型和平均场假定 , 发展了高分子溶液的统计热力 学理论 ,得到了高分子溶液的混合熵 、 混合焓及混合自由能的表达式 。该表达式可以进一步推广到 两组分或多组分高分子共混物体系中 。在高分子溶液混合熵的推导过程中假设高分子链是柔顺的 ,即主 链键内旋转产生的各种微观构象具有相同的能量 。事实上 , 大多数高分子链并不是柔顺的 , 不同微观构 象之间有能量差 。1956 年 ,Flory 进一步发展了半柔顺链的格子统计热力学理论