全合成综述
黄连素全合成综述

Hans Journal of Chemical Engineering and Technology 化学工程与技术, 2020, 10(4), 306-313Published Online July 2020 in Hans. /journal/hjcethttps:///10.12677/hjcet.2020.104039Summary of Total Synthesis of BerberineXuesong LiuShanghai Gengcai New Material Technology Co., Ltd., Anshan Hifichem Co., Ltd. Shanghai R&D Center,ShanghaiReceived: Jun. 29th, 2020; accepted: Jul. 13th, 2020; published: Jul. 20th, 2020AbstractBerberine, also known as berberine hydrochloride, was isolated from the rhizomes of the Coptis chinensis plant Coptis chinensis. Berberine is an isoquinoline alkaloid bearing a quaternary am-monium group. Berberine shows a variety of biological activities, such as anti-infection, regulating blood lipids, lowering blood sugar, lowering blood pressure, increasing insulin sensitivity, an-ti-arrhythmia, immune regulation, anti-tumor etc. Therefore, many efforts have been made to im-prove the synthesis berberine. This review covers all recent advances achieved in the synthesis of berberine.KeywordsBerberine, Total Synthesis, Antibacterial, Clinical黄连素全合成综述刘雪松上海庚彩新材料科技有限公司,鞍山七彩化学股份有限公司上海研发中心,上海收稿日期:2020年6月29日;录用日期:2020年7月13日;发布日期:2020年7月20日摘要黄连素亦称盐酸小檗碱,是从中药黄连等根茎中分离的一种季铵生物碱,也是黄连抗菌的主要有效成分。
药物合成综述

药物合成反应综述前言“药物合成反应”是我国药学教育发展中的一个亮点,至今尚未找到国外药学本科中类似的课程实例。
这门课程指导学生在学习完有机化学基础上继续学习的化学药物以及中间体制备中重要的有机合成反应和合成设计原理,为药学的学习打下基础。
药物合成技术是以有机合成药物作为研究对象,主要任务是研究药物合成反应的机制、反应物结构、反应条件与反应方向和反应产物之间的关系,反应的主要影响因素,试剂特点,应用范围与限制等;探讨药物合成反应的一般规律和特殊性质以及各基本反应之间的关系。
这为制药以及药物的研究做出了很大的贡献,同时也为新药的开发以及改进生产工艺打下坚实基础。
关键词:药物合成反应反应机理药物合成反应分类应用特点1药物合成反应分类按官能团的演变规律分类经过化学反应,有机化合物分子中引入某些原子或原子团。
根据引入的原子或基团的不同,药物合成反应可分为卤化、烃化、酰化、缩合、氧化、还原、重排等反应类型。
下面介绍几个在药物合成中的典型的反应,这是合成药物的基本方法。
1.1卤化反应一、卤化反应的概念在有机化合物分子中建立碳-卤键的反应称为卤化反应。
卤素原子的引入可以使有机化合物的理化性质、生理活性发生一定变化,同时它又能容易地转化成其他官能团,或者被还原除去。
因此,卤化反应在药物合成中的应用非常广泛。
二、卤化反应的类型1.加成反应:氯或溴素对烯烃的加成是药物合成中最重要的卤素加成反应。
2.取代反应:有机化合物分子中的氢原子被其他原子或基团所代替的反应称为取代反应。
3.置换反应:有机化合物分子中,氢以外的原子或基团被其他原子或基团所代替的反应称为置换反应。
三、常用卤化剂及其特点1.氯化亚砜:是常用的良好试剂,反应活性较强,可用于醇羟基和羧羟基的氯置换反应,因为反应中生成的氯化氢和二氧化硫均为气体,易挥发除去而无残留物,产品易纯化。
但是,大量的氯化氢和二氧化硫逸出,会污染环境,需进行三废除理。
2.五氯化磷:可将脂肪酸或芳香酸转化成酰氯。
第九章天然产物全合成简介

Selected landmark total syntheses of natural products from 1901-1939.
Selected syntheses by the Woodward Group (1944 - 1981).
Selected syntheses by the Corey Group (1961 -1999).
TESO COCl2 CH2Cl2, Py, EtOH TBSO 75 % O OCO2Et
N LDA THF TBSO Davis Reagent O S O2
TESO OCO2Et O OH 85 %
NaAlH2(OCH2CH2OCH3)2 (Red Al) toluene Global reduction COCl2 CH2Cl2, Py TBSO
TESO O O OH 97 % O
TESO
DMSO, (COCl)2 Et3N Swern oxidation
TESO
O
TBSO
O O 95 %
O
LiTMP THF TBSO Li N
O OH O O 90 %
TOTAL SYNTHESIS OF (-)-TAXOL (Holton)
TESO SmI2 THF, rt, 4 h TBSO O OH O
C-ring formation about 20 steps
TBSO O
OH
A, B-ring formation 6 steps
O
12 steps
OH 12 steps O (+)-belta-patchoulene 9%
t-BuLi hexane, reflux, 5 h OH
天然产物Thienamycin的全合成研究进展

内酰胺转 化 法 , 子 内成环 法 。 分
1 2+2环 加 成 法
D vdB R.ontn等 [采 用 的 是 乙 酰 氧 丁二 ai . Jhso 1 ]
在 N S D F作用下生成化合物 2 , B 和 M 7 利用氟化钾 脱去硅烷基和 甲酰基生成 中间体 2 , 8 再经过一系列 反应可得到最终产物硫霉素 1图 4 。 ( )
第 3 9卷 第 3期 21 0 0年 3月
化
工
技
术
与
开
发
V 0 .9 No 3 13 .
M a.0 0 r2 1
Teh oo y & De eo me to e i lI d sr c n lg v lp n fCh m c n u ty a
天然产物 T i a c he my i n n的全 合 成 研 究 进 展
烯 2与氯磺酰异氰酸酯 ( S ) C I进行 ( +2 环加成反 2 )
应得到 . 内酰胺 3 脱去氮上取代基得到 4 4经过 , , 催化加氢, 然后在碱性条件下水解生成化合物 5 5 ,
作者简 介 : 胡昆 , ,0岁 , 男 3 讲师 , 现从 事天然产物全合成 与药物合成研究 通讯联 系人 : 任杰 , ,0岁 , 女 3 讲师 , 现从事 药理 学研究
环 方法 , 大体 上 可 以分 为 3类 : +2环 加 成 法 , 丁 2 环
亚胺反应合成环丁内酰胺 , 此方法的产率较高。 Jh B ya 【 以炔 烃 为 原 料进 行 反 应 , onD. unk等 3 】
具体 步 骤如下 : 炔烃 2 化为 含有 硫取 代基 的丙 将 2转 二烯 型化 合物 2 ,3与氯 磺 酸 异 氰 酸 酯 ( S ) 应 32 C I反 生成 环 丁 内酰胺 2 ,4在 22二 甲氧基丙 烷作 用下 42 ,. 生成 双环 化合 物 2 , 瑞 尼 镍 催 化 还 原得 到 2 ,6 5用 6 2
普拉格雷合成综述

普拉格雷合成综述万风云【摘要】作为新一代血小板P2Y12受体阻断剂普拉格雷,与噻氯匹啶、氯吡格雷相比,能更快速、更有效地抑制血小板聚集,因此研究普拉格雷全合成具有非常重要的意义,综述了抗血栓药物普拉格雷的全合成方法,按照起始原料和方法的不同有下面几条路线,并分别对路线加以叙述总结.【期刊名称】《浙江化工》【年(卷),期】2016(047)009【总页数】3页(P4-6)【关键词】普拉格雷;抗血栓;合成;综述【作者】万风云【作者单位】浙江工业大学药学院,浙江杭州 310014【正文语种】中文普拉格雷(Prasugrel),化学名为2-乙酰氧基-5(α-环丙基羰基-2-氟苄基)4,5,6,7-四氢噻吩并[3、2-c]吡啶;是由日本第一制药三共公司和美国礼来公司联合开发的新一代血小板二磷酸腺苷受体阻断剂。
于2009年2月和2009年7月分别获得欧盟委员会和FDA批准上市,商品名为Effient,适应症为心力衰竭、中风、不稳定心绞痛等心脑血管疾病。
它是一种前体药物,在体内经过代谢后形成活性分子,与血小板P2Y12受体结合而发挥抗血小板聚集的活性[1]。
随着人们生活水平的提高和生活节奏的加快,同时也没有合理的体育锻炼,越来越多的人,特别是老年人出现血栓的概率非常高。
据世界卫生组织统计,全世界每年因疾病死亡的人数大约有5100万人,其中有30%的人是死于心脑血管疾病,因此,对抗血栓药物的研究具有非常重要的意义。
目前,文献[2-4]中报道关于普拉格雷的合成方法比较多,我们按照起始原料的不同可分为以下几种方法合成普拉格雷。
程兴栋等[5](如图1)以邻氟苯乙酸(1)和环丙基甲酸甲酯为原料合成1-环丙基-2-氟苄基酮(2),然后再经溴化铜溴代得到1-环丙基羰基-2-氟苄溴(3),化合物3再与5,6,7,7a-四氢噻吩并[3,2-c]吡啶-2(4H)-酮盐酸盐(5)缩合后乙酰化得到普拉格雷(8),总收率为72.9%。
山竹醇全合成-概述说明以及解释

山竹醇全合成-概述说明以及解释1.引言1.1 概述概述部分的内容如下:山竹醇是一种具有重要生物活性的天然产物,具有广泛的应用价值。
其结构独特,具有多种药理活性,如抗菌、抗炎、抗氧化等。
由于山竹醇在医药、化妆品等领域的广泛用途,其全合成研究备受关注。
本文将重点关注山竹醇的结构、合成途径和全合成方法,以期为山竹醇的研究和应用提供参考。
文章结构部分主要是对整篇文章进行整体的概述和安排,以帮助读者更好地理解文章的脉络和内容。
在本文中,文章结构如下:1. 引言1.1 概述1.2 文章结构:本部分介绍了文章的整体结构安排,包括引言、正文和结论三个部分的内容安排和重点展示。
1.3 目的2. 正文2.1 山竹醇的重要性:介绍了山竹醇在化学领域中的重要性和应用价值。
2.2 山竹醇的结构与合成途径:对山竹醇的结构特点进行介绍,并阐述了目前已知的合成途径及相关研究成果。
2.3 山竹醇的全合成方法:详细介绍了山竹醇的全合成方法,包括反应步骤、关键中间体和合成路线等内容。
3. 结论3.1 总结全文内容:对整篇文章的主要内容和结论进行总结和概括。
3.2 展望未来研究方向:探讨了山竹醇研究领域的未来发展方向和可能的研究重点。
3.3 结论:对文章的整体思想和观点进行总结,并提出作者的观点和看法。
1.3 目的本文旨在系统介绍山竹醇的全合成方法,通过对山竹醇的结构与合成途径的深入探讨,探讨其重要性及全合成方法的可行性。
通过详细的分析和讨论,旨在为相关研究提供参考,并为未来山竹醇的全合成方法研究提供新的思路和方向。
同时,通过本文的撰写,也有助于加深读者对山竹醇的了解,对相关领域的研究和应用有所帮助。
2.正文2.1 山竹醇的重要性山竹醇是一种重要的天然产物,具有多种药用价值和广泛的应用领域。
首先,山竹醇具有抗氧化和抗炎作用,可以帮助预防和治疗炎症性疾病。
其次,山竹醇被证明具有抗癌活性,可以通过抑制肿瘤细胞的生长和扩散来帮助治疗癌症。
此外,山竹醇还具有抗菌和抗病毒作用,可以用于治疗感染性疾病和传染病。
天然产物化学的全合成

R. B. Woodward的贡献: (1). 将物理方法用到有机化学:UV,IR,x-Ray (2). 将反应机理的电子理论用于解决结构和合成。 (3). 全合成工作: (A). 能判断一项任务是否能实现,有细致周到的计划。
(B). 将合成目的放在研究新反应之前。
(C). 先形成一个环状体系,以确定功能团的位置和立 体化学。 (D). 常选择一个由天然有机物降解而成的中间物作为 合成路线的中间体。
二. 全合成发展过程(四位化学家):
Wohler:1827年尿素的人工合成。合成的历史开端。 Robinson (1947年诺贝尔化学奖):有机结构电子理论的 发展和逐步完善 。1917年脱品酮 (tropanone) 全合成。全合 成的开始。 Woodward (1965年诺贝尔化学奖):1944年合成了奎宁 (quinine),1954年全合成马钱子碱(strychnine),两项工作 是重要里程碑,1973年又与Eschenmoser合作实现了维生 素B12的人工合成。复杂结构天然产物全合成 。 Corey (1990年诺贝尔化学奖 ):逆合成分析法和合成子 概念,为现代合成化学理论和方法的理性化认识和天然产 物化学合成的广泛、深入开展奠定了基础。
三.合成实例
1.Woodward的利血平(reserpine)合成路线:
O O + O MeOOC Br HO
d
6 8 5
OH
b
H
a
O
H H OH
c
ቤተ መጻሕፍቲ ባይዱH OH O
O
H H
H H
MeOOC
O OH O
f
H MeO
O H H H
e
OH H H
g
Spiroapplanatumine K及其衍生物的全合成研究

Spiroapplanatumine K及其衍生物的全合成研究Spiroapplanatumine K及其衍生物的全合成研究摘要:自然产物一直以来都是有机合成领域的研究热点。
Spiroapplanatumine K是一种新颖的天然产物,具有潜在的医药价值。
本文对其全合成研究进行了综述,并介绍了一种成功的合成方法。
1. 引言Spiroapplanatumine K是一种由蕈藻目真菌Applanatia cinnamomea产生的天然产物,具有复杂的化学结构和潜在的生物活性。
该化合物结构中独特的螺环结构对其生物活性起着至关重要的作用,因此引起了有机合成领域的广泛关注。
为了深入了解Spiroapplanatumine K的生物活性,并在药理学和药物开发中进行进一步应用,全合成研究变得尤为重要。
2. 研究方法为实现Spiroapplanatumine K的全合成,需要设计合理的合成路线,并进行关键中间体的合成、立体选择性的控制和结构验证等方面的研究。
本研究选择了基于多步反应的合成策略,在反应条件和试剂选择上进行了仔细的考虑。
具体来说,我们利用了环氧化-环开反应合成了螺氧杂环结构,利用一系列活化试剂和选择性还原试剂引入基团,通过精确的立体控制,成功地合成了Spiroapplanatumine K。
3. 合成结果与讨论本研究根据前人已有研究成果,设计了一种有效的全合成路线。
通过多步反应,我们首先合成了中间体A,并通过NMR和质谱等手段进行了结构鉴定。
接下来,我们将中间体A经过环氧化-环开反应,获得了螺氧杂环结构的中间体B,并通过NMR和X 射线单晶衍射等方法验证了其结构。
最后,经过一系列活化试剂的引入和选择性还原试剂的使用,我们成功地合成了Spiroapplanatumine K,并通过NMR、质谱和元素分析等方法验证了其结构和纯度。
4. 结论本研究采用了一种有效的合成路线,成功地合成了Spiroapplanatumine K。
天然产物结构改造与全合成

天然产物结构改造与全合成天然产物是指存在于自然界中的有机化合物,具有多样的结构和广泛的生物活性。
这些化合物通常具有复杂的结构,因此其全合成一直是有机化学领域的研究热点之一。
通过天然产物的结构改造和全合成,我们可以深入了解其生物活性机制,同时也为新药物的发现和开发提供了重要的思路和方法。
天然产物的结构改造是指通过有机合成化学手段对其结构进行改变,以获得更具生物活性或药理活性的衍生物。
这种方法可以通过调整分子中的官能团、环结构或手性中心等来实现。
例如,通过引入不同的官能团或改变其位置,可以改变分子的溶解性、稳定性以及与靶点的相互作用方式,从而提高其活性或选择性。
此外,通过合成不同的环结构,也可以改变分子的立体构型和空间排列,进而影响其生物活性。
通过这种结构改造的方法,研究人员可以设计和合成一系列结构类似但具有不同活性的化合物,从而深入探究其结构与活性之间的关系。
与结构改造相比,全合成更具挑战性。
全合成是指从简单的起始物质出发,通过一系列有机合成反应,逐步构建目标天然产物的分子骨架和功能团。
全合成的过程需要考虑反应的选择性、高效性以及产物的纯度和收率等因素。
在全合成中,化学家们经常面临着复杂的分子结构和多步反应的困难。
为了解决这些问题,他们需要不断探索新的反应方法和策略,提高反应的效率和选择性。
同时,他们还需要充分发挥有机合成化学的创造性,灵活运用各种合成方法和技术,以克服合成的难题。
天然产物的结构改造和全合成不仅对于药物研发具有重要意义,也为有机合成化学提供了重要的研究对象和挑战。
通过天然产物的结构改造和全合成,我们可以深入了解天然产物的结构和活性之间的关系,揭示其生物活性机制,为新药物的发现和开发提供重要的线索。
同时,结构改造和全合成也为有机合成化学的发展提供了新的方向和动力。
通过不断探索新的反应方法和策略,提高反应的效率和选择性,有机化学家们可以不断推动有机合成化学的发展,为人类的健康和生活质量做出更大的贡献。
化学全合成文献汇报怎么写

化学全合成文献汇报怎么写
化学全合成文献汇报的撰写可以根据以下步骤进行:
1. 选择主题:首先,你需要选择一个你感兴趣并且有一定了解的化学全合成主题。
这个主题可以是一个具体的化合物,也可以是一个更广泛的领域。
2. 查阅文献:查阅相关的学术文献是撰写文献汇报的基础。
你需要查找并阅读关于这个主题的最新研究论文,综述文章,以及任何其他可能相关的资料。
你可以使用各种学术数据库,如PubMed,Google Scholar,Web of Science等,来查找这些文献。
3. 整理资料:在查阅了大量文献后,你需要整理和分析这些资料。
这包括总结不同研究的发现,找出研究的共识和分歧,以及识别研究的空白和未来的研究方向。
4. 撰写汇报:在整理完资料后,你可以开始撰写文献汇报。
汇报应该包括以下几个部分:介绍(包括主题的重要性和背景),相关研究综述(包括主要发现和研究的共识与分歧),以及总结和未来研究方向。
在撰写过程中,应尽量保持客观和中立,避免个人主观意见。
5. 校对和修改:完成初稿后,你需要仔细校对和修改汇报。
这包括检查语法错误,调整句子和段落结构,以及确保所有的引用都正确无误。
6. 准备汇报:如果你要在会议或者课堂上进行口头汇报,你还需要准备一个幻灯片演示文稿。
这个演示文稿应该包含你文献汇报的主要观点和结论,以及一些相关的图表或图片。
以上是一般性的步骤和建议,实际撰写过程可能会根据具体主题和要求有所不同。
总的来说,撰写化学全合成文献汇报需要耐心和细心,需要反复阅读和整理文献,以确保你的汇报尽可能准确和全面。
天然产物化学的全合成
1978年,我国学者蔡俊超等的合成路线如下:
第3章 天然产物的全合成
一、天然产物全合成的意义 二、天然产物全合成发展过程 三、天然产物全合成实例
一、天然产物全合成的意义
全合成是确定复杂天然产物精确化学结构的直接和有力 的方法。全合成的意义:
(1) 由合成决定结构;
早期研究工作所需,对所推测结构验证。
(2) 突破天然材料的限制; 例如:可的松,需用2万只牛的肾上腺作原料才可分
N
N
HH
H
O H
MeO2C
O OMe
MeO
OMe
OMe
a.PhH, 回流. b. Al(Oi-Pr)3,i-PrOH.
c. ①Br2; ②NaOH, MeOH. d. NBS(aq)·H2SO4, 30 ℃.
e. ①CrO3/HOAc; ② Zn/HOAc 。
f.①CH2N2; ②Ac2O; ③OsO4, NaClO3, H2O.
g. ①HIO4; ②CH2N2. h. PhH.
i. NaBH4/MeOH. j. ①POCl3; ②NaBH4.
k. ①KOH/MeOH; ②HCl; ③DCC/Py.
l. t-BuCO2H, 二甲苯.
m. ①NaOMe, MeOH, 回流; ②3,4,5(MeO)3C6H2COCl, 吡啶.
共十三步
2.喜树碱( Camptothecin )的合成
对溴苯胺的全合成报告

对溴苯胺的全合成报告溴苯胺是一种重要的有机化合物,广泛应用于医药、化工、染料和农药等领域。
其全合成过程具有一定的复杂性,需要考虑反应条件和反应品的选择。
下面将对溴苯胺的全合成过程进行详细介绍。
一、溴苯胺的常用合成方法目前常见的合成溴苯胺的方法包括三种:1)偶氮化法;2)Grignard试剂法;3)Reductive Amination法。
其中,偶氮化法是溴苯胺的主要制备方法,但该方法反应条件较苛,产物的纯度较低;Grignard试剂法反应条件相对温和,但该法需要制备Grignard 试剂,难度较大;而Reductive Amination法是一种高效、简便和环保的制备方法,已成为制备溴苯胺的首选方法之一。
二、使用Reductive Amination法制备溴苯胺的流程1. 制备胺的前体溴苯胺的制备需要先制备苯甲醛和对溴苯乙胺(或其它对卤代苯胺)的前体。
常见的制备苯甲醛的方法是苯乙烯直接羟甲基化。
制备对溴苯乙胺的方法包括碘铜法,氢溴酸钾与过量溴代乙醇反应法,以及金属铜的还原氯化法等。
2. 进行Catalytic Hydrogenation将前体苯甲醛、对溴苯乙胺等加入反应体系中,在有机溶剂和酸催化剂的催化下,利用氢气进行还原胺和醛的羰基,产生aminobenzyl alcohol。
在还原过程中,需要注意反应温度的控制,反应时间的掌握和反应物浓度的配比等因素的影响。
利用亚硫酸钠、氢气和催化剂(镍或铁)催化Reductive Amination反应,使aminobenzyl alcohol与溴苯胺进行反应,生成溴苯胺。
反应完成后,通过蒸馏或其它方式得到纯度较高的溴苯胺。
三、存在的问题及解决方法在使用Reductive Amination法提高产率和纯度等方面存在一定的问题,采取一些对策可以解决这些问题:1. 反应物质量的控制:在Reductive Amination反应中,反应物浓度的配比是影响产物纯度和产率的重要因素,尤其是质量初始比例的错误会导致产物纯度下降、副反应增加。
全合成 方法学

全合成方法学全合成方法学是有机合成化学中的一种方法学,用于合成复杂有机分子。
全合成方法学是通过有机合成化学家的智慧和创新能力,利用有机合成化学的基本原理和反应,以及合成反应的顺序和条件,从简单的起始物质开始,逐步合成目标分子的方法。
全合成方法学的核心是设计合成路线。
在设计合成路线时,有机合成化学家需要考虑到反应的适用性、选择性、反应条件、步骤的顺序等因素。
通过合理的设计,合成路线可以使合成过程更加高效、经济、环保,并且可以避免副反应和废物的产生。
全合成方法学的第一步是确定目标分子的结构和性质。
有机合成化学家需要了解目标分子的结构、功能和用途,以及相关文献中已有的合成方法和研究成果。
然后,根据已有的合成方法和研究成果,有机合成化学家可以选择合适的起始物质和反应条件,设计出合成路线。
在设计合成路线时,有机合成化学家需要考虑反应的顺序和条件。
合成路线中的每一步反应都需要选择合适的反应试剂和条件,以及合适的催化剂和溶剂。
有机合成化学家还需要考虑反应的选择性,即在多个可能的反应路径中选择最合适的路径,以获得目标分子。
在合成过程中,有机合成化学家需要进行反应的优化和调节。
通过改变反应条件、反应试剂和催化剂的用量,有机合成化学家可以提高反应的产率和选择性。
有机合成化学家还可以利用现代有机合成化学的技术和方法,如保护基策略、合成方法学的应用等,来解决合成路线中的难题和挑战。
全合成方法学的最终目标是合成复杂有机分子。
在合成过程中,有机合成化学家需要进行多步反应,每一步反应都需要选择合适的反应条件和试剂。
有机合成化学家还需要进行反应的监控和分析,以确保反应的进行和产物的纯度。
最后,有机合成化学家还需要对合成的产物进行分离和纯化,以获得目标分子。
全合成方法学是有机合成化学中的一种方法学,通过有机合成化学家的智慧和创新能力,利用有机合成化学的原理和反应,以及合理的合成路线和反应条件,来合成复杂有机分子。
全合成方法学的发展对于有机合成化学的进步和发展具有重要意义,可以为药物研发、材料科学等领域提供丰富的化合物资源。
天然产物全合成最新版详解

天然产物全合成最新版详解天然产物全合成是指通过人工合成方法合成天然产物的过程。
天然产物是指生物体内自然合成的有机化合物,具有丰富的结构和生物活性。
在过去的几十年里,天然产物全合成已经成为有机合成化学的重要分支,对于开发新药和探索生物活性分子的机制具有重要意义。
天然产物作为药物和生物活性分子的源头,具有广泛的药理活性和潜在的药物发现价值。
然而,由于天然产物的合成复杂性和数量有限性,使得研究人员在进一步探索这些化合物的生物活性和开发新药方面面临挑战。
因此,天然产物全合成成为一种重要的策略,通过人工合成天然产物,可以扩展天然产物结构空间,提供更多有机化合物用于生物活性研究和新药开发。
此外,天然产物全合成还可以揭示天然产物的生物合成途径和结构-活性关系,为有机化学研究提供重要的参考。
综上所述,天然产物全合成在药物发现、药物开发以及研究生物活性分子的机制方面具有重要意义。
通过人工合成天然产物,可以制备更多结构多样化的有机化合物,推动药物研发的进展,并推动有机化学的发展和创新。
概述天然产物全合成的基本原理和方法。
天然产物全合成是一个旨在合成天然产物的化学领域,它是基于天然产物结构和活性的全面理解和研究的基础上进行的。
全合成的目标是合成复杂的天然产物,使其成为可获取且可应用的化合物。
在天然产物全合成中,基本原理包括寻找合适的出发物(起始原料)、设计合成路径、选择合适的转化方法和优化反应条件。
通过有序的化学反应步骤,将简单的化合物逐步转变为复杂的中间体,最终生成目标天然产物。
在合成路径设计时,考虑到反应的高效性和选择性是非常重要的。
全合成的方法包括但不限于:1)有机合成化学方法,如羧酸衍生物的活化、碳碳键形成反应等;2)金属催化反应,例如氧化还原反应、偶联反应等;3)生物合成方法,如酶催化反应和微生物发酵等;4)物理方法,如光合成等。
天然产物的全合成在药物研发、农业化学品生产和材料科学等领域具有重要应用价值。
通过全合成,可以扩大天然产物的供应量,在研究和应用中提供更多的机会和灵活性。
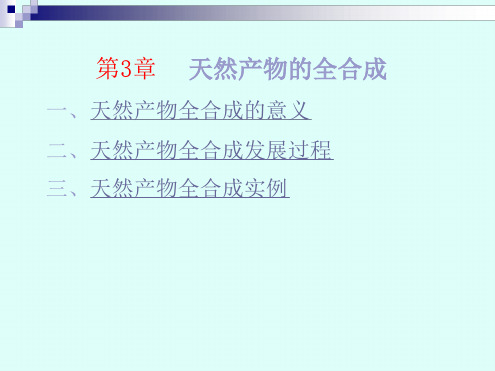
全合成|《Nature》(-)-Bilobalide
全合成|《Nature》(-)-Bilobalide引言Bilobalide(白果内酯),为银杏叶的主要功效成分之一,是从银杏科植物银杏中提取出来的唯一的倍半萜物质,白果内酯对心绞痛、心肌梗死、冠心病、高血压和心律失常等心脑血管性疾病有良好的疗效。
该化合物在临床上,目前主要用于治疗神经病、脑病和脊髓病、抗衰老。
该化合物结构复杂,具有多个稠合五元环体系,六个相连的手性中心,并且有三个连续的季碳中心。
除了一个环戊烷和戊内酯结构,还有由缩醛酯形成的两个并五元环结构。
此外,骨架上还有大位阻的叔丁基、活泼的羟基等。
这些结构特征,使得该化合物在合成上极具挑战。
虽然如此,Corey和Crimmins团队分别完成了消旋Bilobalide 的全合成,随后,Corey团队还完成了(-)-Bilobalide的全合成。
近期,Shenvi等人再次通过更为简洁的合成路线完成了(-)-Bilobalide的全合成。
该研究成果发表在顶级期刊《Nature》上下面,我们一起看看这些有机合成大家如何造Bilobalide1 Corey团队Corey作为世界顶级的有机合成化学家,不仅提出了逆合成分析理论,并且在全合成领域也作出了非常多漂亮的工作。
他带领团队,在1987年,即完成了(±)-bilobalide的全合成。
其合成过程如下所示:1988年,他们根据之前研究的成果,开发了对映选择性全合成(-)-Bilobalide的方法:通过CBS对映选择性还原烯酮,制备关键的手性烯丙基醇,以此作为对映选择性合成的关键。
经过总共24步反应,最终完成了(-)-Bilobalide全合成。
2Crimmins团队Crimmins等人在1993年,完成了(±)-bilobalide的全合成。
相比于Corey团队,Crimmins等人开发的合成路线缩短为17步,虽然个别步骤收率不够理想,但是总体路线缩短还是很鼓舞人心的。
3Shenvi团队Shenvi等人,根据Corey和Crimmins团队的研究成果,分析后发现:Corey团队在后续转化阶段有11步涉及氧化还原操作;而Crimmins团队也使用了8步氧化还原操作。
美罗培南侧链的合成工艺综述

CON(CH3)2
N PNZ
61.4% total yield
Sunagawa M , Matsumura H, Inoue T et al. A novel carbapenem antibiotic, SM-7338 structure activity relationships [J]. J Antibiotics, 1990, 43 (5) :519-532
COOH
COOH
O
HO
PNZ-Cl HO
NH
NaOH
N PNZ
PMB-Cl ,NEt3
HO
O PMB
O
N PNZ
N C OEt 2
O
O PMB
89%yield (93%)
89%yield (88%)
PPh3 ,AcSH AcS
N PNZ
100%yield (99%)
TFA PhOCH3AcS
O OH
COOMs
MsCl ,NEt3 MsO
a)
COOH
-25 ℃
N PNZ
H2S , NEt3 MsO
HO
2
-25 ℃
1
N PNZ
1) ClCOOi-Pr ,NEt3
b) 2) MsCl ,NEt3
MsO
COOCOOi-Pr
COSH
N PNZ
5
-25 ℃
N PNZ
NEt3 CH2Cl2
6refluxFra bibliotekO胡来兴, 刘浚, 金洁.美罗培南全合成的改进. 中国医药工业杂志. 2000, 31 (7):290-292
HO MsO
COOH PNZ-Cl HO
青蒿素的化学全合成.总结

青蒿素的合成与研究进展摘要:青蒿素是目前世界上最有效的治疗疟疾的药物之一,存在活性好、毒副作用小、市场需求大、来源窄等特点。
目前,青蒿素的获取途径主要有直接从青蒿中提取、化学合成和生物合成。
本综述将针对近年来青蒿素的发展特点及合成方法进行论述。
关键词:青蒿素;合成方法;研究进展青蒿素是中国学者在20世纪70年代初从中药黄花蒿(Artem isia annua L1 )中分离得到的抗疟有效单体化合物,是目前世界上最有效的治疗脑型疟疾和抗氯喹恶性疟疾的药物, 对恶性疟、间日疟都有效,可用于凶险型疟疾的抢救和抗氯喹病例的治疗.青蒿素还具有抑制淋巴细胞的增殖和细胞毒性的用1;具有影响人体白血病U937细胞的凋亡及分化的作用2;还具有部分逆转MCF-7/ARD细胞耐药性作用3;还具有抑制人胃癌裸鼠移植瘤的生长的作用4;还具有一定的抗肿瘤作用5等.除此之外,青蒿素及其衍生物还具有生物抗炎免疫作用、生物抗肿瘤作用、抑制神经母细胞瘤细胞增殖的作用等。
世界卫生组织确定为治疗疟疾的首选药物,具有快速、高效、和低毒副作用的特征.6。
因在发现青蒿素过程中的杰出贡献,屠呦呦先后被授予2011年度拉斯克临床医学研究奖和2015年诺贝尔医学奖.1 青蒿素的理化性质及来源青蒿素的分子式为C15H22O5, 相对分子质量为282。
33.是一种含有过氧桥结构的新型倍半萜内酯,有一个包括过氧化物在内的1,2,4-三烷结构单元,它的分子中还包括7个手性中心,合成难度很大.中国科学院有机所经过研究,解决了架设过氧桥难题,在1983年完成了青蒿素的全合成.青蒿素也有一些缺点,如在水和油中的溶解度比较小,不能制成针剂使用等。
2青蒿中提取青蒿素青蒿素是从菊科植物黄花蒿中提取出来的含有过氧桥的倍半萜内酯类化合物,在治疗疟疾方面具有起效快、疗效好、使用安全等特点。
目前主要的提取方法有溶剂提取法、超临界提取法、超声波萃取法、微波萃取法、其他萃取法等。
苦参碱类生物碱全合成研究概况

酝葬灶凿藻造造 蕴 等在前期研究基础上,首次报道了苦参碱的化 学全合成( 杂糟澡藻皂藻 远 )[圆缘,圆远]。起 始 原 料 猿鄄氧鄄庚 二 酸 二 乙 酯 (猿苑)与丙氨酸酯在苯液中高温回流,中间产物通过添加亚当 氏催化剂在乙醇、冰醋酸溶液中发生还原得到产物 猿愿,该产 物在真空条件下经高温发生内酰胺化作用形成单环结构。猿愿 在氢化钠作用下通过 阅蚤藻噪皂葬灶灶 反应得到二环中间产物 猿怨, 然后与丙烯腈发生 杂贼燥则噪 烯胺反应得到二元腈衍生物 源园,源园 经钯催化两条氰基侧链闭合得到终产物苦参碱 员。该合成途
烃合成为四环含氮化合物。其明显的缺陷是路径长、经多步 催化、终产物得率非常低,难以实现工业化等,因而实际应用 价值不大,但该路径为后续研究开辟了先河。
杂糟澡藻皂藻摇 员
摇 摇 日本学者 韵噪怎凿葬 杂 以三环含 晕 化合物 员圆 为原料[圆员],氧 化铂为氢化反应催化剂,氢化产物在 源豫 碱液中反应后,氯仿 萃取部分经盐酸酸化,再以甲醇鄄丙酮重结晶得到 员源,其环上 氰基被羧基所取代,然后在甲基化试剂重氮甲烷作用下,得到 酯化产物 员缘,接着与乙酸酐反应,其相邻环上的 晕 发生乙酰 化取代,得到产物 员远。员远 与 晕葬鄄泽葬灶凿 在二甲苯溶液中发生闭
中分离得到,随后从槐属其他植物中也陆续分离纯化到该化 合物,其结构式在 员怨猿远 年由 运燥灶凿燥 匀 予以确认[圆园]。本文按 文献发表顺序对苦参碱和苦参碱衍生物的合成研究进行综 述。
栽泽怎凿葬 运 研究小组[圆园]在 员怨缘远 年以 圆 为起始原料,与甲醛 和盐酸甲胺经 皂葬灶灶蚤糟澡 则藻葬糟贼蚤燥灶 得到含 晕 的六元单环产物 猿,猿 在氨水中继续环化得到二环中间产物 源,每个环上各含一个 晕 原子,然后在 悦怎圆 垣 的催化下,通过加氢得到三环化合物 缘, 接着经溴化氰取代脱去 晕 上的甲基形成氰化物,在盐酸作用 下化合物 远 的鄄悦晕 被取代得到 苑,与化合物 员 相比缺少一个与
mesembrine合成方法综述

mesembrine合成方法综述
mesembrine是一种从南非草药Sceletiumtortuosum中提取的生物碱,具有抗抑郁、抗焦虑、镇静、镇痛等药理作用。
近年来,mesembrine得到了广泛关注,成为研究热点。
目前,mesembrine的主要来源是从Sceletium tortuosum中提取,但其提取量较低,需要大量准备原料。
因此,研究mesembrine的合成方法具有重要的意义。
本文综述了mesembrine的合成方法,主要包括三种方法:1、从Sceletium tortuosum中提取;2、天然产物的半合成;3、全合成。
其中,从Sceletium tortuosum中提取的方法是最常用的,但其成本较高,提取量有限;天然产物的半合成方法可以减少准备原料的量,但是操作复杂,产率较低;全合成方法虽然操作简单,但是需要更多的前置反应,成本也更高。
总体而言,mesembrine的合成方法需要综合考虑成本、操作难度、产率等因素。
未来的研究可以探索更加高效、经济的mesembrine 合成方法,以满足其在药物研发和医学应用方面的需求。
- 1 -。
全合成(7)
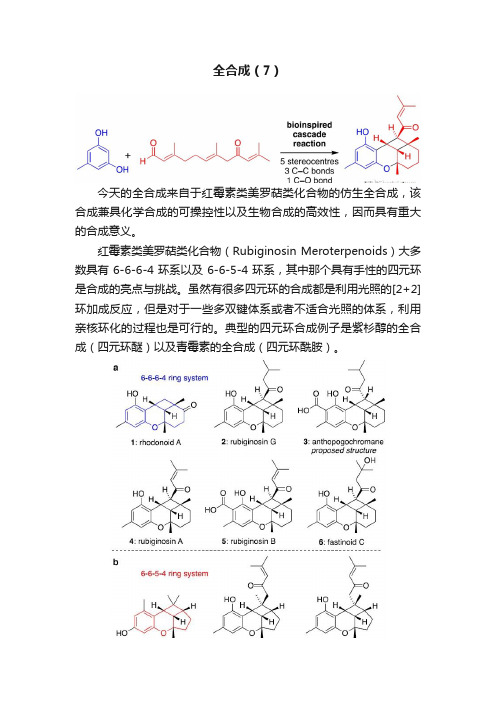
全合成(7)
今天的全合成来自于红霉素类美罗萜类化合物的仿生全合成,该合成兼具化学合成的可操控性以及生物合成的高效性,因而具有重大的合成意义。
红霉素类美罗萜类化合物(Rubiginosin Meroterpenoids)大多数具有6-6-6-4环系以及6-6-5-4环系,其中那个具有手性的四元环是合成的亮点与挑战。
虽然有很多四元环的合成都是利用光照的[2+2]环加成反应,但是对于一些多双键体系或者不适合光照的体系,利用亲核环化的过程也是可行的。
典型的四元环合成例子是紫杉醇的全合成(四元环醚)以及青霉素的全合成(四元环酰胺)。
在生物体系中,可能经历如下的可能过程,包括氧化、脱羧与环加成:
在化学合成中采用的长链烯烃是商业不可得的,因此需要进行偶联与修饰。
首先进行烯丙基溴的类Sonogashira偶联,接着发生Meyer−Schuster重排,产生一个不饱和羰基化合物。
环的构建利用了一锅的串联反应,请横屏观看:
由于底物甲基间苯二酚具有多个亲核位点,因而涉及区域选择性的问题。
这些化合物可以通过核磁共振氢谱进一步确证:。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
阿枯米灵生物碱(−)-Vincorine的全合成分析
阿枯米灵生物碱(−)-Vincorine具有复杂的多环结构以及重要的生物活性一直吸引着众多化学合成工作者的关注。
早在2009年,秦勇课题组率先完成了(−)-Vincorine的全合成工作,最近马大为课题组应用分子内氧化偶联的方法成功地以18步,总收率5%的路线合成了(−)-Vincorine。
本文将对马课题组合成该化合物的方法进行简单的介绍。
一:(−)-Vincorine的逆合成分析
先将N4-C21键断开得到化合物11,化合物11可由12得到,12可由化合物13经分子内氧化偶联而得,化合物13则由14与15经迈克尔加成得到(Figure1)。
Figure1:(−)-Vincorine的逆合成分析
二:(−)-Vincorine的合成路线
该课题组以市售的5 - 甲氧基色胺为原料(16)经(Boc)2O及Pd(OAc)2的催化作用得到1,2,3,5四取代吲哚17,后烯烃双键氢化加成,酯基还原得到化合物18。
18在IBX氧化下得到醛与丙二酸二甲酯反应得到化合物14,后经迈克尔加成得到化合物20.化合物20为一非对映异构体,将其混合物进行反应,氧化消除
得烯烃化合物21,该化合物为E式和Z混合物。
选择性还原醛基,加上TBS保护,移除Boc即得化合物13。
(Figure2)
Figure2
得到关键化合物13后便可尝试分子内氧化偶联反应,经反复实验确定在LiHMDS, I2, THF, −40 °C to r.t.条件可以成功得到只有一种构型的目标产物23且
产率为67%。
后经Krapcho的反应条件成功去除一个酯基,再通过氯化,环合,甲基化成功合成得到(−)-Vincorin。
(Figure3)
Figure3
三:关键反应的应用
在全合成的路线中,用到了一些关键反应,正是这些反应的精妙使用,使这个复杂的分子的合成得以实现。
1:钯催化的C-H功能化反应
反应广泛应用于吲哚等的芳基化和烯烃化反应中,它不需要以往所需的卤素等离去集团的参与,直接在C-H键上交叉耦合。
其普遍形式如Figure4。
Figure4
2:迈克尔加成
迈克尔加成反应必须在碱的催化下进行,常用的碱有:乙醇钠、
氢化钠、氨基钠和有机碱等。
根据反应物的反应活性来选择合适的碱,如果反应物双方均有较高的反应活性时,用较弱的碱也能使反应进行:迈克尔加成反应有一定的区域选择性。
加成时,烃基化的位置总是在取代较多的碳原子上。
通式如:Figure5。
Figure5
3:分子内氧化偶联反应:
该小组曾做过类似的分子内氧化偶联反应,并取得成功。
见:Figure6
Figure6
故以此为参考设计了(−)-Vincorine的合成思路,经过反复实验成果获得目标产物,且化合物构型单一。
化合物13为一混合物,因为酯基和吲哚部分的排斥作用,pathB不能反应,故反应只能按照pathA进行,因而得到了构型单一的化合物23。
见figure7
Figure7
参考文献:
Weiwei Zi, Weiqing Xie, and Dawei Ma*.Total Synthesis of Akuammiline Alkaloid (−)-Vincorine via Intramolecular Oxidative Coupling. J. Am. Chem. Soc. 2012, 134, 9126−9129。