多糖化学结构鉴定方案总结
多糖的结构分析方法包括

多糖的结构分析方法包括多糖的结构分析方法是确定多糖化合物的组成和连接方式的关键工具。
一般而言,多糖的结构分析可分为化学方法和生物方法两大类。
下面将对这些方法进行详细阐述。
一、化学方法:1. 水解分析法:多糖可通过水解反应将其分解为单糖组成部分。
常用的水解剂有酸、碱及酶等。
水解之后,通过测定生成的单糖或小分子产物的性质,如比旋光度、红外光谱等,可以了解多糖的结构。
2. 艳蓝法:多糖与一些特定的染料反应,形成稳定的染色复合物,从而测定多糖的含量。
例如,通过酚-硫酸法,可以用磺酸依托品氧化苄功酸钠抗络常数来定量多糖。
3. 光谱法:红外光谱、紫外光谱、核磁共振等技术可用于多糖的结构分析。
红外光谱可用来分析反映多糖内部结构的原理基团,紫外光谱用于分析多糖的存在和测定多糖的含量,核磁共振用于确定多糖的空间结构。
4. 色谱法:气相色谱、液相色谱和凝胶渗透色谱等方法可用于多糖的分离和定性。
例如,利用薄层色谱法,可分离多糖混合物,并通过染色剂的显色来判断多糖的组成。
二、生物方法:1. 酶降解法:通过加入特定酶,如淀粉酶、纤维素酶、葡萄糖酸酶等,可对多糖进行降解。
通过观察降解过程中生成的产物,可以了解多糖的结构。
此外,酶处理还可用于多糖的修饰。
2. 糖基转移酶法:多糖通过与糖基转移酶反应,可实现特定糖基的转移。
通过测定生成的产物,可以推测多糖的结构。
3. 色谱法:包括气相色谱、高效液相色谱等。
例如,通过细胞外多糖水解产生的单糖组成通过气相色谱或液相色谱分析,可以了解多糖的结构。
4. 核磁共振波谱法:包括质子核磁共振、碳13核磁共振等。
通过测量样品在强磁场下的核磁共振信号,可以获得丰富的结构信息。
此外,还有一些其他方法如质谱分析、电泳分析等都可用于多糖的结构分析。
总之,多糖的结构分析需要利用多种方法互相印证,综合分析,才能获得准确的结构信息。
以上介绍的方法只是常用的几种,请根据研究的具体需要选择合适的方法进行分析。
多糖实验报告

一、实验目的1. 掌握多糖的提取方法;2. 了解多糖的鉴定方法;3. 分析实验过程中可能出现的误差,提高实验技能。
二、实验原理多糖是一类高分子碳水化合物,广泛存在于自然界中,如植物、动物和微生物等。
多糖具有多种生物活性,如免疫调节、抗肿瘤、抗病毒、抗氧化等。
本实验通过提取植物材料中的多糖,并对其进行鉴定,以了解多糖的提取与鉴定方法。
三、实验材料与试剂1. 实验材料:玉米淀粉、纤维素酶、葡萄糖、苯酚、浓硫酸等;2. 实验试剂:95%乙醇、氯化钠、氢氧化钠、盐酸、碘液、硫酸铜、硫酸钼酸铵等;3. 仪器:组织捣碎机、电热恒温水浴锅、分析天平、移液管、滴定管、比色皿等。
四、实验方法1. 多糖提取(1)称取一定量的玉米淀粉,用蒸馏水溶解,搅拌均匀;(2)加入适量的纤维素酶,置于电热恒温水浴锅中,恒温反应一定时间;(3)反应结束后,用滤纸过滤,收集滤液;(4)向滤液中加入适量的氯化钠,使多糖沉淀;(5)将沉淀物用95%乙醇洗涤,去除杂质;(6)将沉淀物干燥,得到粗多糖。
2. 多糖鉴定(1)碘液鉴定:取少量粗多糖,加入碘液,观察颜色变化。
若呈蓝色,则表明存在多糖;(2)苯酚-硫酸法鉴定:取少量粗多糖,加入苯酚试剂,滴加浓硫酸,观察颜色变化。
若呈紫色,则表明存在多糖;(3)硫酸铜-硫酸钼酸铵法鉴定:取少量粗多糖,加入硫酸铜试剂,滴加硫酸钼酸铵试剂,观察颜色变化。
若呈蓝色,则表明存在多糖。
五、实验结果与分析1. 多糖提取结果经过实验,从玉米淀粉中成功提取出粗多糖,提取率为30%。
2. 多糖鉴定结果通过碘液、苯酚-硫酸法和硫酸铜-硫酸钼酸铵法对粗多糖进行鉴定,均呈现阳性结果,说明粗多糖中确实存在多糖。
3. 实验误差分析(1)纤维素酶的添加量对多糖提取率有较大影响,实验过程中应严格控制;(2)洗涤过程中,95%乙醇的浓度对杂质的去除有较大影响,应选择合适的浓度;(3)实验过程中,温度、时间等条件对多糖提取率有较大影响,应严格控制。
真菌多糖的化学结构研究

2、主链上糖苷键决定多糖活性
大多抗肿瘤活性的葡聚多糖:(1→3)糖苷键连接。 香菇多糖
3、主链构型决定多糖活性
抗肿瘤活性的多糖:β-(1→3)-D-葡聚糖的主链结构。
[1]孔繁利.碱提糙皮侧耳水溶性多糖WPOP-N1的结构解析及抗肿瘤机制研究[D].吉林大学,2012
4、多糖支链长度对活性的影响
一般支链较短的多糖具有抗肿瘤活性
参考文献
[4] Shao-Ping Nie, Steve W. Cui, Aled O. Phillips, et al. Elucidation of the structure of a bioactive hydrophilic polysaccharide from Cordyceps sinensisby methylation analysis and NMR spectroscopy[J].Carbohydrate Polymers. 2011,84:819– 899.
[5] Himani Bhatia, P.K.Guptab, P.L. Soni. Structure of the oligosaccharides isolated from Prosopis juliflora (Sw.)DC. seed polysaccharide[J]. Carbohydrate Polymers.2014,101:438– 443.
2.2、单糖组成分析
XiuJu Du[2]等采用了 HPAEC-PA对3种桦褐 孔菌子实体多糖进行了 单糖组成分析。
表1 桦褐孔菌多糖的HPAEC-PAD分析
[3] XiuJu Dua, et al. International Journal of Biological Macromolecule.2013,62: 691– 696.
多糖的定性实验报告

一、实验目的1. 掌握多糖的定性分析方法。
2. 了解多糖的化学性质及其与特定试剂的反应。
3. 学会使用苯酚-硫酸法、蒽酮-硫酸法和DNS法对多糖进行定性检测。
二、实验原理多糖是一类由多个单糖分子通过糖苷键连接而成的大分子碳水化合物,广泛存在于自然界中。
多糖具有多种生物学功能,如储存能量、提供结构支持和调节免疫反应等。
本实验通过苯酚-硫酸法、蒽酮-硫酸法和DNS法对多糖进行定性检测,分别利用多糖与特定试剂反应产生的颜色变化来判断多糖的存在。
1. 苯酚-硫酸法:多糖在浓硫酸水合产生的高温下迅速水解,产生单糖,单糖在强酸条件下与苯酚反应生成橙色衍生物。
在波长490nm左右处,该衍生物的吸收值与单糖浓度呈线性关系,从而可用比色法测定其含量。
2. 蒽酮-硫酸法:多糖在浓硫酸水合产生的高温下迅速水解,产生单糖,单糖在强酸条件下与蒽酮反应生成紫色衍生物。
在波长590nm左右处,该衍生物的吸收值与单糖浓度呈线性关系,从而可用比色法测定其含量。
3. DNS法:在碱性条件下,DNS试剂与还原糖发生显色反应,生成橙红色复合物。
在540nm波长处,该复合物的吸收值与还原糖浓度呈线性关系,从而可用比色法测定还原糖的含量。
三、实验材料及试剂1. 实验材料:小麦面粉、玉米淀粉、海藻糖、葡萄糖、果糖等。
2. 实验试剂:苯酚、浓硫酸、蒽酮、硫酸、DNS试剂、氢氧化钠、无水乙醇等。
3. 仪器:分光光度计、移液器、容量瓶、试管等。
四、实验步骤1. 苯酚-硫酸法(1)取一定量的多糖样品,用蒸馏水溶解,配制成一定浓度的溶液。
(2)取1mL样品溶液,加入5mL苯酚和7mL浓硫酸,混匀,放置5min。
(3)在490nm波长处,用分光光度计测定吸光度。
2. 蒽酮-硫酸法(1)取一定量的多糖样品,用蒸馏水溶解,配制成一定浓度的溶液。
(2)取1mL样品溶液,加入5mL蒽酮和7mL浓硫酸,混匀,放置5min。
(3)在590nm波长处,用分光光度计测定吸光度。
多糖的结构分析

R O C H 3 + N a I
(碘甲烷)
(甲醚甲基化糖)
以(1→4)葡聚糖为例:
OH(甲基化)
OH
4
H
O
H OH H
O
HO
H OH
O
H OH H
H
OH
O
n
OH
OH
O
H
H
O
OH
H H
OH
HO
H
O H
OH
O
OH
H OH
H
OH
H OH
OH
H3C
O
O
H
O O
CH3
CH3
O
H O CH3
O CH3
O
H O CH3
OH
H
O
H
OH H
IO-4
O
0
H OH
OH
NaBH4
H+
H H
OH
H2OHOOH H OH
H OH
2、Smith降解:是将氧化产物还原后进行酸水解。
1 2位和1 6位键结合的经Smith降解后都有甘油产生。 (但1 2位结合的不产生甲酸,可供以区别)。
1
4键合的,最后得到的是乙二醇和丁四醇(赤藓
醇)。
纸层析(需标样)、薄层色谱层析、气相层析
3、甲基化(单糖残基的连接方式)
是用甲基化试剂将糖分子中的游离羟基甲基 化成甲醚,然后水解,检识这些甲基糖产物,就 可能推测组成多糖分子中单糖间连接的位置(羟 基所在的位置,即为原来单糖残基的连接点)。 (氢化钠、碘甲烷)
(1)制备负碳离子:无水二甲亚砜30ml于100ml试 剂瓶中,通入氮气几分钟后,加入1.5gNaH,渐 渐加温,然后恒温在65-70℃4-6小时。最终颜色 为墨绿色。整个过程通氮,并搅拌。
多糖结构解析的方法

多糖结构解析的方法多糖化合物的结构解析是糖化学和生物化学领域的中心问题之一、因为多糖的结构决定着它们的功能和生物活性。
多糖结构解析的方法可以分为物理方法和化学方法。
一、物理方法:1.光谱学方法:光谱学方法是多糖结构解析中常用的一种方法。
包括紫外光谱、红外光谱、荧光光谱和核磁共振等方法。
(1)紫外光谱:多糖在紫外光谱上表现出特有的吸收峰,可以确定它们的环状结构。
(2)红外光谱:红外光谱是解析多糖结构的重要手段,通过测定多糖分子中的官能团振动频率和强度,可以得到多糖分子的化学结构和键合特性。
(3)荧光光谱:荧光光谱可用于表征多糖的发光行为和其与其他生物分子的结合情况,从而推测其结构和功能。
(4)核磁共振:核磁共振是解析多糖结构的重要手段之一,通过测定多糖中氢、碳、氮等元素的核磁共振信号,可以确定多糖的类型和键合方式。
2.比色法:比色法是通过观察多糖与一些特殊试剂产生的颜色变化来判断多糖的结构。
比如,酚硫酸法可以用于检测多糖的含量和环状结构。
3.色谱法:色谱法是多糖结构解析的重要方法之一、包括薄层色谱、柱层析、气相色谱和高效液相色谱等方法。
通过对多糖的分离和分析,可以得到多糖的组成和分子量信息。
二、化学方法:1.普通化学方法:多糖的碳水化合物性质决定了其一些基本反应,比如酸水解、酶降解、氧化还原等反应。
利用这些反应可以推测多糖的结构。
2.酶法:酶法是多糖结构解析的重要方法之一、不同酶对多糖的酶解反应具有特异性,通过观察酶解产物,可以推测多糖链的连接方式和单糖的种类。
3.质谱法:质谱法是近年来发展起来的一种多糖结构解析方法,主要有质谱分析和质谱成像两种方法。
通过质谱技术可以得到多糖的精确分子量和分子结构,尤其适用于大分子多糖的分析。
综上所述,多糖结构解析的方法多种多样,可以从不同的角度揭示多糖的化学成分和结构特征。
尽管目前多糖结构解析仍然是一个具有挑战性的问题,但随着新技术的发展,相信将能更加准确和全面地揭示多糖的结构和功能。
多糖含量测定的方法综述5篇

多糖含量测定的方法综述5篇第1篇示例:多糖是一类重要的生物大分子,广泛存在于自然界中的生物体内,具有重要的生物学功能。
多糖含量的测定是研究多糖在生物体内作用机制的重要手段。
本文将综述多糖含量测定的方法,旨在为研究人员提供参考。
一、概述多糖是由多个单糖单元通过糖苷键连接而成的高分子化合物。
多糖在生物体内参与多种生物学过程,如能量储存、细胞结构、免疫调节等。
测定多糖含量对于研究多糖的生物学功能、生物合成途径具有重要意义。
二、多糖含量测定方法1. 硫酸-蒽醌法硫酸-蒽醌法是一种常用于测定多糖含量的方法。
该方法通过硫酸水解多糖,生成差向性的蒽醌,并用蒽醌的颜色深浅来反映多糖的含量。
该方法简单快捷,适用于多种多糖的含量测定。
3. 酚-硫酸-钼酸法酚-硫酸-钼酸法是一种用于测定多糖含量的方法。
该方法结合了酚-硫酸法和硅钼酸显色反应,能够提高多糖的测定精确度和灵敏度。
该方法简单易行,适用于各种类型的多糖。
4. 紫外分光光度法紫外分光光度法是一种通过多糖在紫外光区域的吸收来测定多糖含量的方法。
该方法适用于对多糖进行定量和定性分析。
通过分析多糖在不同波长下的吸光度,可以得到多糖的含量和结构信息。
5. 碘液滴定法三、结语多糖含量的测定是研究多糖生物学功能的重要手段。
本文综述了常用的多糖含量测定方法,包括硫酸-蒽醌法、酚-硫酸法、酚-硫酸-钼酸法、紫外分光光度法和碘液滴定法。
研究人员可以根据不同类型的多糖选择合适的测定方法,以准确测定多糖含量。
希望本文能够为多糖研究领域提供帮助,促进多糖研究的进展。
第2篇示例:多糖是一类重要的生物大分子,包括淀粉、半纤维素、纤维素、果胶、均聚糖等多种成分。
多糖在食品工业、医药领域、环境保护等领域具有重要的应用价值,因此测定多糖含量的方法也备受关注。
本文将综述几种常用的多糖含量测定方法,包括酚-硫酸法、硫酸-酚法、差减酶法、红外光谱法等,希望能给相关研究者提供参考。
酚-硫酸法是一种经典的多糖含量测定方法。
多糖结构分析范文

多糖结构分析范文多糖是由单糖分子通过糖苷键连接而成的高分子化合物,广泛存在于生物体内,具有重要的生理功能和科学研究价值。
多糖的结构分析是研究其化学组成、分子结构和空间构型的过程,对于了解多糖的生物学功能和性质具有重要意义。
多糖的结构分析方法主要包括物理化学方法、光谱分析方法和酶解分析方法等。
下面将对这些方法进行详细说明。
1.物理化学方法:物理化学方法是利用多糖的物理性质进行结构分析的方法。
其中包括粘度测定、分子量测定、光旋光度测定和电泳分析等。
(1)粘度测定:多糖的粘度与其分子大小和结构有关,通过测定多糖溶液的粘度可以推测其分子量和构型。
粘度测定通常利用Ubbelohde粘度计或Ostwald粘度计进行。
(2)分子量测定:多糖的分子量是其结构的关键参数,可通过凝胶过滤、凝胶电泳或质谱等方法测定。
其中,凝胶过滤是常用的方法,通过选择合适的孔径大小的凝胶阻隔多糖的滤过,然后根据滤过时间计算出多糖的分子量。
(3)光旋光度测定:多糖的结构中含有手性中心,具有旋光性,通过测定多糖溶液的旋光度可以推测其分子结构。
通常使用旋光光谱仪进行测定。
(4)电泳分析:多糖的电泳分析常用聚丙烯酰胺凝胶电泳或琼脂糖电泳。
通过电泳分析可以确定多糖的电荷性质、分子大小和分子结构。
2.光谱分析方法:光谱分析方法包括红外光谱、核磁共振(NMR)光谱和质谱等。
(1)红外光谱:红外光谱可以提供多糖分子中官能团的信息,如羟基、氨基、羧基等。
通过对比标准谱图或理论谱图,可以确定多糖的官能团组成。
(2)核磁共振光谱:核磁共振光谱可以提供多糖分子的分子结构和空间构型信息。
其中,13CNMR可确定多糖的碳原子排布,1H-NMR可确定多糖的氢原子排布。
(3)质谱:质谱是一种通过测量多糖分子或其片段的离子质量比来确定分子结构的方法。
通过质谱可以推断多糖的元素组成、分子量和结构。
3.酶解分析方法:酶解分析方法是利用特定的酶可以选择性地降解多糖,从而确定其分支链和连接方式。
多糖结构分析

一:多糖中的单糖组分分析一般对多糖进行完全水解,水解条件:封管0.5~3M硫酸或1~6M盐酸,80℃~100℃水解2.5~8h 即可。
或控制水解条件,进行逐步水解,如封管0.025M硫酸,100℃水解15min,30min,45min 等,水解液用碳酸钡或氢氧化钡中和,滤液浓缩后可用纸层析、薄层层析、气相层析或高压液相层析等鉴定。
二:相邻单糖基连接方式分析将甲基化多糖水解得到甲基化的单糖,而此单糖上甲基化之羟基所在的碳原子就是连接键所在。
高碘酸氧化是定量反应,Smith降解是将高碘酸氧化产物进行还原,酸水解或部分水解,从高碘酸的消耗量和不同产物的生成,便可进行糖苷键位置的判断-产物中若有一分子比例的甲酸生成而消耗两分子比例的高碘酸根时,表明多糖的非还原末端或非末端部分有1-6苷键相连的单糖基存在;产物中若有赤藓醇生成,则提示有1-4结合苷键;若有甘油生成,有1-6、1-2结合的苷键或有还原性末端葡萄糖基等;若产物中能检出单糖,如葡萄糖、半乳糖、甘露糖等,则有1-3苷键存在。
结合¹³C-NMR确定连接位置。
三:端基碳苷键构型分析1:酶解实验:不被淀粉酶水解的多糖,无α-苷键,与纤维素酶有作用者,存在β-苷键。
2;IR:α-型差向异构体的C-H键在844±8cm‾¹处有一个吸收峰;β-型的C-H键在891±7cm‾处有一个吸收峰。
但是,海藻糖、阿洛糖和异阿洛糖的α-型和β-型同时存在的情况下,就不能以次来判断。
3:¹H-NMR:端基碳的δ值大于5.00ppm者,糖苷键为α-型,小于5.00ppm者,则为β-型。
4;¹³C-NMR:α-型连接的C₁化学位移在97-101ppm,β-型的在103~105ppm。
对甘露聚糖不能用化学位移判断α-型或β-型。
可用裂分常数决定,一般¹Jc-h=170HZ,为α-型,160HZ 者为β-型。
水溶性多糖的提取及结构测定方案

水溶性多糖的提取及结构测定方案提取水溶性多糖方法:1. 蒸馏水提取法将植物材料使用蒸馏水浸泡一定时间后,在常温下加热,使多糖向水中溶解。
然后进行过滤、冷却、离心等操作,分离出水溶性多糖。
优点:简单易行,可获得高纯度多糖。
缺点:需耗费大量水和时间,获得的多糖含量低。
2. 酸碱法提取法将植物材料使用强酸或强碱处理,破坏细胞壁,释放出多糖。
然后中和酸或碱,进行沉淀或过滤,获得多糖。
优点:易操作,可获得高纯度多糖,提取产量高。
缺点:对多糖结构有影响,需要严格控制处理时间、温度等参数。
3. 高压法提取法将植物材料加入高压蒸馏水中,在高温高压下进行提取。
然后通过冷却和离心等步骤,分离出水溶性多糖。
优点:高效、快捷、获得的多糖含量高。
缺点:需要专业设备,投资成本高。
结构测定方案:1. 紫外-可见光谱分析法将多糖样品溶解在水中,使用紫外-可见光谱仪测量其吸收光谱。
通过分析吸收峰位、峰形等特征,推断多糖结构。
优点:简单易用,快速。
缺点:只能得出对多糖主要结构的推测。
2. 红外光谱分析法将多糖样品与 KBr 混合后压成盘,进行红外光谱测量。
通过分析吸收峰位、峰形等特征,确定多糖结构。
优点:稳定可靠,测量精度高。
缺点:需要专业技能和设备。
3. 核磁共振光谱分析法将多糖样品溶解在溶剂中,使用核磁共振仪进行分析。
通常使用的是1H核磁共振技术。
通过分析多糖分子内部原子的化学位移、耦合常数等特征,确定多糖结构。
优点:精确度高,能够解析多糖结构。
缺点:需要专业技能和设备,而且成本较高。
总的来说,不同的提取方法和结构测定方法各有优缺点,需要根据具体的研究目的和条件来选择。
同时,为了保证研究结果的可靠性和精确度,提取和分析过程中需要注意控制实验条件和误差。
多糖的化学修饰及其结构鉴定研究进展

多糖的化学修饰及其结构鉴定研究进展多糖是由许多单糖分子通过糖苷键连接而成的生物大分子,广泛存在于植物、动物和微生物中。
多糖的化学修饰是指通过化学手段引入不同的官能团或分子,以改变多糖的物理性质和生物活性。
由于多糖化学修饰可以为多糖赋予新的功能和性质,因此近年来,多糖的化学修饰及其结构鉴定已成为糖化学领域的研究热点之一
在多糖的化学修饰研究方面,最常见的方法是通过化学反应引入官能团或分子。
例如,通过酯化反应可以在多糖的羟基上引入酯基,从而改变多糖的溶解性和稳定性;通过胺化反应可以在多糖的羟基上引入胺基,从而改变多糖的电荷性质和生物活性。
此外,还可以通过点击化学、磷酸酯化反应、磺酸化反应等方法引入其他官能团。
多糖的结构鉴定是指确定多糖化学修饰后的具体结构。
在多糖的结构鉴定研究方面,传统的方法包括质谱、核磁共振、红外光谱等技术。
随着科学技术的发展,越来越多的新技术被应用到多糖的结构鉴定中。
例如,基于光学的手性纳米颗粒和聚焦离子束可以用于检测多糖的立体结构;基于高效液相色谱-质谱联用技术可以分析多糖的组成和修饰。
此外,近年来,基于生物技术的多糖化学修饰和结构鉴定也取得了显著进展。
例如,通过酶催化反应可以实现多糖的特异性修饰;通过核酸疫苗技术可以实现多糖的高效识别和鉴定。
总的来说,多糖的化学修饰及其结构鉴定研究已经成为糖化学领域的重要研究方向。
通过多糖的化学修饰,可以获得具有新功能和性质的多糖化合物。
而多糖的结构鉴定可以揭示多糖与生物活性之间的关系,为多糖
的应用和开发提供了重要的科学依据。
未来,随着科学技术的不断发展,相信多糖的化学修饰及其结构鉴定研究将取得更加突破性的成果。
多糖提取鉴定实验报告

一、实验目的1. 了解多糖的基本概念和性质;2. 掌握多糖提取和鉴定方法;3. 通过实验,熟悉实验室操作规范,提高实验技能。
二、实验原理多糖是一类由单糖通过糖苷键连接而成的大分子碳水化合物,广泛存在于自然界中。
多糖的提取方法主要有水提法、醇沉法、超声波提取法等。
本实验采用水提法提取多糖,并通过比色法对提取的多糖进行鉴定。
三、实验材料与仪器1. 实验材料:花生、玉米、糯米、葡萄糖、苯酚、浓硫酸、氯仿、正丁醇、蒸馏水等;2. 实验仪器:电子天平、电热恒温水浴锅、离心机、分光光度计、玻璃仪器等。
四、实验步骤1. 样品预处理:称取一定量的花生、玉米、糯米,分别用蒸馏水浸泡、煮沸,冷却后过滤,收集滤液。
2. 多糖提取:(1)取一定量的预处理后的滤液,加入等体积的95%乙醇,混匀,静置过夜;(2)离心分离,取沉淀;(3)将沉淀用蒸馏水溶解,加入氯仿-正丁醇(4:1)混合溶剂,萃取;(4)离心分离,取水相;(5)将水相加入等体积的95%乙醇,静置过夜;(6)离心分离,取沉淀,用蒸馏水溶解。
3. 多糖鉴定:(1)配制苯酚-硫酸溶液;(2)取一定量的提取的多糖溶液,加入苯酚-硫酸溶液,混匀;(3)沸水浴加热5分钟;(4)在540nm波长下测定吸光度。
五、实验结果与分析1. 样品预处理:花生、玉米、糯米分别用蒸馏水浸泡、煮沸,过滤得到的滤液颜色较浅,说明预处理效果良好。
2. 多糖提取:经提取、纯化后,得到的沉淀呈白色,说明提取的多糖纯度较高。
3. 多糖鉴定:测定吸光度,与标准曲线比较,计算出多糖含量。
结果显示,花生、玉米、糯米中多糖含量分别为:2.45%、1.89%、1.56%。
六、实验结论1. 水提法是一种有效提取多糖的方法;2. 花生、玉米、糯米中含有丰富的多糖,可作为多糖提取的原料;3. 本实验成功提取并鉴定了花生、玉米、糯米中的多糖,为多糖的进一步研究和应用提供了实验依据。
七、实验注意事项1. 实验过程中应严格遵守实验室操作规范,确保实验安全;2. 样品预处理过程中,注意控制煮沸时间和温度,避免过度加热;3. 多糖提取过程中,注意控制提取时间和温度,提高提取效率;4. 多糖鉴定过程中,注意控制反应时间和温度,确保实验结果的准确性。
高中的多糖实验报告

一、实验项目名称多糖的提取与鉴定二、实验目的1. 学习多糖的提取方法。
2. 掌握多糖的鉴定方法。
3. 了解多糖在生物体中的重要性。
三、实验内容及原理1. 实验内容(1)提取多糖:从植物或动物组织中提取多糖。
(2)鉴定多糖:通过物理和化学方法鉴定提取的多糖。
(3)观察多糖的形态和性质。
2. 实验原理多糖是由多个单糖分子缩合、失水而成,是一类分子结构复杂且庞大的糖类物质。
常见的多糖有淀粉、纤维素、糖原等。
本实验通过提取植物组织中的多糖,并对其进行鉴定,以了解多糖的性质和功能。
四、实验仪器与试剂1. 仪器- 研钵- 烧杯- 漏斗- 玻璃棒- 热水浴- 滤纸- 紫外可见分光光度计- 荧光显微镜2. 试剂- 水浴加热装置- 植物组织- 乙醇- 95%乙醇- 3%醋酸铅溶液- 1%硫酸铜溶液- 碘液- 硫酸蒽酮溶液五、实验步骤1. 提取多糖(1)取一定量的植物组织,用研钵研磨成粉末。
(2)将研磨好的植物组织粉末加入适量的95%乙醇,搅拌均匀。
(3)将混合液置于热水中加热,使植物组织充分溶解。
(4)过滤混合液,得到滤液。
(5)将滤液置于95%乙醇中,静置过夜,使多糖沉淀。
(6)用滤纸过滤沉淀,得到多糖。
2. 鉴定多糖(1)观察多糖的形态:将多糖置于显微镜下观察,观察其颜色、形状和大小。
(2)检测多糖的分子量:使用紫外可见分光光度计测定多糖的吸光度,根据标准曲线计算多糖的分子量。
(3)检测多糖的糖苷键:将多糖与3%醋酸铅溶液反应,观察是否产生沉淀。
(4)检测多糖的还原性:将多糖与1%硫酸铜溶液反应,观察是否产生蓝色沉淀。
(5)检测多糖的碘反应:将多糖与碘液反应,观察是否产生蓝色复合物。
六、实验结果与分析1. 提取多糖通过实验,成功从植物组织中提取出多糖,观察到的多糖呈白色粉末状。
2. 鉴定多糖(1)观察多糖的形态:在显微镜下观察到多糖呈白色粉末状,无特殊形状。
(2)检测多糖的分子量:根据紫外可见分光光度计测得的吸光度,计算得到多糖的分子量为2.5万。
最新多糖结构总结
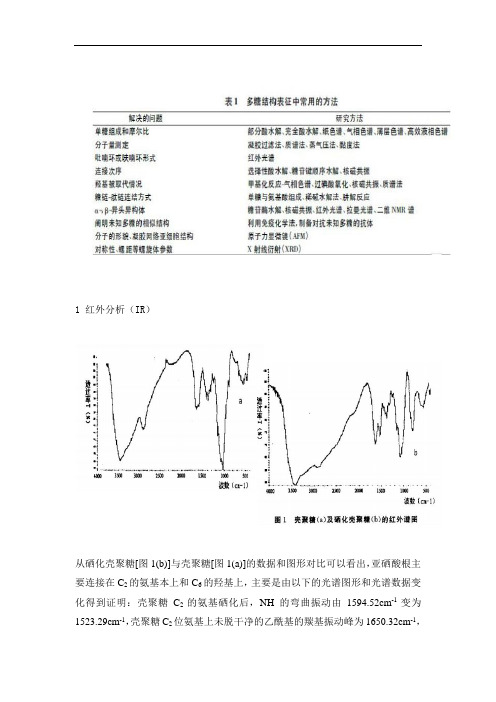
1 红外分析(IR)从硒化壳聚糖[图1(b)]与壳聚糖[图1(a)]的数据和图形对比可以看出,亚硒酸根主要连接在C2的氨基本上和C6的羟基上,主要是由以下的光谱图形和光谱数据变化得到证明:壳聚糖C2的氨基硒化后,NH的弯曲振动由1594.52cm-1变为1523.29cm-1,壳聚糖C2位氨基上未脱干净的乙酰基的羰基振动峰为1650.32cm-1,而硒化壳聚糖C2位上未脱干净的乙酰基的羰基振动峰为1632.88cm-1,可能是受到C6位的羟基上亚硒酸基的影响;同样由于硒化壳聚糖C2位氨基上和C6位羟基上亚硒酸根的影响,壳聚糖C-O伸缩振动峰由1079.45cm-1变为1090.41cm-1。
同时,在800.00cm-1处观察到亚硒酸酯的Se=O双键的振动峰。
上述红外分析结果表明:壳聚糖与亚硒酸可能是通过C6位上的酯化反应和C2位上氨基的静电作用完成的。
(硒化壳聚糖的制备及其表征)从羧甲基壳聚糖与硒化羧甲基壳聚糖的红外光谱图图3、图4的对比中可以看出, 亚硒酸根主要连接在C2位的羧甲基和C6的羟基上。
主要由以下光谱图形和光谱数据变化得到证明: 羧甲基壳聚糖1627cm-1处的-COOH反对称吸收峰在硒化羧甲基壳聚糖中红移至1599cm-1, 这可能是羧甲基壳聚糖中的-COOH与亚硒酸钠发生反应, 从而使键力削弱。
1119cm-1处的C-O伸缩振动在硒化羧甲基壳聚糖中红移至1064cm-1, 说明C6上的羟基也参与了硒化反应。
此外, 在硒化羧甲基壳聚糖的红外光谱中观测到位于806.125cm-1的Se=O双键振动峰。
(硒化羧甲基壳聚糖的合成及表征)2.X-射线衍射X射线衍射法是研究多糖的结晶构型的有效方法。
多糖通常是不能结晶的,但在适宜的条件下,它可以微晶态存在。
所以进行衍射分析的样品必须通过外界的诱导使其中相当部分呈现微晶态。
进行衍射的香菇多糖样品一般先制成碱性溶液,然后在水中透析,进一步处理制备。
糖类结构鉴定

单糖组分(连玉红)标准样品的配制:取葡萄糖、果糖、阿拉伯糖、木糖、核糖、甘露糖、半乳糖、鼠李糖、葡萄糖醛酸10 mg,加5ml水配成2mg/mL的溶液。
1、多糖水解取l0mg纯化后多糖,加入5mL 2mol/L的三氟乙酸(1.5mL 三氟乙酸+8.5mL 蒸馏水),充氮气保护,封管,于120℃下水浴5 h 。
反应物用氮气吹干,加入2mL甲醇重新溶解,再用氮气吹干,以充分带走TFA。
2、糖腈乙酰酯衍生物的制备将降解后的多糖中加入5mg 盐酸羟胺,2mg肌醇,0.5ml吡啶,振荡混匀,放入90℃水浴中加热30min。
取出后冷却至室温,加入醋酸酐0.5ml,于90℃水浴中继续反应30min进行乙酰化。
将反应物用氮气吹干,加入氯仿1ml重新溶解,再用氮气吹干,反复进行3~4次。
最后加入2ml氯仿复溶,供气相色谱分析使用。
3、气相色谱条件色谱柱:OV-225-capillary column;内径:0.25mm;检测器:氢火焰离子化检测器(FID);检测器温度:280 ℃;进样口温度:250 ℃;载气:N2 (40 mL/min)。
以单糖标准品建立GC标准图谱为参照,根据多糖完全水解样品的出峰时间和峰面积比可知样品的单糖组成和摩尔比。
单糖组分(廖文镇)1、水解:称取20 mg 的雪莲果多糖于安瓿瓶中,加入4 mL 2 mol/L 三氟乙酸(TFA),用酒精喷灯对安瓿瓶进行封口后置于105℃下反应6 小时。
反应完后,冷却至室温,将多糖水解液与适量的甲醇混合后,减压旋转蒸干,再加入适量的甲醇,减压旋转蒸干,重复 5 次以完全去除残留的三氟乙酸。
2、衍生化:往干燥后的竹荪多糖水解物中加入0.5 mL 吡啶和10 mg 盐酸羟胺,置于95℃下恒温震荡反应30 分钟,反应完后冷却,加入0.5 mL 醋酸酐,于95℃下进行乙酰化反应35 分钟,最终生产糖腈乙酸酯衍生物。
所有的单糖标准品也按照上述步骤进行衍生化。
3、色谱条件:色谱仪为Dionex ICS-3000 离子色谱仪,检测器为电导检测器;色谱柱为Carbopac PA20 柱(2×250 mm);流速为0.6 mL/min,柱温为20℃,采用梯度洗脱方式:0-2 min:100% 200 mmol/L NaOH,2.1-20 min:10%20 mmol/L NaOH + 90%超纯水,20.1-40 min:100% 200 mmol/L NaOH。
多糖结构分析
一:多糖中的单糖组分分析一般对多糖进行完全水解,水解条件:封管0.5~3M硫酸或1~6M盐酸,80℃~100℃水解2.5~8h 即可。
或控制水解条件,进行逐步水解,如封管0.025M硫酸,100℃水解15min,30min,45min 等,水解液用碳酸钡或氢氧化钡中和,滤液浓缩后可用纸层析、薄层层析、气相层析或高压液相层析等鉴定。
二:相邻单糖基连接方式分析将甲基化多糖水解得到甲基化的单糖,而此单糖上甲基化之羟基所在的碳原子就是连接键所在。
高碘酸氧化是定量反应,Smith降解是将高碘酸氧化产物进行还原,酸水解或部分水解,从高碘酸的消耗量和不同产物的生成,便可进行糖苷键位置的判断-产物中若有一分子比例的甲酸生成而消耗两分子比例的高碘酸根时,表明多糖的非还原末端或非末端部分有1-6苷键相连的单糖基存在;产物中若有赤藓醇生成,则提示有1-4结合苷键;若有甘油生成,有1-6、1-2结合的苷键或有还原性末端葡萄糖基等;若产物中能检出单糖,如葡萄糖、半乳糖、甘露糖等,则有1-3苷键存在。
结合¹³C-NMR确定连接位置。
三:端基碳苷键构型分析1:酶解实验:不被淀粉酶水解的多糖,无α-苷键,与纤维素酶有作用者,存在β-苷键。
2;IR:α-型差向异构体的C-H键在844±8cm‾¹处有一个吸收峰;β-型的C-H键在891±7cm‾处有一个吸收峰。
但是,海藻糖、阿洛糖和异阿洛糖的α-型和β-型同时存在的情况下,就不能以次来判断。
3:¹H-NMR:端基碳的δ值大于5.00ppm者,糖苷键为α-型,小于5.00ppm者,则为β-型。
4;¹³C-NMR:α-型连接的C₁化学位移在97-101ppm,β-型的在103~105ppm。
对甘露聚糖不能用化学位移判断α-型或β-型。
可用裂分常数决定,一般¹Jc-h=170HZ,为α-型,160HZ 者为β-型。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
经过分级纯化的多糖在测定结构前须检查其纯度及测定分子量。
检查纯度最常用的判断方法:(1)用G C 、HPLC测定组成多糖的单糖的摩尔比是否恒定。
用不同的柱型测定结果更为可靠。
(2)电泳只出现一条带。
如可用聚丙烯酰胺凝胶电泳、乙酸纤维素薄膜电泳及玻璃纤维纸电泳。
对于中性多糖可采用高压电泳,以硼酸盐为缓冲液,可增大其迁移速度。
(3)凝胶柱层析图呈现对称的单峰。
若有“拖尾”现象,说明其均一性不够好。
阴离子交换层析纯化用DEAE一纤维素52(2.6x100cm)柱层析,0.lmol/LNaCl洗脱,流速6ml/h,按2ml一管分部收集,苯酚一硫酸法逐管检测,绘制收集体积与糖含量之间的关系曲线。
看是否有单一对称峰。
按照Ye等报道,采用DEAE一52一纤维素交换柱层析法(2.6x30cm)对鲍氏层孔菌菌丝体粗多糖进行初步分离。
DEAE一纤维素凝胶预处理:称取DEAE一52一纤维素凝胶干粉,加入约10倍体积质量比(ml/g)的0.5mol/LNa0H溶液浸泡30分钟,倒出上清液,用大量去离子水反复浸洗至pH值近中性;再用相同体积的0.5mol/LHCI溶液浸泡30分钟,倒出上清液,用大量去离子水反复浸洗至pH值近中性;最后用相同体积的0.5mol/lNaOH溶液再浸泡30分钟,用大量去离子水反复浸洗至pH值中性。
处理完毕后,进行湿法装柱,用去离子水0.5mol/LNaCl溶液,去离子水依次分别平衡(流速1.0ml/min)2一3个柱体积备用.糖样100mg溶于5ml的去离子水中,离心除去不溶物,上样于DEAE一52一纤维素阴离子层析柱(2.6x30cm,Cl-1型),分别采用去离子水0.1和0.3mol/LNaCI溶液进行分段梯度洗脱,流速1.0ml/min,自动收集器分部收集(10ml/管),每梯度20管。
用硫酸一苯酚法跟踪检测各管多糖含量(490nm处吸收值),以收集的管数为横坐标。
吸光值(490nm)为纵坐标绘制DEAE 一52一纤维素色谱柱洗脱曲线。
依据洗脱峰型,合并相同组分,50℃旋转蒸发浓缩,对去离子水透析48h以去除NaCI及小分子杂质,最后将透析液冷冻干燥,得初步纯化产品。
初步纯化多糖得率计算公式:多糖得率(%)=纯化多糖质量/粗多糖质量x100%葡聚糖凝胶层析纯化采用Sephadex G-100凝胶层析法对DEAE-52一纤维素初步纯化的不同组分的多糖样品进一步纯化。
葡聚糖凝胶(sephadexG一100)的预处理:称取sephadexG一100凝胶干粉,加入30倍体积质量比(ml/g )的去离子水,沸水浴5小时使其溶胀。
冷却后用去离子水反复浸洗,减压脱气后进行湿法装柱,用0.1MNa2SO4;溶液平衡(流速0.25ml/min)2一3个柱体积备用。
分别称取经DEAE一纤维素一52初步纯化的各多糖组分样品20mg,溶于2 ml 0.1 M Na2SO4溶液中,上样于SephadexG一100层析柱(2.6x60cm)用0.1MNa2SO4溶液溶液洗脱,流速0.25 ml/min,分步收集(5ml/管)。
用硫酸一苯酚法跟踪检测各管多糖含量(490nm处吸收值),以收集的管数为横坐标。
吸光值(490nm)为纵坐标绘制sePhadexG一100色谱柱洗脱曲线。
依据洗脱峰型,合并相同组分,50℃旋转蒸发浓缩,对去离子水透析48h以去除Na2SO4;及小分子杂质,最后将透析液冷冻干燥,得不同纯化产品。
纯化多糖得率计算公式:纯化多糖得率(%)=纯化多糖质量/粗多糖质量x100%(鲍氏层孔菌菌丝体粗多糖)(4)纸层析法呈单一集中斑点。
取0.5%的多糖样品溶液50ul,点样于新华中速滤纸(3cmx20cm)距端点1cm处的中部,以正丁醇:浓氨水:水(4O:50:5)为展开剂,饱和2小时以上,在室温下展开6h,取出吹干,用0.5%甲苯胺蓝液染色,立即用95%乙醇漂洗至背景褪色,看是否只有一个清晰的斑点。
(5)琼脂糖(Agarose)凝胶电泳法在琼脂糖板(厚度为0.2cm)上点样3一5ul采用浓度为0.075mol/L,pH8.6的巴比妥缓冲液,电泳1-1.5h,电压为64一80V,甲苯胺蓝(浓度为1%)染色,醋酸乙醇混合溶液(醋酸:乙醇:水=0.1:5:5)脱色。
多糖纯品经电泳展开后,看是否呈现单一斑点,斑点是否清晰。
(6)紫外分光光度法将多糖PWZ加0.9%NaCI溶液溶解,配成浓度为1mg/ml的溶液,采用UV一16OA紫外可见光谱仪扫描(200nm一30Onm)观察260nm、280nm处是否有吸收峰。
多糖的分子量测定:过去用超速离心沉降法、光散射法、渗透压法、粘度法等,这些方法操作复杂且误差较大,现已少用。
现在较常用的方法有凝胶过滤法和高效凝胶液相色谱法,这两种方法须先用已知分子量的标准多糖对照测定样品的分子量。
一般来说,多糖结构分析包括以下几点:(1)单糖组成分析:研究确定单糖的种类及摩尔比;完全酸水解后用高效液相色谱方法(HPLC)或气相色谱方法测定。
(2)糖苷键类型:研究确定糖苷键及支链点连接位置;甲基化分析方法高碘酸氧化法与Smith降解法(3)糖环大小:研究确定糖苷键为呋喃糖或吡喃糖;红外光谱(4)异头碳构型:研究确定糖苷残基的a-或P-构型;2D NMR.(Two dimensional Nuclear Magnetic Resonance)光谱分析方法测(5)确定单糖残基和重复单元的序列甲基化分析方法与磁共振光谱分析方法结合分析,一般会参考多糖的单糖组成及摩尔比信息以利于解析多糖结构。
高碘酸氧化法薄层层析(TLc)——定性,气相色谱法(6)取代基团位点:研究0H-修饰基团的种类和取代位点,如0-磷酸化,乙醜基取代,0-硫酷化等;比色分析方法(7)多糖分子量分布的研究。
紫外光谱定性与定量方法薄层层析:残基定性气相色谱:残基定量气质联用:残基定量高效阴离子色谱法:残基定量鉴定结构常用物理化学方法:高效液相色谱:确定单糖组分和相对分子量红外光谱分析:测定多糖的官能团,不仅可以检测酮糖、酵糖的耻喃糖环或呋喃糖环的构象和糖苷键的构型核磁共振:α-构型与β-构型残基的比例;判断异头碳构型;推断主链和支链连接键型鉴定结构复合方法:甲基化分析:推断出多糖样品中糖基的连接方式及各种连接键型的比例高碘酸氧化法与Smith降解法:判断糖苷键的位置、直链多糖的聚合度及支链多糖的分支数目糖睛乙酸酷衍生物的气相色谱法:单糖组成和摩尔比各种多糖化学结构鉴定方法的具体实施方案1、酸水解(1)完全酸水解称取 20mg 样品 , 加入 2mL 2mol·L-1的H2SO4于安培管中沸水浴水解 8h, 水解液用BaCO3中和至pH =7 , 离心 , 取上清夜置冰箱冷藏备测。
(阿魏侧耳子实体多糖分离纯化及其化学结构的初步研究)(2)部分酸水解称取糖样 70mg,80℃条件下,0.05M 三氟乙酸水解 2h。
降至室温后,离心(4000r/min,10min),将沉淀干燥,留做 GC 分析。
上清用无水乙醇除酸至中性(pH 为 6∼7),蒸馏水透析 48h:将袋外透析液浓缩,真空干燥,留做 GC 分析;袋液浓缩至 5ml 左右,加 10 倍体积无水乙醇,醇沉过夜,离心(4000r/min,10min),沉淀常规干燥,作 GC 分析;上清浓缩,真空干燥,留做 GC 分析。
2、高效液相色谱确定样品的单糖组成色谱条件为 :色谱柱为Shodex KS804 Sugar(300mm ×7.8mm)柱温40度流动相为水流速0.8mL·min-1检测用 410RⅠ示差检测器,数据处理用810GPC软件进行。
同时用鼠糖、阿拉伯糖、甘露糖、半乳糖、木糖、果糖、葡萄糖、岩藻糖8种单糖进行对照, 根据峰值确定样品的单糖组成。
分子量的测定以标准分子量的葡聚糖 Pulluan 作分子量测定标准。
让其通过高压液相色谱柱, 条件同上, 先以分子量对数与对应的保留时间作标准曲线, 从标准葡聚糖Pulluan 的工作曲线上可以求得该成分分子量。
例如:(阿魏侧耳子实体多糖分离纯化及其化学结构的初步研究)将水解后的多糖样品PW2进行HPLC分析,结果如表所示:阿魏侧耳子实体多糖PW2经过酸水解后,得到2种单糖:葡萄糖和半乳糖,摩尔比例1.77∶1。
例如:经HPLC测定后对照标准曲线得多糖PW2的分子量为3.18×104。
(阿魏侧耳子实体多糖分离纯化及其化学结构的初步研究)4、甲基化分析(1)基本原理先将多糖中各种单糖残基中的游离羟基全部甲基化,然后将多糖中的糖苷键进行完全酸水解,水解后得到的化合物,其羟基所在的位置,即为原来单糖残基的连接点。
同时根据不同甲基化单糖的比例,可以推测出此种连接键型在多糖重复结构中所占的比例。
用此种方法得到的羟基及NaBH4还原醛基后产生的羟基,经乙酰化可得到甲基化的糖醇乙酸酯(此产物易挥发,可进行GC分析),再经GC与GC-MS分析,通过气相色谱的出峰顺序和对质谱谱图的主要离子碎片的分析便可以较准确地确定糖的连接键型。
反应通式如下:(2)甲基化反应取充分干燥的多糖样品10 mg,溶解在2.0 ml的二甲基亚矾(DMSO)中。
在N2保护下快速加入干燥的NaOH粉50 mg,用N2排出空气,加盖密封,室温反应1.0 h并间歇振荡。
在N2保护下缓慢滴加碘甲烷1.0 ml,用N2排出空气,加盖密封,室温继续反应1.0 h并间歇振荡,反应完成后加0.5 ml水终止反应。
反应液先用自来水流水透析48 h,再用蒸馏水透析24 h,透析液冷冻干燥得第一次甲基化样品。
第一次甲基化样品继续甲基化,反应步骤同上,得第二次甲基化样品。
如此重复甲基化三次。
甲基化后的样品用红外光谱检测,3700cm-1—3100 cm-1,附近无羟基的特征吸收峰,表明甲基化反应完全。
(3)甲基化样品的衍生化上述完全甲基化多糖样品加入2 mol/1的三氟乙酸(TFA ) 3.0 ml, 120℃密闭水解2.0 h。
冷却后50℃减压蒸发干,加2.0 ml甲醇再蒸发干,重复三次,最后蒸发干得完全甲基化多糖的水解产物。
加蒸馏水2.0 ml使其溶解,再加硼氢化钠25 mg,振荡后室温还原反应2.0 h。
反应完后滴加0.1 mol/1醋酸分解过量的硼氢化钠并调pH值至5.5~7.0弱酸性。
反应液50℃减压蒸发蒸干,加2.0 ml甲醇再蒸干,重复三次,最后蒸发干。
上述蒸发干样品加入1.0 ml醋酸酐和1.0 ml吡啶,封管后在沸水浴中反应1.0 h。
反应液50℃减压蒸发干,加2.0 ml甲醇再蒸发干,重复三次,最后蒸发干。
加入丙酮1.0 ml,用0.45ul尼龙微孔滤膜过滤,取样进行GC/MS分析。