产品中残留溶剂控制申明模板(国外审计)
美国药典通则467残留溶剂的检测

美國藥典通則<467>殘留溶劑的檢測<467> RESIDUAL SOLVENTSSpectrum 质检部斯百全化学(上海)有限公司800.720.5720 通則適用範圍•所有藥品原料,輔料,製劑中可能存在的溶劑通則目標•為病人的安全,規定藥品中可接受的殘留溶劑量殘留溶劑分類•一類(class 1): 已知會產生不可接受毒性的一些溶劑(Solvents that are know to cause unacceptabletoxicities)•二類(class 2):與並不嚴重毒性相關的一些溶劑(Solvents associated with less severe toxicity)•三類(class 3):較少毒性的溶劑(Less toxic solvents )•其他殘留溶劑:無足夠毒理學數據(Other residual solvents)未列在本通則的溶劑怎麼處理?•當工廠使用經法定監管機構批准使用的未在本通則中羅列的新溶劑時,工廠有責任告知USP 在該品種標準中這一溶劑的鑒定、限度、測試方法,USP將會在個論中加上這一項。
•ICH 認可的新溶劑,USP將直接加在該通則中。
測試範圍•使用過的溶劑•生產或純化過程中產生的溶劑測試方式•直接測最終產品•或者各成份累加:如果各成分累加的值大于本通则的限度,则需测最终产品。
Colchicine秋水仙堿•Colchicine•C22H25NO6 399.44•Acetamide, N-(5,6,7,9-tetrahydro-1,2,3,10-tetramethoxy-9-oxobenzo[a]heptalen-7-yl)-, (S)-.Colchicine[64-86-8].•»Colchicine is an alkaloid contained in various species of Colchicum and in other genera. It contains not less than 94.0 percent and not more than 101.0 percent of C22H25NO6, calculated on the anhydrous, solvent-free basis.•Caution—Colchicine is extremely poisonous.•Packaging and storage—Preserve in tight, light-resistant containers.•USP Reference standards 11—USP Colchicine RS.•Identification, Infrared Absorption 197K—[note—Disregard any peak occurring at 1735 cm1.]•Specific rotation 781S: between 240 and 250, calculated on the anhydrous, solvent-free basis. Test solution: 10 mg per mL, in alcohol. •Water, Method I 921: not more than 2.0%.•Limit of colchiceine—To 5 mL of a solution (1 in 100) add 2 drops of ferric chloride TS: no definite green color is produced. •Limit of ethyl acetate—Internal standard solution—Dilute 0.5 mL of n-propyl alcohol with water to 100.0 mL. •Standard solution—Pipet1 mL of ethyl acetate and 0.5 mL of n-propyl alcohol into a 1000-mL volumetric flask, add water to volume, and mix. Each mL of Standard solution contains 0.90 mg of ethyl acetate.•Test solution—Place about 250 mg of Colchicine, accurately weighed, in a 10-mL volumetric flask, dissolve in about 8 mL of water, and add 1.0 mL of Internal standard solution. Add water to volume, and mix.•Procedure—Determine appropriate sensitivity settings on a gas chromatograph (see Chromatography 621) fitted with a 4-mm ×1.5-m column packed with 20% (w/v) phase G14 on support S1, maintaining the column temperature at75, using nitrogen as the carrier gas, and using a flame-ionization detector. Inject the Standard solution and the Test solution, determine the peak height for ethyl acetate relative to the peak height for n-propyl alcohol, and calculate the percentage, by weight, of ethyl acetate in the portion of Colchicine taken: not more than 8.0% is found.•Chromatographic purity—The sum of the responses of any peaks other than that due to colchicine, eluting within 1.5 times the retention time for colchicine, is not more than 5.0% of the sum of all responses, obtained as directed in the Assay.•Residual solvents 467: meets the requirements, except that the limit of chloroform is 100 ppm.二類溶劑計算選項•選項1:Dose ≤10g/dayConcentration(ppm)=(1000ug/mg ×PDE)/dose•選項2:根據藥品實際情況直接計算PDE是否符合要求,若超出限度則需測試。
usp 溶剂残留 中英文版

<467>溶剂残留简介:INTRODUCTIONThis general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.本章节适用于现有的原料药,辅料和制剂。
应对原料药或制剂产品中可能存在溶剂的所有原料及制剂产品进行控制。
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs, because the solvents employed may vary from one manufacturer to another.当限值与下面提供的数值相符合,残留溶剂的测试方法一般不会在专论中特别,因为不同制造商所使用的溶剂不同。
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
残留溶剂简短报告模板
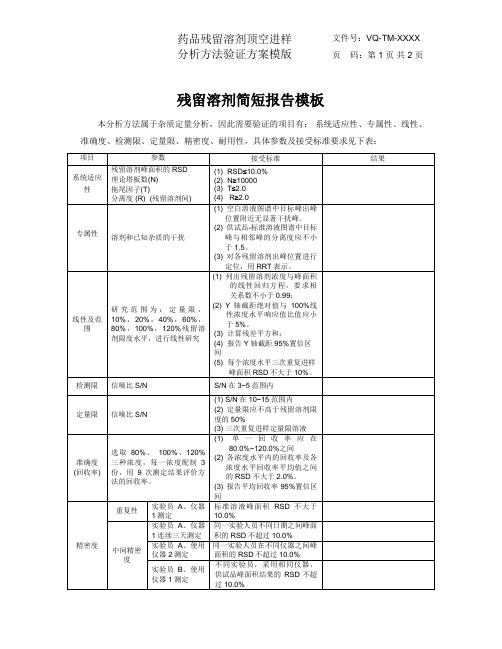
本分析方法属于杂质定量分析,因此需要验证的项目有:系统适应性、专属性、线性、准确度、检测限、定量限、精密度、耐用性,具体参数及接受标准要求见下表:
项目
参数
接受标准
结果
系统适应性
残留溶剂峰面积的RSD
理论塔板数(N)
拖尾因子(T)
分离度(R) (残留溶剂间)
(1) RSD≤10.0%
(1)列出残留溶剂浓度与峰面积的线性回归方程,要求相关系数不小于0.99;
(2) Y轴截距绝对值与100%线性浓度水平响应值比值应小于5%。
(3)计算残差平方和;
(4)报告Y轴截距95%置信区间
(5)每个浓度水平三次重复进样峰面积RSD不大于10%。
检测限
信噪比S/N
S/N在3~5范围内
定量限Байду номын сангаас
信噪比S/N
耐用性
1.改变流速(±XX%)
同“系统适应性要求”
2.改变初始柱温(±XX℃)
3改变色谱柱的批号
(3)报告平均回收率95%置信区间
精密度
重复性
实验员A、仪器1测定
标准溶液峰面积RSD不大于10.0%
中间精密度
实验员A、仪器1连续三天测定
同一实验人员不同日期之间峰面积的RSD不超过10.0%
实验员A、使用仪器2测定
同一实验人员在不同仪器之间峰面积的RSD不超过10.0%
实验员B、使用仪器1测定
不同实验员,采用相同仪器,供试品峰面积结果的RSD不超过10.0%
(1) S/N在10~15范围内
(2)定量限应不高于残留溶剂限度的50%
(3)三次重复进样定量限溶液
准确度
(回收率)
Q3C残留溶剂中英文

杂质:残留溶剂的指导原则1.介绍本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。
在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。
纯度和溶解度。
因此.有时溶剂是合成中非常关键的因素。
本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。
出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。
制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。
一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。
使用低毒溶剂(第三类,表3)较为理想。
附录1中列出了指导原则中的全部溶剂。
表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。
第一、二类溶剂的建议限度或溶剂的分类会随着。
新的安全性资料的获得而调整。
含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。
2. 指导原则的范围指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。
应进行残留溶剂的检验。
也只有在上述情况下,才有必要作溶剂的检查。
虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。
如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。
美国对残留溶剂的要求指南文件

Guidance for Industry Residual Solvents in Drug Products Marketed in theUnited StatesU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)November 2009CMCGuidance for Industry Residual Solvents in Drug Products Marketed in theUnited StatesAdditional copies are available from:Office of CommunicationDivision of Drug InformationCenter for Drug Evaluation and ResearchFood and Drug Administration10903 New Hampshire AvenueBldg. 51, rm. 2201Silver Spring, MD 20993-0002(Tel) 301-796-3400/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)November 2009CMCTABLE OF CONTENTSI.INTRODUCTION (1)II.BACKGROUND/POLICY (2)III.RECOMMENDATIONS (2)pendial Drug Products Approved Under an NDA or ANDA (2)pendial Drug Products Not Approved Under an NDA or ANDA (3)C.Non-compendial NDA or ANDA Drug Products (3)Guidance for Industry1Residual Solvents in Drug Products Marketed in the United States This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.I. INTRODUCTIONThis guidance is intended to assist manufacturers in responding to the issuance of the United States Pharmacopeia (USP) requirement2 for the control of residual solvents in drug products marketed in the United States. Specifically, this guidance makes recommendations on the following:1. How new drug application (NDA) and abbreviated new drug application (ANDA)applicants for noncompendial drug products should limit residual solvents as described in the International Conference on Harmonisation (ICH) guidance for industry Q3CImpurities: Residual Solvents (Q3C). This guidance contains recommendations onsolvent classification and permitted daily exposure.32. How manufacturers of compendial drug products that are not marketed under anapproved NDA or ANDA can comply with USP General Chapter <467> “ResidualSolvents” and the Federal Food, Drug, and Cosmetic Act (the Act).3. How holders of NDAs or ANDAs for compendial drug products should report changes inchemistry, manufacturing, and controls specifications to FDA to comply with GeneralChapter <467> and 21 CFR 314.70.For recommendations on solvent classification and permitted daily exposure, please refer to the ICH Q3C.FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are1 This guidance has been prepared by the Office of Pharmaceutical Science in the Center for Drug Evaluation and Research (CDER) at the Food and Drug Administration.2 USP; General Chapter <467> “Residual Solvents.”3 The levels in ICH Q3C and General Chapter <467> should also be considered for products that are not subject to an NDA or ANDA (e.g., over-the-counter monograph products).cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.II. BACKGROUNDOn July 1, 2008, the USP implemented a requirement for the control of residual solvents in drug products marketed in the United States. The requirement, General Chapter <467> “Residual Solvents,” replaced General Chapter <467> “Organic Volatile Impurities.” The effective date of this change was July 1, 2008.III. RECOMMENDATIONSFDA makes the following recommendations concerning implementation of the USP requirement General Chapter <467> “Residual Solvents.”A. Compendial Drug Products Approved Under an NDA or ANDA1.Beginning July 1, 2008, U.S. marketed drug products with an official USPmonograph were required to meet the requirements for control of residual solvents asdescribed in General Chapter <467>.4General Chapter <467> requires control of residual solvents in finished drug products. Although manufacturers may choose to test the drug product, General Chapter <467> also provides options for evaluating the active pharmaceutical ingredient and excipient components of the finished drug product for residual solvents. FDA can accept residual solvent test data on components from tests performed by the drug product manufacturer or the manufacturer may provide test data or, if applicable, appropriate statements obtained from properly qualified suppliers as described in 21 CFR 211.84(d)(2). For example, reports of analysis can be accepted from a properly qualified5 supplier of a drug product component and will be used by the drug product manufacturer to determine whether the finished drug product complies with the General Chapter <467> defined limits. If the test limits are met for the drug product components, finished product testing is unnecessary.2.FDA will accept the use of appropriate analytical procedures other than thoseincluded in General Chapter <467>.The USP General Notices section on “Tests and Assays – Residual Solvents” references the use of “suitable methods” other than the specific analytical methods included in General Chapter<467>. FDA will accept the use of such other analytical procedures as referenced in 21 CFR 314.50(d) provided that all such procedures are properly described and validated and their4 The Federal Food, Drug, and Cosmetic Act, section 501(b) (21 U.S.C. 351).5 As part of ongoing supplier management, a drug product manufacturer is expected to monitor a supplier to assure that the component it produces continues to be of consistent quality and laboratory results reported on the COA remain reliable.suitability verified under actual conditions of use as described in the current good manufacturing practices (CGMPs) regulations at 21 CFR 211.165(e) and 211.194(a)(2).For compendial drug products approved under an NDA or ANDA, changes made to the specifications in the approved application regarding General Chapter <467> should be in accordance with applicable regulations described in 21 CFR 314.70 and the recommendations in the guidance for industry on Changes to an Approved NDA or ANDA.Generally, an annual report, if needed, can be used to report changes such as adding a test to a finished product specification or adding an alternative analytical procedure to a specification to comply with the USP. The annual report must contain the information described in 21 CFR 314.70(d)(3). As described in 21 CFR parts 210 and 211, detailed data and information related to control of residual solvents and compliance with General Chapter <467> should be documented and kept available at the manufacturing site for the Agency to review upon request during an inspection.The annual report should be submitted in accordance with applicable regulations described in 21 CFR 314.70 and the recommendations in the guidance for industry on Changes to an Approved NDA or ANDA.3.Applicants can submit an amendment to their pending NDA or ANDA to documentany changes made to implement General Chapter <467> if the drug products are thesubject of an official USP monograph and the applicants have already submittedNDAs or ANDAs to the Agency for approval.The amendment should be submitted as soon as possible. Similarly, this same information should be included in all new NDAs and ANDAs submitted for compendial drug products.pendial Drug Products Not Approved Under an NDA or ANDAMarketed compendial drug products not approved under an NDA or ANDA (e.g., over-the-counter (OTC) drug products marketed under an FDA OTC monograph) are also subject to the provisions of the Act, General Chapter <467>, and CGMP documentation requirements described in 21 CFR parts 210 and 211. Manufacturers can use appropriate analytical procedures other than those in General Chapter <467> provided they properly describe and validate their procedures and verify their suitability under actual conditions of use as described in 21 CFR 211.165(e) and 211.194(a)(2).C.Non-compendial NDA or ANDA Drug ProductsGeneral Chapter <467> does not apply to noncompendial drug products. However, FDA recommends that NDA and ANDA applicants for noncompendial drug products limit residual solvents as described in guidance for industry Q3C Impurities: Residual Solvents. Applicants who have not included limits for residual solvents in their NDA or ANDA, as described in 21 CFR 314.50(d), should amend their pending applications as soon as possible.。
002关于现报品种中“残留溶剂”的相关问题及处理建议的讨论会会议纪要

发布日期20011216栏目化药药物评价>>化药质量控制标题关于现报品种中“残留溶剂”的相关问题及处理建议的讨论会会议纪要作者审评管理与协调部部门正文内容时间:2001年5月19日;2001年6月18日地点:药审中心第四会议室主持人(略)参加人员(略)一、目前申报资料中有关“残留溶剂”的研究资料的情况。
药品中的“残留溶剂”是指合成原料药、辅料和制剂在生产的过程中使用或产生的但未能完全被清除的挥发性有机化学物质。
近年来,药品中残存的有机溶剂的毒性和致癌作用日益引起各方面的重视。
目前,申报资料中关于“残留溶剂”部分的研究资料各品种间有较大差别。
其原因是由于国内对有关“残留溶剂”的研究资料尚未提出明确的要求,导致不同时期、不同的申报单位以及不同初审单位间对此方面的要求差异较大。
例如,早期申报资料中对使用的有机溶剂(包括一类溶剂)或检查、或没有检查,且极少订入质量标准;近期的申报资料中也未区分溶剂类别,仅作检查但未订入标准、或仅将个别溶剂订入标准。
二、关于“残留溶剂”的国内外管理情况。
从1994年起,ICH(人用药物注册技术要求国际协调会议)开始着手制定《Q3c杂质:残留溶剂的指导原则》。
该原则中将常用的溶剂分为第一类、第二类和第三类,对各类溶剂的限度有不同要求。
此原则于1997年12月24日正式生效,在欧盟、日本、美国实施。
2000年版英国药典中收载了“残留溶剂”指导原则。
未见其他国家和地区对残留溶剂管理情况的报道。
中国药典2000年版制定了“有机溶剂残留量测定法”,要求对生产过程中引入的有害有机溶剂残留量进行检查。
这些有机溶剂包括苯、氯仿、二氧六环、二氯甲烷、吡啶、甲苯和环氧乙烷。
生产过程中如涉及其它需要检查的有害有机溶剂,则应在各品种项下另作规定。
此版中国药典未对有机溶剂进行分类,未对上述几种溶剂外的其他有机溶剂及其限度进行规定。
三、关于目前审评工作中对“残留溶剂”的技术要求的情况。
目前,我国尚未对新药研究或生产过程中所使用的有害有机溶剂的残留量的检查提出管理原则和明确的技术要求。
EP2.4.24残留溶剂的鉴定与控制中英文对照版(带图)

残留溶剂的鉴定及控制该方法适用于: .1.在活性物质、赋形剂或药品中的未知的一类或二类溶剂残留量的鉴定;2.在活性物质、赋形剂或药品中的一类或二类溶剂残留量的限量试验;3.当二类溶剂的限度大于1000ppm(0.1%)或要求检测三类溶剂时的限量试验。
—类溶剂、二类溶剂、三类溶剂的分类见5.4以下给出了三种样品溶液的稀释方法,以及气相色谱顶空进样的系统条件。
还给出了两种气相色谱的系统条件,系统A为首选方法,同时系统B适用于一般的鉴别试验。
样品溶液的制备方法由样品的溶解性和待测的溶剂种类决定。
下列溶剂不适于用顶空进样法测定:甲酰胺、2—乙氧基乙醇、2—甲氧基乙醇、乙二醇、N-甲基吡咯烷酮、环丁砜(四氢噻吩砜),但可采用其他的适当的方法测定。
当采用其他的方法定量测定有机残留量时,必须进行方法验证采用静态顶空进样法测定样品溶液制备1:适用于在水中易溶的物质的残留溶剂的测定样品溶液(1):取0.200g待测物质,用水溶解并稀释至20m1样品溶液制备2:适用于在水中不溶的物质的残留溶剂的测定样品溶液(2):取0.200g待测物质,用N,N--二甲基甲酰胺(DMF)溶解并稀释至20ml样品溶液制备3:适用于测定N,N---二甲基乙酰胺或N,N--二甲基甲酰胺的残留量,当怀疑他们存在时。
样品溶液(3):取0.200g待测物质,用1,3—二甲基-2-咪唑啉酮(DMI)溶解并稀释至20ml如果上述方法均不适宜,那么所用稀释方法及静态顶空进样条件均必须验证其合理性。
溶剂溶液(a):吸取一类残留溶剂标准溶液1.0ml,用水稀释至100.0ml,再吸取该溶液1.0ml,用水稀释至10.0ml。
溶剂溶液(b):吸取适量二类溶剂溶于二甲基亚砜,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格二)。
溶剂溶液(c):称取1.00g溶剂或待测物质中存在的,用二甲基亚砜或水溶解,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格一或二)。
国外客户GMP审计整改报告模板

、
Annex1-1:偏差调查报告与相关人员培训记录/Deviation report and related personnel training record
报告完成日期
企业名.
企业地址City ZhejiangP.C. 317016,China
Fax: +86(576)xxx, Phone: + 86(576)xxxxx,Email:xxx@
Part one:Correction Report
Serial No.
Annex10-1
11
Minor
一般
仓库
Warehouse
Aug.26,2015
Annex11-1
12
Minor
一般
仓库
Warehouse
July.20,2015
Annex12-1
13
Minor
一般
仓库
Warehouse
July.20,2015
Annex13-1
14
Minor
一般
仓库、QC
Warehouse
文件(整改证明)
1
a.Mismatching of theidentificationnumber on the weighingbalance in the warehouse as per calibration certificate it wasxxx;however as per status label on the balance it wasxxx
USP-467 溶剂残留

[467]有机挥发性杂质残留溶剂限度根据药典要求,药品中残留溶剂定义:在药物或赋形剂制造或使用过程中,或药物制剂生产过程中残存的有机挥发性物质。
目前制药技术不能完全去除残留溶剂。
药物或赋形剂合成中选择适当的溶剂可提高得率,或获得某些特性如晶型、纯度和溶解性。
因此,合成中所用溶剂有时可能是危险因素。
本章节不包括涉及用于组分的溶剂或溶剂化物。
尽管如此,以上产品仍应标明所含溶剂含量并证明对人体安全。
由于残留溶剂不用于治疗用途,因此应尽可能除去,以符合药物、赋形剂和产品规格要求、GMP或其他质量标准。
药物制剂中所含溶剂不得高于安全性评价规定限度。
众所周知,药物、赋形剂、药物制剂不应含能引起不可逆毒副作用(Ⅰ级,见表1)的残留溶剂,除非证明有很好的风险-效益比。
应对引起次级严重毒副作用的残留溶剂(Ⅱ级,见表2)规定限度,以防止病人发生潜在的不良反应。
理想状态下,应尽可能使用低毒性溶剂(Ⅲ级,见表3)。
本章节所有溶剂列表见附录1。
下述表格及限度并不代表全部。
当医药工业发展需使用其他溶剂情况下,应将新溶剂添加到列表中。
当药物、赋形剂、药物制剂生产和纯化过程中残留已知有机溶剂时,应依法检查溶剂限度。
本限度检查仅检查用于制造和纯化加工所用的溶剂。
虽然制造商可能选择测试药物,我们可采用累积法从制造工艺水平计算产品中残留溶剂。
如残留溶剂计算结果等于或低于本章节规定限度,可不进行残留溶剂检查。
如残留溶剂计算结果高于本章节规定限度,仍需检查残留溶剂限度以确定是否精加工降低了有关溶剂水平至可接受量。
如残留溶剂为制造所用溶剂,必须检查药物制剂残留溶剂限度。
附录2为有关残留溶剂的相关背景资料。
根据安全评估残留溶剂分类国际化学安全机构用术语“可忍受日摄食量”(TDI)描述有毒化学物质残留限度。
世界卫生组织和其他国家和国际卫生机构用术语“可接受日摄食量”(ADI)来描述残留限度。
术语“允许日接触量”(PDE)定义为根据药效学残留溶剂可接受摄入量,避免与ADIs混淆。
残留溶剂培训讲义——USP467

• 2类混合A标准溶液:移取1.0ml2类标准贮备液A到合适的顶空瓶中,加
5.0ml水,密封,加盖 。 • 2类混合B标准溶液:移取5.0ml2类标准贮备液B到合适的顶空瓶中,加
1.0ml水,密封,加盖。
• 供试贮备液——精密称取供试品约250mg到25-ml容量瓶,用水溶解并稀 释到刻度,摇匀。
• 供试液——移取5.0ml供试贮备液到适宜的顶空瓶中,加入1.0ml水,
残留溶剂 ——USP467
绪
言
——与国外的质控理念接轨
逐渐采用先进的分析方法和分析理念
专属的HPLC方法替代传统的容量法与生物法 重视对组分、相关物质(包括光学异构体)的控制 重视对残留溶剂的控制 重视对粒度的控制 重视对晶型的控制 用细菌内毒素检查法替代热原检查法 基本取消了异常毒性检查
气相色谱系统(见色谱法<621>) [注——若操作方法B得到的色谱图比操作方法A的色谱图好, 则采用操作方法B的色谱系统。]
• 色谱柱:0.32-mm × 30-m的弹性石英色谱柱,液膜厚1.8-µ m的 G43,或0.53-mm × 30-m的多孔性色谱柱,液膜厚3.0-µ m的 G43 • 载气:氮气(或氦气)线速度约为35cm/s • 分流比为1:5 (为了优化灵敏度,可适当调节) • 柱温:先在40℃维持20分钟,再以10℃/min的升温速率升至 240℃,并维持20分钟。 • 进样口温度: 140℃ • FID检测器温℃:250℃
在药品生产过程中使用过;
在药品生产过程中应当去除而未除尽; 特指有机挥发性化合物。
药品中残留溶剂的特点
种类相对固定,ICH规定了69种;
在具体样品中具有不确定性;
残留量相对较低,一般在痕量或微量范围
残留溶剂测试方法验证方案模板

检验方法验证复核方案(实验部分)方法名称编制人/日期:复核人/日期:批准人/日期:Tabel of Contentsl内容表1.目的PURPOSE (3)2.范围SCOPE (3)3.引言INTRODUCTION (3)4.参考资料REFENRENCE (3)5.分析方法TEST METHOD (3)6.验证的特性指标和接受标准VALIDATION CHARACTERISTICS AND ACCEPTANCE CRITERIA (4)6.1准确度Accuracy (4)6.2线性范围LINEARITY RANGE (5)6.3检测限/定量限LIMIT OF DETECTION / LIMIT OF QUANTITATION (5)6.4专一性SPECIFICITY (5)7.附件APPENDIX (5)1.目的PURPOSE对盐酸文拉法辛清洁残留的测试方法进行方法复核。
2.范围SCOPE此复核方案包含了盐酸文拉法辛清洁残留测定方法的准确度(包括不锈钢;塑料;橡胶三种材料的回收率);线性范围;检测限/定量限;专一性测试。
3.引言INTRODUCTION根据SOPQC-67要求,我们将对生产过程中盐酸文拉法辛清洁残留测定方法的准确度、线性范围、检测限/定量限、专一性进行复核。
4.参考资料REFENRENCE方案编号 :QC-PQ-304 Revision 1 -----见附件1方案编号:02-CV-PKG-OC.IMAC90.001 Revision 0 -----见附件25.分析方法TEST METHOD5.1试剂:(1)水:纯化水(2)乙腈:色谱级,Fisher Scientific公司。
(3)磷酸(85%): 色谱级,TEDIA公司。
(4)三乙胺:分析纯,国药集团化学试剂有限公司。
三乙胺缓冲液:取三乙胺2ml,加水稀释至800ml,用85%磷酸调节pH至3.0。
(5)盐酸文拉法辛(Venlafaxine HCL):Lot2000129753 100.0% Wyeth ReferenceStandard。
农药残留物检测申请书模板

尊敬的XX检测机构:您好!我代表XX公司,向您提交一份农药残留物检测申请。
我们希望通过您的专业检测,了解我司产品中农药残留物的实际情况,以确保产品符合我国及国际相关标准,保障消费者健康。
一、申请检测产品简介1. 产品名称:XX蔬菜(或其他农产品)2. 产品类型:新鲜蔬菜(或其他农产品类别)3. 产地:XX地区4. 生产日期:XX年XX月XX日5. 产品规格:XX公斤/箱(或其他规格)二、检测项目及标准1. 检测项目:根据我国GB 2763-2019《食品安全国家标准食品中农药最大残留限量》以及其他相关国际标准,包括但不限于以下农药残留物:- 有机磷农药:水胺硫磷、甲基对硫磷、甲胺磷、乙酰甲胺磷、杀螟硫磷、三唑磷、对硫磷、马拉硫磷等;- 有机氯农药:六氯苯、五氯硝基苯、七氯、艾氏剂、狄氏剂、硫丹等;- 其他农药残留物:如菊酯类农药、新烟碱类农药等。
2. 检测标准:GB 2763-2019、《食品安全国家标准食品中农药最大残留限量》、CIPAC分析方法等。
三、检测目的1. 确保产品中农药残留物符合我国及国际相关标准,保障消费者健康;2. 提高产品质量和市场竞争力;3. 为企业制定合理的农药使用政策提供科学依据。
四、检测方式及流程1. 请您根据申请检测的产品,按照相关标准和方法进行检测;2. 检测过程中,如有需要,请及时与我们沟通,以便我们提供相关协助;3. 检测完成后,请您出具检测报告,并告知我们检测结果;4. 我们将根据检测结果,采取相应措施,确保产品安全。
五、申请日期本申请于XX年XX月XX日提交。
六、联系方式联系人:XX电话:XX邮箱:XX地址:XX在此,我们感谢您对我司产品的关注和支持,期待与您携手共创美好未来!此致敬礼!XX公司代表:XXXX年XX月XX日。
产品中残留溶剂控制申明模板(国外审计)

产品中残留溶剂控制申明模板(国外审计)
Statement of Residual solvents for产品名称/简称
For “对方企业名称(英文)”
Name of the material:化学名称
,short name:有产品名简称时适用(与出厂标签上一致)
Following are the solvents used in the manufacturing process of +化学名称/简称
ClassⅠResidual Solvents:
一类溶剂的名称/
如未用到一类溶剂的填N/A
ClassⅡResidual Solvents:
二类溶剂的名称/
如未用到二类溶剂的填N/A
Class ⅢResidual Solvents:
三类溶剂的名称/
如未用到三类溶剂的填N/A
Other Residual Solvents:
除了上述三类溶剂之外的溶剂名称
Other than above mentioned none of the residual solvents are used.
We confirm that,if tested the “产品名称/简称” will comply with the standards of current USP and supplements for Residual Solvents<467>.
QA经理或质量负责人签字/日期
Mr./Mrs.xxxx,(上述签字人员名字拼音)
职务,Quality Assurance
“自己企业名称(英文)”。
国外供应商审计GMP外部检察报告书(日本)

文件
核对/备注SOP文件编号
1)制造管理基准书
2)制造卫生管理基准书ቤተ መጻሕፍቲ ባይዱ
3)品质管理基准操作书
4)产品标准操作书
5)验证标准操作书
6)苦情处理操作书(投诉)
7)回收处理操作书
8)自我检点操作书
9)教育训练操作书
4、GMP其他书类
文件
核对/备注
1)生产指示书、记录表
2)原料、资材、中间品、最终产品的保管、确认
3)化验设备是否完善?
12、文件
项目
核对/备注
1)制造指示书是否完善?
2)制造记录书是否正确记录?
3)工艺发生异常时的操作程序是否制定?
13、验证
项目
核对/备注
1)各验证对象是否有目的明确的实施计划书?
2)要验证的工艺是否进行了充分的化验?
3)验证的结果是否作正确的评价?
14、包装
项目
核对/备注
1)包装机器为避免交叉污染或混同,是否作适当的隔离?
3)洗净后的机器、容器是否分类可供识别?
4)是否作机器清洗记录?
5)机器在使用时,是否检查事先有无清洗?
6)清洗检查是否有作记录?
7)批号更换时是否清洗?
6、称量
项目
核对/备注
1)称量物的确认交叉污染是否特别注意?
2)容器、道具是否适用,干净与否?
7、精制工程
项目
核对/备注
1)精制液的调整、管理是否妥善?
3)批号管理
4)设备、机械器具的检查、计量仪器的校正
5)防止污染、混同
6)确认标签、包装
5、验证的实施计划、报告书
文件
核对/备注
EP2.4.24残留溶剂的鉴定与控制中英文对照版(带图)

残留溶剂的鉴定及控制该方法适用于: .1.在活性物质、赋形剂或药品中的未知的一类或二类溶剂残留量的鉴定;2.在活性物质、赋形剂或药品中的一类或二类溶剂残留量的限量试验;3.当二类溶剂的限度大于1000ppm(0.1%)或要求检测三类溶剂时的限量试验。
—类溶剂、二类溶剂、三类溶剂的分类见5.4以下给出了三种样品溶液的稀释方法,以及气相色谱顶空进样的系统条件。
还给出了两种气相色谱的系统条件,系统A为首选方法,同时系统B适用于一般的鉴别试验。
样品溶液的制备方法由样品的溶解性和待测的溶剂种类决定。
下列溶剂不适于用顶空进样法测定:甲酰胺、2—乙氧基乙醇、2—甲氧基乙醇、乙二醇、N-甲基吡咯烷酮、环丁砜(四氢噻吩砜),但可采用其他的适当的方法测定。
当采用其他的方法定量测定有机残留量时,必须进行方法验证采用静态顶空进样法测定样品溶液制备1:适用于在水中易溶的物质的残留溶剂的测定样品溶液(1):取0.200g待测物质,用水溶解并稀释至20m1样品溶液制备2:适用于在水中不溶的物质的残留溶剂的测定样品溶液(2):取0.200g待测物质,用N,N--二甲基甲酰胺(DMF)溶解并稀释至20ml样品溶液制备3:适用于测定N,N---二甲基乙酰胺或N,N--二甲基甲酰胺的残留量,当怀疑他们存在时。
样品溶液(3):取0.200g待测物质,用1,3—二甲基-2-咪唑啉酮(DMI)溶解并稀释至20ml如果上述方法均不适宜,那么所用稀释方法及静态顶空进样条件均必须验证其合理性。
溶剂溶液(a):吸取一类残留溶剂标准溶液1.0ml,用水稀释至100.0ml,再吸取该溶液1.0ml,用水稀释至10.0ml。
溶剂溶液(b):吸取适量二类溶剂溶于二甲基亚砜,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格二)。
溶剂溶液(c):称取1.00g溶剂或待测物质中存在的,用二甲基亚砜或水溶解,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格一或二)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Statement of Residual solvents for产品名称/简称
For “对方企业名称(英文)”
Name of the material:化学名称
,short name:有产品名简称时适用(与出厂标签上一致)
Following are the solvents used in the manufacturing process of +化学名称/简称
ClassⅠResidual Solvents:
♦一类溶剂的名称/
♦如未用到一类溶剂的填N/A
ClassⅡResidual Solvents:
♦二类溶剂的名称/
♦如未用到二类溶剂的填N/A
Class ⅢResidual Solvents:
♦三类溶剂的名称/
♦如未用到三类溶剂的填N/A
Other Residual Solvents:
♦除了上述三类溶剂之外的溶剂名称
Other than above mentioned none of the residual solvents are used.
We confirm that,if tested the “产品名称/简称” will comply with the standards of current USP and supplements for Residual Solvents<467>.
QA经理或质量负责人签字/日期
Mr./Mrs.xxxx,(上述签字人员名字拼音)
职务,Quality Assurance
“自己企业名称(英文)”。