紫杉醇的分离工艺
紫杉醇提取纯化工艺的研究进展

紫杉醇提取纯化工艺的研究进展摘要:紫杉醇以其独特的抗癌作用机制及显著的疗效,被认为是近十几年来天然抗癌药物研究领域最重大的发现。
由于紫杉醇在红豆杉树皮中含量极低,提取精制困难等原因,导致紫杉醇纯品价格昂贵。
因此完善紫杉醇的提取纯化工艺,降低生产成本,对保障人类健康都具有重要的意义。
本文对紫杉醇提取纯化工艺的研究进展进行了探讨。
关键词:紫杉醇;提取;纯化;工艺;研究紫杉醇(Paclitaxel,商品名Taxo1)是红豆杉属植物中独有的一种抗肿瘤天然药物,由于其独特的抗癌作用机制及显著的疗效,而被认为是近十几年来天然抗癌药物研究领域最重大的发现。
目前,国内外紫杉醇的商业化规模生产多以红豆杉植物为原料,通过一系列的分离纯化获得。
一、紫杉醇提取纯化工艺概述由于紫杉醇在红豆杉树皮中含量极低(<0.06%),提取精制困难等原因,导致紫杉醇纯品价格昂贵。
因此,完善紫杉醇的提取纯化工艺,降低生产成本,得到更加便宜的原料对保障人类健康都具有重要的意义。
表1 .紫杉醇提取纯化工艺中各个步骤所得纯度和回收率目前,工业生产中制备紫杉醇还局限于植物提取法。
因此研制出一套操作简单、提纯高、提取率高的紫杉醇规模制备技术无疑对改善紫杉醇原料药产业现状发挥积极作用。
一般来说,从红豆杉中提取紫杉醇的分离工艺,可以分为粗提和纯化两个阶段。
紫杉醇粗提阶段的目的,在于从原料液中尽可能多的提取目标化合物,所得物料再进行后续提纯直至获得纯品。
粗提过程可分为初级萃取和次级萃取,两种过程因所用溶剂不同而除去不同的杂质。
将粗提物进一步纯化是获得纯品紫杉醇的关键步骤,通常用色谱法完成。
在最后获得医用级纯品之前,色谱过程也必不可少。
用色谱法分离纯化紫杉醇时,柱色谱被用于紫杉醇的预分离和最终分离,而薄层色谱则被用于紫杉醇的检测和纯化。
目前,随着分离工艺的不断完善,除了柱色谱法以外,还有高效液相色谱、高速逆流色谱等。
不同工艺中各个步骤的纯度、回收率见表1。
紫杉醇提取分离方法研究

一、提取:
紫杉醇的提取方法,其效果直接影响紫杉醇分 离、纯化的难度及最终产量。
粗提多采用甲醇、乙醇、甲醇一二氯甲烷(1: 1),但所得浸膏中杂质量较高。研究发现,在 乙酸乙酯、乙醚等溶剂中,乙酸乙酯一丙酮(1: 1)混和溶剂提取紫杉醇效果最佳,浸膏中紫杉 醇含量高达甲醇提取的3倍。 常用的方法有冷浸、渗漉和索氏提取法,又有 人在提取过程中引入超声和微波技术 ,从而 大大缩短了提取时间。
通过Ⅱ-Ⅲ临床研究,紫杉醇主要适用于卵巢 癌和乳腺癌,对肺癌、大肠癌、黑色素瘤、头 颈部癌、淋巴瘤、脑瘤也都有一定疗效。
【分 子 式】 C47H51NO14 【结构式】
发现历史
1963年美国化学家瓦尼(M.C. Wani)和沃尔 (Monre E. Wall)首次从一种生长在美国西部大森林 中称谓太平洋杉(Pacific Yew)树皮和木材中分离到 了紫杉醇的粗提物。在筛选实验 中,Wani和 Wall发 现紫杉醇粗提物对离体培养的鼠肿瘤细胞有很高活性, 并开始分离这种活性成份。由于该活性成份在植物中 含量极低,直到1971年,他们才同杜克(Duke)大 学的化学教授姆克法尔(Andre T. McPhail)合作, 通过x-射线分析确定了该活性成份的化学结构——一 种四环二萜化合物,并把它命名为紫杉醇(taxol)。
虽然紫杉醇的全合成巳获成功,但就目前临床 供药而言,主要还是依靠从植物中提取。无论 是从天然的红豆杉植物还是从培养的植物细胞 组织中提取紫杉醇,都要涉及到如何能分离并 去掉与紫杉醇结构相似的其它紫杉烷类化合物。 由于这些紫杉烷类化合物在化学结构和极性等 方面都同紫杉醇极为相似,因此给分离工作带 来很大的困难。
通过文献调研可知,近年来,人们就紫杉醇的 提取和分离研究出了许多不同的方法,从而提 高了紫杉醇提取分离的效率,降低了其生产成 本。在本文中,根据已有的基础条件,结合本 学期的专业实验,给出了优化实验的基本思路。 这项研究已取得了长足的进展,一些新的技术 和手段不断应用于这一领域,并显示出诸多优 势和良好前景。相信,随着人们对这领域的进 一步研究,紫杉醇的提取和分离方法将会变得 更加完善。
从红豆杉树皮浸膏中提取紫杉醇初分离工艺的研究

粗品22.50 0.146 0.65 100 1
液-液萃取11.79 0.145 1.23 99.3 1.9
固相萃取2.27 0.146 6.44 100 9.9
硅胶柱层析0.98 0.144 14.63 98.9 22.5
氧化铝柱层析0.79 0.205 26.3 140.5 40.5
3结论
本工艺主要目的是简便除去紫杉醇类似物及其它生物碱,获得初步纯化的紫杉醇。过程
采用价格低谦的氧化铝为层析介质,省略了液-液萃取步骤,简化了分离工艺,提高了分离效
率,有利于工业放大。
氧化铝柱层析处理云南红豆杉针叶浸膏,紫杉醇的回收率大于140%,这使有限的红豆杉
资源能得以充分的利用。
采用优化的初分离工艺,从浸膏中有效地分离出了紫杉醇。样品经此初分离工艺处理后
存在许多问题。首先,紫杉醇来源匮乏,其主要存在于红豆杉树皮和针叶中,其次,紫杉醇
在植物中含量极低,大约为0.010%~0.013%,而紫杉醇与其它紫杉烷化合物在化学结构和极性
等方面又极为相似,要将它们完全分离困难很大。
关于紫杉醇提取分离方法,已有过不少的研究。其中以液-液萃取应用最为广泛,在文献
报道的每一种工艺中,几乎都采用过它。Willey等和Mattina等在测定样品中紫杉醇浓度时,
薄层层析1.05 0.146 13.91 99.7 21.4
-------------------------------------------------------------------------
2.7初分离工艺的提出
HPLC分析表明,粗品经过氧化铝柱层析处理后,极性比紫杉醇弱的物质基本被除
紫杉醇提取技术

紫杉醇提取技术
紫杉醇提取技术是一种从红豆杉树(Taxus brevifolia)中提取的一种抗肿瘤药物。
以下是简要的提取步骤:
1. 切片:将红豆杉树干切成薄片。
2. 干燥:将切好的树干片放入干燥设备中,保持适当的温度和湿度,以减少水分。
3. 粉碎:将干燥后的树干片研磨成粉末。
4. 提取:将粉末与有机溶剂(如甲醇或乙醇)混合,进行超声波辅助提取。
提取次数和时间根据实验条件而异。
5. 过滤:将提取液与固体废物分离,使用滤纸或其他过滤设备。
6. 浓缩:将过滤后的提取液进行旋转蒸发或减压浓缩,去除大部分有机溶剂。
7. 回收:利用柱层析或其他分离技术,从浓缩液中分离出紫杉醇。
8. 纯化:通过结晶、重结晶等方法对紫杉醇进行纯化,得到高纯度的紫杉醇。
需要注意的是,实际操作过程中可能涉及到的设备和条件会根据不同实验室和研究者的方法而有所不同。
此外,提取紫杉醇的过程中要严格遵守实验安全规程,因为紫杉醇和其代谢产物具有毒性。
紫杉醇的提取

1、紫杉醇的提取——溶剂萃取法溶剂萃取法常用于紫杉醇的粗提阶段。
紫杉醇的粗提阶段又可分为初级萃取和次级萃取。
在这两级萃取过程中,溶剂的选择对于精提产物的质量和过程经济性具有重要影响。
初级萃取和次级萃取一般采用的溶剂系统不同。
各个时期的研究者对这两个过程的溶剂系统的研究结果已有详细的总结。
最近、日本学者对紫杉醇提取的溶剂种类进行了详细的研究,结果表明:在乙酸乙酯、乙醚、乙腈、丙酮、甲醇、已烷、异丙醇、乙酸乙酯-甲醇、乙酸乙酯-二氯甲烷、乙酸乙酯-丙酮、乙酸乙酯-乙醚等溶剂中,以乙酸乙酯-丙酮(1:1)混和溶剂提取的效果最好,所得浸膏仅为植物干重的7.70%,紫杉醇的含量高达浸膏的0.084%,而用甲醇提取所得浸膏为植物干重的20.98%,紫杉醇的含量为浸膏的0.027%,尚需要多次抽提才能得到紫杉醇含量较高的浸膏。
现在看来利用乙酸乙酯-丙酮(1:1)一次便可以使紫杉醇提取量高于以往常用溶剂所能得到的量,这就为后序的分离纯化工作带来很大的方便,由于乙酸乙酯-丙酮(1:1)的价格与甲醇的价格相当,且可回收再利用,因此,这一提取方法的经济性较为合理。
在初级萃取过程中引入超声技术,可大大缩短初级萃取过程的时间。
例如Xu采用甲醇-二氯甲烷(95.5)作初级溶剂,所需萃取时间约为35-60min。
在溶剂系统不变的情况下,将原料与溶剂的混和物进行超声振荡,萃取达到平衡的时间缩短到仅5min,与此对比,Hoke 等人采用纯甲醇作为初级溶剂,无超声振荡,所需时间长达16-48h。
超声技术的引入,除可大大缩短萃取平衡时间外,还可以使初级萃取在低温下进行,从而避免了紫杉醇在高温下转化为其它物质而造成收率降低。
2、紫杉醇的提取——色谱法早期的色谱纯化紫杉醇工艺是采用多根硅胶层析柱串联的一种操作,因为硅胶对紫杉醇的不可逆吸附造成的损失很大,使得紫杉醇的收率很低,仅为0.004%左右。
近年来,随着色谱技术的进步,不断有新的色谱技术被引入到紫杉醇的分离提取过程中来。
紫杉醇的生产工艺

紫杉醇的生产工艺第七章7.4 紫杉醇的生产工艺7.4.1 概述紫杉醇(Paclitaxel,Taxol?,图1),最早是由美国化学家Wani 和Wall于1971年从太平洋红豆杉的树皮中提取到的一种具有抗肿瘤活性的物质。
它具有独特的抗癌机制,其作用位点为有丝分裂和细胞周期中至关重要的微管蛋白。
紫杉醇能促进微管蛋白聚合而形成稳定的微管,并抑制微管的解聚,从而抑制了细胞的有丝分裂,最终导致癌细胞的死亡。
紫杉醇1992年12月被美国FDA批准用于治疗晚期卵巢癌。
1994年,批准用于治疗转移性乳腺癌,1997年FDA批准使用紫杉醇治疗爱滋病关联的Kaposi's恶性肿瘤;1998年和1999年,FDA 又分别批准半合成紫杉醇与顺铂联合使用作为治疗晚期卵巢癌和非小细胞肺癌的一线用药。
紫杉醇是近几年国际公认的疗效确切的重要的抗肿瘤药物之一。
-五羟基-紫杉-11-烯-9-酮-4-乙酸酯-2-苯甲酸酯-10-乙酰基-13-α,13β,7α,4α,2β,20-环氧-1β紫杉醇具有复杂的化学结构,整个分子由三个主环构成的二萜核和一个苯基异丝氨酸侧链组成。
分子中有11个手性中心和多个取代基团。
分子式C47H51NO14,分子量853.92,元素百分比C:66.41,H:6.02,N:1.64,O:26.23。
化学名:5 [(2′R,3′S) -N-苯甲酰基-3′-苯基异丝氨酸酯]紫杉醇的来源最初以天然提取为主,主要是从由红豆杉属植物的树皮中分离得到。
红豆杉植物是生长极为缓慢的乔木或灌木,其树皮中紫杉醇的含量平均为万分之一点五,从中提取紫杉醇的收率大约为万分之一,这样制取1 kg紫杉醇就需树皮10吨,这种生产紫杉醇的方法严重破坏资源和环境。
目前,包括我国在内的许多国家都已经禁止或严格限制用这种方法来生产紫杉醇。
为解决紫杉醇的大量供应问题,人们曾探索通过组织和细胞培养、化学合成等方法制取紫杉醇。
其中,化学合成是人们首先想到的解决紫杉醇药源问题的一条途径。
从紫杉植物中提取紫杉醇的简化方法

从紫杉植物中提取紫杉醇的简化方法红豆杉Taxus又名紫杉,也称赤柏松,生于海拔1000~1200m处的山地,是世界上公认的濒临灭绝天然珍稀植物,从其根、皮、茎、叶中提取的紫杉醇taxol是目前世界上最有效的抗癌药物之一。
全球每年大约需要1500~2500kg紫杉醇,而1 kg树皮仅能提取50~100mg。
10-脱乙酰巴卡亭Ⅲ又称10-脱乙酰基浆果赤霉素Ⅲ,10-deacetylbaccatinⅢ,10-DABⅢ为有抑制肿瘤作用的紫杉烷二萜类化合物。
Bissery等报道,可利用10-DABⅢ合成具有比紫杉醇更高抗癌活性的多烯他赛docetaxel。
紫杉醇主要存在于树杆和树皮中,10-DABⅢ主要存在于树叶中,其含量大大高于紫杉醇的含量。
红豆杉是国家珍稀保护植物,生长缓慢,如果直接从红豆杉树皮中提取紫杉醇,不仅资源有限,而且不利于资源保护。
以10-DABⅢ为原料采用酶催化半合成工艺方法来制备紫杉醇,可大大简化合成过程,使紫杉醇骨架修饰所需步骤更少,操作更简单,提高了紫杉醇合成的选择性和生产率,进而为在更大规模上进行紫杉醇生产提供了技术支持,最终使紫杉醇的化学合成半合成的产业化有了实现的可能。
目前文献报道从各种紫杉植物中提取紫杉醇的方法,均需经过繁冗的分离过程。
本实验采用了一种适合于以各种紫杉植物树叶或树枝做原料,通过极性梯度溶剂萃取的方法逐步脱除大量不相干杂质,得到合成紫杉醇的前体10-DABⅢ的方法,然后通过反相层析柱加成,即可获得抗癌活性成分紫杉醇;材料与方法1 材料与仪器南方红豆杉Taxus mairei枝叶取自浙江宁海红豆杉种植基地,8年树龄。
10-DABⅢ对照品为Sigma公司产品,纯度98%;所用甲醇;乙醇、乙酸乙酯、乙酸丁酯、二氯甲烷、氯仿、正己烷、石油醚、乙腈等均为分析纯试剂。
JJ一1精密增力电动搅拌仪,江苏金坛市江南仪器厂;SENCO R一201旋转蒸发仪,上海申顺生物科技有限公司;玻璃硅胶柱为2cm×40cm,杭州常盛科教器具厂;UV一2802PC/PCS型分光光度计,UNICO上海仪器有限公司;Sigma一3K18低温离心机4℃,转速18000rmin;LabAlliance高效液相色谱仪美国SSI公司。
制药工艺学:紫杉醇生产工艺

解决办法(二)
– 生物方法 • 组织和细胞培养
• 微生物发酵
优点:1、摆脱自然因素,可长期稳定 生长
2、适应市场、方便调节 3、成分简单,有利于分离纯化 4、成本低、生长周期短 5、为半合成提供原料 6、有望工业化生产
• 生物合成
缺点:1、产量低、不稳定 2、工业化放大研究
研究阶段
红豆杉生物合成途径基本明确
将天然产物蒎烯(pinene)的氧化物verhenon(化合 物27)为起始原料,化合物27含有A环结构且可提供 紫杉醇母核骨架中20个碳中的1O个碳原子。经过若 干步反应将27转化为化合物31,再将化合物31转化 为化合物32,完成了AB环的构建。然后通过在C.3 位上设计的反应以及氧化反应得到化合物34、35, 进一步通过醇醛缩合得到化合物36、37,这样就完 成了c环的构建。再通过C.5位溴代、c4和C-20臭 氧化完成了含氧D环的构建(化合物38),最后得到了 Baccatin III(2),再完成C.10乙酰化及与侧链的 加成反应等,最终完成了紫杉醇的全合成(图7)
缺点:1、紫杉醇含量低 生长缓慢
2、提取工艺复杂
紫杉醇生产工艺
2)生物工程方法生产紫杉醇
Phyton克服细胞培养过程的各种 技术障碍,突破细胞培养生产紫杉 醇技术,生产过程得到FDA认证;
2002 年 7 月 10 日 --Phyton, Inc. 与 Bristol-Myers Squibb 签署 长期合作协议用细胞培养生产紫杉 醇;
verhenon
五、 Kuwajima全合成路线 (1998)
日本东京科技研究院Isao Kuwajima教授领导 的紫杉醇全合成研究小组采用了A+c—Ac—ABc —ABcD的会聚法合成路线 ∞ ,其类似于Nico— laou法和Danishefsky法。由
抗癌药物紫杉醇的天然提取与分离技术

反渗透又称逆渗透,一种以 压力差为推动力,从溶液中 分离出溶剂的膜分离操作。 对膜一侧的料液施加压力, 当压力超过它的渗透压时, 溶剂会逆着自然渗透的方向 作反向渗透。从而在膜的低 压侧得到透过的溶剂,即渗 透液;高压侧得到浓缩的溶 液,即浓缩液。
早期的色谱纯化紫杉醇工艺是采用多根硅胶层析 柱串联的一种操作,但是因为硅胶对紫杉醇的不 可逆吸附造成了巨大的损失,使得紫杉醇的回收 率很低,仅为0.004%左右。近年来,随着色谱技 术的进步,不断有新的色谱技术被引入到紫杉醇 的分离提取过程中来。除了高速液相色谱法 (HPLC,其中包括正相-HPLC、反相-HPLC)外, 还有薄层色谱法(TLC)、胶束电动毛细管色谱法 (MECC)和高速逆流色谱法(HSCCC)等。
经实验后的HPLC色谱分析可 以看出SFE法可以高效的分离 出紫杉醇,且效率高于传统的 使用各种有机溶剂的提取方法, 具有高效,周期短,废渣无残 留,能最大程度保证各组分原 有活性,不产生三废,绿色环 保等诸多优点。
实 验 结 果
江苏无锡有中国 最大的紫杉醇培 植基地 人工培植的紫杉醇幼苗
下面以复旦大学的实验设备为例,讲解CO2超临界流体萃取的原理及其设备。
CO2和修饰剂分别由CO2泵和修饰剂泵 打入各泵的锥形腔体中,再经流体混合 其按设定的比例混合后,流入萃取器中 的集流腔。在达到萃取设定的温度和压 力后,萃取器开始萃取。动态萃取时, 超临界CO2流体经限流管流入甲醇收集 瓶后减压排放,流体带出的萃取所得物 则溶于收集液中。
超滤膜,是一种Leabharlann 径规格一致, 额定孔径范围为0.001-0.02微米的 微孔过滤膜。在膜的一侧施以适 当压力,就能筛出小于孔径的溶 质分子,以分离分子量大于500道 尔顿、粒径大于2~20纳米的颗粒。 超滤膜是最早开发的高分子分离 膜之一,在60年代超滤装臵就实 现了工业化。
紫杉醇的提取工艺研究资料讲解

紫杉醇的提取工艺研究紫杉醇提取纯化方法的研究进展紫杉醇是最早从红豆杉属植物中分离出来的三环二菇类化合物,是继阿霉素和顺铂之后最热点的抗癌新药。
紫杉醇具有复杂的化学结构,分子由3个主环构成二菇核,分子中有11个手性中心和多个取代基团,母环部分是一个复杂的四环体系,有许多功能基团和立体化学特征。
分子式C47H51NO14,分子量853.92。
同位素示踪表明, 紫杉醇只结合到聚合的微管上, 不与未聚合的微管蛋白二聚体反应。
细胞接触紫杉醇后会在细胞内积累大量的微管,这些微管的积累干扰了细胞的各种功能,特别是使细胞分裂停止于有丝分裂期,阻断了细胞的正常分裂。
通过Ⅱ-Ⅲ临床研究,紫杉醇主要适用于卵巢癌和乳腺癌,对肺癌、大肠癌、黑色素瘤、头颈部癌、淋巴瘤、脑瘤也都有一定疗效。
紫杉醇属于有丝分裂抑制剂,它的独特机制在于可以诱导和促进微管蛋白聚合,促进微管装配及阻止微管的生理解聚,由此抑制癌细胞纺锤体的形成,阻止有丝分裂的完成,使其停留在G2期和M期直至死亡,从而起到抗癌的作用。
迄今为止紫杉醇是唯一促进微管聚合的新型抗癌药。
这一新的发现引起了各国医药界的极大兴趣。
现在已有包括我国在内的十多个国家批准了紫杉醇类药物的正式生产。
目前有关紫杉醇研究的几个主要问题是:紫杉醇的提取;紫杉醇的人工合成;紫杉醇的临床应用(水不溶性问题的解决);紫杉醇的构效关系;紫杉醇的抗癌机理。
紫杉醇的抗癌机理1971年,Wani等报道了紫杉醇在一些实验体系中具有抗癌活性。
1978年,Schiff等发现紫杉醇在极低的浓度下(0.25μM)可以完全抑制Hela细胞的分裂,而且在对细胞4小时的培养过程中,对DNA、RNA和蛋白质的合成没有明显影响。
Hela细胞在与紫杉醇共同温育20小时后被阻断在G2后期和M期。
1979年Schiff等用浊度法进行了研究,发现紫杉醇能缩短微管在体外的聚合时间,使平衡向微管聚合方向移动,从而减小微管聚合临界浓度。
在有GTP时,紫杉醇可以和PC-tubulin结合,计量比为1:1。
紫杉醇的分离与纯化

摘要目的:探索红豆杉中紫杉醇的提取纯化工艺。
方法:将新鲜的红豆杉树皮干燥后用甲醇浸泡,陶瓷膜进行固液分离,纳滤膜浓缩,再用大孔树脂HZ818层析,洗脱液浓缩结晶,再活性炭脱色后甲醇重结晶,再硅胶正向层析,洗脱液浓缩后正已烷结晶,再真空干燥得成品。
结论:按本方法从红豆杉中提取的紫杉醇纯度为97.5%,收率为十万分之八。
关键词红豆杉,紫杉醇,提取纯化,树脂,硅胶目录一、紫杉醇目前的一些分离纯化方法。
................................................. 错误!未定义书签。
(一)液相萃取。
.................................................................................... 错误!未定义书签。
(二)固相萃取法。
............................................................................... 错误!未定义书签。
(三)树脂层析法。
............................................................................... 错误!未定义书签。
(四)活性炭脱色。
............................................................................... 错误!未定义书签。
(五)硅胶正向层析。
.......................................................................... 错误!未定义书签。
(六)结晶纯化。
.................................................................................... 错误!未定义书签。
紫杉醇提取

紫杉醇最新提取工艺
摘要:紫杉醇为著名的抗肿瘤天然产物,来源有限,化学合成是制备紫杉醇的可能途径之一。
本文主要综述了紫杉醇新的提炼方法主要是在天然提取分离、细胞培养、有机合成、紫杉醇的全合成基础上取得重要成就,从而改进得出的最新提取工艺路线。
其最大的改进是少用了很多有机溶剂,为分离与纯化提供便捷。
关键词:紫杉醇;天然提取分离;细胞培养;合成
1 简介
1.1 发展史
从植物中寻找有效抗癌成分的历史至少可以追溯到公元前1500年关于纸莎草(papryrus)的研究,但真正对天然产物进行系统的科学研究却是从本世纪 50年代才开始的。
50年代,Hartuell及其同事着重研究了抗癌制剂鬼臼毒素(PgdoPhyllotoxin)及其衍生物的应用。
同时一系列寻找天然抗肿瘤成分的研究导致了象长春花碱(Vinblastlnc)、长春新碱(Vincristine)及秋水仙碱(Colchicine)等一系列具有代表性的抗癌药物的产生。
1971年,Wanj及其同要从欧洲短叶红豆杉中分离得到一种具有细胞毒性的新化合物,命名为紫杉醇(Paclltaxel,现已商品化,其注册名为Taxol),药理实验证明,它具有广谱抗癌作用,但由于其天然含量极低,故而在当时并未引起人们的注意,直到1977年,HorwitZ博士发现其抗癌机理在于能够与微管蛋白结合,促进微管蛋白聚合装配成微管二聚体,从而抑制细胞中微管的正常生理解聚,使细胞有丝分裂停止在G2期及M期,阻止了癌细胞的快速繁殖,这一机理与上述纺锤体毒性的抗癌药物(如长春新碱与秋水仙碱)的作用机制恰好相反,从而引起随后20多年关于该属植物的广泛研究。
1.2 自然资源。
紫杉醇提取工艺研究

紫杉醇提取工艺研究1.紫杉醇简介紫杉醇(C47H51NO14)是红豆杉属植物中的一种复杂的次生代谢产物,是目前所了解的惟一一种可以促进微管聚合和稳定已聚合微管的药物。
同位素示踪表明, 紫杉醇只结合到聚合的微管上, 不与未聚合的微管蛋白二聚体反应。
细胞接触紫杉醇后会在细胞内积累大量的微管,这些微管的积累干扰了细胞的各种功能,特别是使细胞分裂停止于有丝分裂期,阻断了细胞的正常分裂。
通过Ⅱ-Ⅲ临床研究,紫杉醇主要适用于卵巢癌和乳腺癌,对肺癌、淋巴瘤、脑瘤等也都有一定疗效[1]。
紫杉醇是一种无臭,无味的白色结晶体粉末,熔点为213-216℃,不溶于水,易溶于氯仿、丙酮等有机溶剂,主要由红豆杉的树皮中提取。
由于紫杉醇的质量分数极低(仅为干质量的0.01%左右),杂质多,为后续的分离纯化带来困难,因此,紫杉醇初分离除杂过程是分离纯化工艺的关键步骤。
国内外提取紫杉醇的方法主要有溶剂浸提法、索氏提取、固相提取法等。
这些方法能耗少、紫杉醇回收率高、易于操作,但工艺周期长、溶剂用量大、提取选择性差,且紫杉醇在长时间的受热过程中易引起结构变化[2]。
因此,近年来国内外科研人员纷纷研究开发快速高效的紫杉醇提取方法,研发的重点集中在沉淀法、超声波、柱层析、超临界流体萃取等新型方法。
2.紫杉醇提取的工艺流程红豆杉枝叶、树皮、树枝——干燥与粉碎——有机溶剂提取——浸膏——固液萃取——液液萃取——己烷沉淀——硅胶柱层析——结晶——TLC 检测——高效液相色谱检测3.常见提取操作介绍3.1有机溶剂提取紫杉醇的粗提阶段常用有机溶剂进行提取。
李春斌等人[3]研究发现,在氯仿、甲醇、乙醇、V(甲醇):V(乙醇)=1:1、V(氯仿):V(丙酮)=1:1和V(乙酸乙酯):V(丙酮)=1;1溶剂中,以V(乙酸乙酯):V(丙酮)=1:1的提取效果最好,基本无毒且可回收再利用,因此有望用于紫杉醇的工业化生产中。
通过综合对比普通浸提、冷浸法、渗漉法和索氏回流法,发现采用索氏提取方法提取其浸膏得率达20.7%。
抗癌药物紫杉醇的提取与分离纯化技术

3:A 水分的乙醇溶液浸提未经干燥的新鲜枝叶 $ 然后再加入占浸
提液体积 4AB3A 的活性 炭 处 理 浸 提 液 $ 最 后 经 过 一 系 列 的 硅 胶 柱层析就可以得到纯度为 C::A 的紫杉醇 # 这一方法具有一定的 创新性 $ 其优越之处为可免去干燥枝 叶 这 一 步 骤 $ 且 去 除 了 色 素 % 脂类等杂质 # 另外 $ 紫杉醇除了以游离的状态存 在 于 植 物 体 内 外 $ 还 可 以 与糖基结合 $ 存在于植物体内 # 由于紫杉醇的糖基化可以使其
./0123 ’/14 5678 9::;
豆 杉 !!"#$% &’()*+,-*" " 树 皮 中 的 紫 杉 醇 大 部 分 都 能 得 到 有 效 的 提取 $ 回收率达 \3A 以上 $ 对紫杉醇的选择性也比传统 的 单 纯 乙 醇溶剂好得多 # 此外 $.6HO6H6 等 D9C@利用超临界的 ’9) 以及 ’9) 与乙醇 的 混 合物 $ 从短叶红豆杉树皮中提取紫杉醇 # 由于超临界流体萃取技 术对仪器设备要求较高 $ 因此限制了其应 用 $ 但 国 内 外 学 者 正 积 极开发 $ 且已证实 S)9M%^" 是一种很有前途的分离萃取方法 #
# 紫杉醇以其独特的抗癌机理得 到 世 界 各 国 研 究 人 员 的 重
紫杉醇的分离工艺
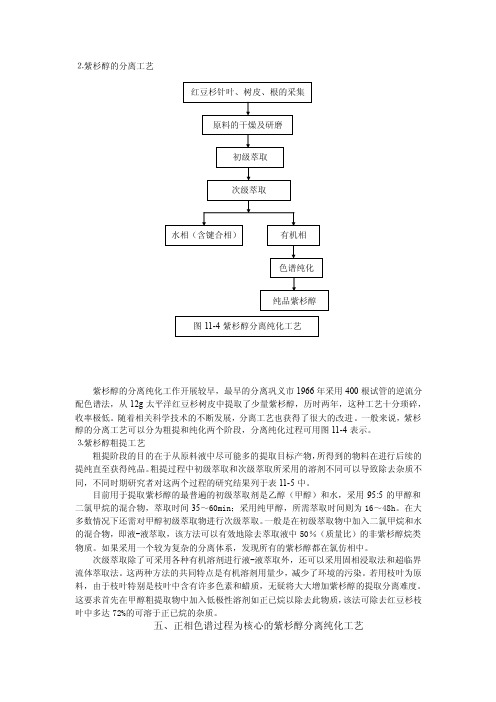
⒉紫杉醇的分离工艺红豆杉针叶、树皮、根的采集原料的干燥及研磨初级萃取次级萃取水相(含键合相)有机相色谱纯化纯品紫杉醇图11-4紫杉醇分离纯化工艺紫杉醇的分离纯化工作开展较早,最早的分离巩义市1966年采用400根试管的逆流分配色谱法,从12g太平洋红豆杉树皮中提取了少量紫杉醇,历时两年,这种工艺十分琐碎,收率极低。
随着相关科学技术的不断发展,分离工艺也获得了很大的改进。
一般来说,紫杉醇的分离工艺可以分为粗提和纯化两个阶段,分离纯化过程可用图11-4表示。
⒊紫杉醇粗提工艺粗提阶段的目的在于从原料液中尽可能多的提取目标产物,所得到的物料在进行后续的提纯直至获得纯品。
粗提过程中初级萃取和次级萃取所采用的溶剂不同可以导致除去杂质不同,不同时期研究者对这两个过程的研究结果列于表11-5中。
目前用于提取紫杉醇的最普遍的初级萃取剂是乙醇(甲醇)和水,采用95:5的甲醇和二氯甲烷的混合物,萃取时间35~60min;采用纯甲醇,所需萃取时间则为16~48h。
在大多数情况下还需对甲醇初级萃取物进行次级萃取。
一般是在初级萃取物中加入二氯甲烷和水的混合物,即液-液萃取,该方法可以有效地除去萃取液中50%(质量比)的非紫杉醇烷类物质。
如果采用一个较为复杂的分离体系,发现所有的紫杉醇都在氯仿相中。
次级萃取除了可采用各种有机溶剂进行液-液萃取外,还可以采用固相浸取法和超临界流体萃取法。
这两种方法的共同特点是有机溶剂用量少,减少了环境的污染。
若用枝叶为原料,由于枝叶特别是枝叶中含有许多色素和蜡质,无疑将大大增加紫杉醇的提取分离难度。
这要求首先在甲醇粗提取物中加入低极性溶剂如正已烷以除去此物质,该法可除去红豆杉枝叶中多达72%的可溶于正已烷的杂质。
五、正相色谱过程为核心的紫杉醇分离纯化工艺正相色谱是紫杉醇分离纯化工艺中普遍采用的方法,在早期紫杉醇分离纯化的研究中占有主导地位,至今仍在广泛应用。
在紫杉醇分离纯化过程中,正相色谱的突出优点是固定相价格廉价,用普通的硅胶即可,而且洗脱用流动相多为挥发性很强的有机溶剂,溶剂回收简单、能耗低。
紫杉醇的提取

10-去乙酰紫杉醇
DMAP,乙醇 乙酸酐酰化
粗产品
制备型薄层层析分离
二甲胺 处理
溶于乙醇的紫杉醇
重结晶
紫杉醇纯品产率88%
3.21 方法评估
1)来源没有10-去乙酰巴卡亭广 2)合成产率比较高没有上一种方法高
4 紫杉醇的提取工艺和评估
液液萃取法 固液萃取法 超临界流体萃取法 柱切换技术法
4.1 液液萃取法
上230~400目硅胶-60(Merck)色谱柱 有机浓缩液 二氯甲烷-甲醇(v∶v=99∶1)洗脱
化合物1.03 g,收率:86%.
3.15 合成紫衫醇
溶于无水甲苯70 mL,搅拌下加入侧链1.3 g, 7-三乙基硅烷巴卡亭1g
再加入DCC 470 mg,反应5分钟
加入DMAP 350 mg,搅拌4 min,加 热至75℃,反应10 h.
4.2 方法评估
简单易行但所获产物杂质较多固液萃取 法具有省时省溶剂萃取效率高有良好的 选择性的特点 ,但同时也可以看出,所 消耗的提取液比较多,得率比不高的缺点
Ph Co
Ac Ac
Ac
SiEt3 SiEt3
CO2CH2CCl3
3.12 实验理论流程图HOO NhomakorabeaOH
HO
O
HO
O
SiEt3
HO
O
O AcO HO
SiEt3 O
OH OBz O O
10-去乙酰卡巴亭
O AcO
OH OBz O O
7-三乙基硅烷-10-去乙酰卡巴亭
SiEt3
OH OBz O O
NHBz O
红豆杉树皮粉末2g 95%乙醇20ml,超声萃取30min
滤渣 重复上步操作
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
⒉紫杉醇的分离工艺红豆杉针叶、树皮、根的采集原料的干燥及研磨初级萃取次级萃取水相(含键合相)有机相色谱纯化纯品紫杉醇图11-4紫杉醇分离纯化工艺紫杉醇的分离纯化工作开展较早,最早的分离巩义市1966年采用400根试管的逆流分配色谱法,从12g太平洋红豆杉树皮中提取了少量紫杉醇,历时两年,这种工艺十分琐碎,收率极低。
随着相关科学技术的不断发展,分离工艺也获得了很大的改进。
一般来说,紫杉醇的分离工艺可以分为粗提和纯化两个阶段,分离纯化过程可用图11-4表示。
⒊紫杉醇粗提工艺粗提阶段的目的在于从原料液中尽可能多的提取目标产物,所得到的物料在进行后续的提纯直至获得纯品。
粗提过程中初级萃取和次级萃取所采用的溶剂不同可以导致除去杂质不同,不同时期研究者对这两个过程的研究结果列于表11-5中。
目前用于提取紫杉醇的最普遍的初级萃取剂是乙醇(甲醇)和水,采用95:5的甲醇和二氯甲烷的混合物,萃取时间35~60min;采用纯甲醇,所需萃取时间则为16~48h。
在大多数情况下还需对甲醇初级萃取物进行次级萃取。
一般是在初级萃取物中加入二氯甲烷和水的混合物,即液-液萃取,该方法可以有效地除去萃取液中50%(质量比)的非紫杉醇烷类物质。
如果采用一个较为复杂的分离体系,发现所有的紫杉醇都在氯仿相中。
次级萃取除了可采用各种有机溶剂进行液-液萃取外,还可以采用固相浸取法和超临界流体萃取法。
这两种方法的共同特点是有机溶剂用量少,减少了环境的污染。
若用枝叶为原料,由于枝叶特别是枝叶中含有许多色素和蜡质,无疑将大大增加紫杉醇的提取分离难度。
这要求首先在甲醇粗提取物中加入低极性溶剂如正已烷以除去此物质,该法可除去红豆杉枝叶中多达72%的可溶于正已烷的杂质。
五、正相色谱过程为核心的紫杉醇分离纯化工艺正相色谱是紫杉醇分离纯化工艺中普遍采用的方法,在早期紫杉醇分离纯化的研究中占有主导地位,至今仍在广泛应用。
在紫杉醇分离纯化过程中,正相色谱的突出优点是固定相价格廉价,用普通的硅胶即可,而且洗脱用流动相多为挥发性很强的有机溶剂,溶剂回收简单、能耗低。
鉴于正相色谱的这些优点,综合一些已有的工艺过程,针对我国的红豆杉资源给出了一个分离纯化紫杉醇的工艺。
该生产工艺突破已有专利的保护,具有明显的特色。
⒈试验物系用甲醇、乙醇或二氯甲烷-甲醇(1:1)对云南红豆杉树皮浸提得到起始物浸提膏粉,溶剂回收,所得到浸膏用二氯甲烷-水或氯仿-水进行分配萃取,最后处理有机相,得到含紫杉醇大约1%的浸膏粉,将试验用的浸膏粉用HPLC检测,分析谱图发现有紫杉醇物质峰,溶解浸膏粉的溶剂为甲醇(色谱纯)。
因有一些杂质在227nm波长处没有被吸收,造成谱图中紫杉醇的面积百分比分数高于实际情况,外标法测定浸膏粉中紫杉醇的含量为1.02%。
作为工业放大的需要,实验中涉及的有机溶剂均匀为市售的工业级,使用时蒸馏除去不挥发残留。
试验用硅胶300-400目,拌样用硅藻土采用Ceite公司产品,150目;色谱柱是订做的玻璃柱,柱参数在涉及时作具体说明。
分析检测用TLC法结合HPLC法,TLC法采用E.MERCK公司生产的铝制60F254硅胶板,班上硅胶厚度0.2mm,HPLC流动相为甲醇-水(65:35)或乙腈-水(47:53),流速1.0mL/min,双波长检测,检测波长227nm、233nm。
⒉样品预处理浸膏样品中含有大量杂质,它们在液相色谱固定向上的竞争性吸附或不可逆吸附会导致固定相用量增加,目标产物的分离度降低,分离效率低下等。
因此,在分离纯化的早期阶段就使用色谱过程是不经济的。
对样品选择合适的方法进行预处理,提高目标产物的浓度,在提高效率和操作控制方面都会带来方便。
⑴固-液浸取浸膏用品来源于树皮、枝叶或植株体的其他部位,除含量很低的目标产物紫杉醇外,还含有各种类型的极性和非极性杂质。
在获得浸膏的粗提过程中,以去除大部分杂质,但仍有相当部分滞留在浸膏中。
这部分杂志中,一些低极性或非极性类杂质如焦油等,会粘附在作为色谱固定向的硅胶上,是硅胶失去吸附力,而且随流动相运动被洗脱下来,会给洗脱馏分的干燥带来困难。
对于这类杂质,可用一些低极性的醚类和烷类有机溶剂进行固-液萃取出去。
从石油醚(60%~90%)对浸膏进行固-液萃取的结果可以看出,随石油醚用量的增加,萃取的杂质百分率也在增加,但当石油醚的用量达到原始浸膏的5倍(mL/g),被浸出的萃取的杂质百分数则不再有明显变化。
因此,和原始浸膏的质量相比,5倍体积的石油醚可以认为是一个优化的值。
对浸膏进行匀浆搅拌与否,杂质浸出的百分率差别很大,搅拌的效果显著。
但是,搅拌后沉淀颗粒过细,过滤有困难,需预先在过滤设备上预涂一层硅藻土或其他类型的助滤剂帮助过滤。
固-液浸取中的有机溶剂也可用正已烷或环已烷代替,具有相似的效果。
综合以上研究,可得固-液浸取出去非极性杂质的优化条件是:匀浆搅拌,按体积质量比5倍体积的石油醚可除去10%~12%的杂质。
固-液浸取过程目标产物紫杉醇的收率接近100%。
⑵碱洗将经石油醚固液浸取后的浸膏样品用一定浓度的NaOH溶液洗涤,是本工艺的独到创新之处。
由于植株体中含有大量鞣质酸等其他一些酸类杂质,和碱溶液发生一种类似皂化的反应,生成一些易溶于的物质,可以用水洗涤除去。
用碱性溶液洗涤去除杂质在制作药物中间体环孢素时使用的非常普遍,但在紫杉醇分离纯化过程中的应用至今还没有报道,原因在于紫杉醇在碱性环境中极其不稳定,很开裂解成巴卡亭III和其他一些物质。
用乙酸乙酯作溶剂,在用1mol/L的NaOH溶液裂解紫杉醇制备少量巴卡亭III的过程中发现,紫杉醇没有发生变化(数据未列出),表明用乙酸乙酯作溶剂的水解反应能够屏蔽紫杉醇的裂解反应。
这一结论使浸膏样品的碱洗过程有了理论基础。
不同浓度NaOH溶液对经石油醚固液浸取后浸膏样品进行碱洗的结果,表明浸膏用10倍体积(mL/g)的乙酸乙酯溶解,加入等体积的碱溶液,结果显示,NaOH浓度增加时,被去除杂质的百分率也增加,但当NaOH溶液浓度增加到1mol/L以上后,再增加则无明显影响。
而且,由于酯的水解,溶液过高是有机溶剂消耗量变大,因此,1mol/L较为合适。
同样,碱溶液用量也有类似影响,根据实验结果,以等体积较好。
将碱洗前后浸膏样品的图谱相比较几乎没有变化,可知碱洗主要除去一些在227nm或233nm处没有吸收峰的杂质,而紫杉醇则不受影响。
碱洗过程具体步骤为:用10倍体积(mL/g)乙酸乙酯溶解适量浸膏;加入等体积1mol/L 的NaOH溶液,搅拌10~20min;分相后有机相用0.5倍NaOH溶液体积的蒸馏水洗涤两次。
其中第二次洗涤常伴有乳化的现象,需加入适量盐帮助分相或直接用盐水洗涤,过程中乙酸乙酯因水解大约损失一半;洗涤后有机相用无水硫酸钠进行脱水,根据后面工序的需要减压浓缩至25%~35%有机相体积。
这一步骤可取出最高达60%的杂质(以石油醚固液浸取后的干浸膏量为基准),因乙酸乙酯少量溶于水是紫杉醇有所损失,收率在97%左右。
⑶己烷沉淀将碱洗后得到的浸膏样品溶解在一种极性较强的溶剂中,向其中缓缓滴入己烷(正己烷或环己烷)溶剂,可以使一些石油醚不能萃取除去的低极性杂质留在母液中,而将紫杉醇等目标产物沉淀下来。
溶解浸膏样品的溶剂可以是丙酮、二氯甲烷或乙酸乙酯,为方便工艺的前后衔接,以乙酸乙酯为好。
不同体积比例的正己烷加入碱洗后浸膏样品的乙酸乙酯溶液中的沉淀结果显示,当加入己烷的量较小时,沉淀的量很少,大部分留在母液中,紫杉醇损失较大;在加入己烷体积和溶液体积比在6以上时,沉淀量几乎不再变化,但HPLC 测定发现,体积比在10以下时,紫杉醇不能沉淀完全,仍有少量留在母液当中。
因此,己烷沉淀以加入10倍浸膏样品的乙酸乙酯溶液为好,加入量再大,不但对除去杂质没有多少帮助,还会造成己烷使用量过大,为溶剂回收造成不必要麻烦。
这一步骤也和前一工序的有机相浓缩有关。
浓缩的比例太小会影响去除杂质的量,太大又会造成溶剂用量过大的相同问题。
实验证实,有机相以浓缩到碱洗后乙酸乙酯相体积的25%-30%为佳。
(4)一次层析浸膏样品经己烷沉淀后,目标产物紫杉醇的含量已经接近5%,除一些难以除去的色素外,杂质含量主要是一些紫杉烷类化合物,其中不乏价值很高的紫杉醇类似物如三尖杉宁碱、10-去乙酰基紫杉醇、巴卡亭Ⅲ等,同样需要分离纯化。
因为目标产物的含量还很低,尤其伴随有结构性质相似的类似物三尖杉宁碱一次层析将紫杉醇纯化到一定的纯度难度很大,而且也不经济。
比较合理的方法是,先用一个层析过程粗分一下,使富集的馏分中目标产物有较高的含量,为进一步分离纯化奠定基础。
因为这步层析仅是粗分,对色谱柱高径比的要求不是很大,硅胶用量也不必太高,而且可以使用较高的洗脱速度。
从常州市第二干燥设备厂订做的GXCX50型,能实现柱切换技术的中压高效液相层析设备,通过阀门的开启和关闭进行柱切换;层析段有效床层容量6L,内径100mm,柱高径比7:1;承载样品的预柱容量3L,高径比3:1,设计工作压力为2.5MPa。
①洗脱体系:洗脱过程中可以通过采用极性较强的溶剂和极性较低的溶剂混合调整极性来改变洗脱强度。
因为浸膏样品中紫杉烷类物质种类多,极性差别很大,以梯度洗脱较为合适。
但连续梯度洗脱较为繁琐,考虑到实际操作的方便,采用分段梯度(Step-wise Gradient)操作最为适宜。
洗脱中作为流动相的溶剂可以选择己烷-丙酮、二氯甲烷-丙酮、己烷-乙酸乙酯、二氯甲烷-乙酸乙酯或氯仿-甲醇体系等,但从溶剂回收难易、洗脱强度大小、固定相循环使用和溶剂毒性等方面综合考虑,流动相以己烷-丙酮为优化选择。
②样品制备经己烷沉淀后的浸膏样品在己烷-丙酮作为流动相的洗脱剂中溶解度不是很大,以固态上样法较为合适。
将样品按1:10(g/ml)的比例溶解在丙酮中,然后加入5倍两量(以浸膏样品质量为基准)的硅藻土,拌匀后自然风干,如果量较大,可以将该混合物在40℃下用旋转蒸发仪蒸干,得到固态样品。
用时加到色谱柱上部。
洗脱之前,可在样品上端覆盖一层粗砂或玻璃珠,以防止样品界面被破坏。
③洗脱模式:洗脱模式即分段梯度洗脱过程中的梯度构成,参量标准是固定相床层的有效体积,可先用干柱色谱法进行初步估计。
干柱色谱法师将干的吸附剂装入色谱柱中。
将待分离的样品配成浓溶液或吸附于少量填料上,然后上样。
当洗脱液依靠毛细作用从柱上端流下接近色谱柱底部时,停止洗脱,将吸附剂分段从色带位置挖出或切开,用适当的溶剂洗出。
这种方法溶剂消耗量少,所需时间短,可根据目标产物在柱中的位置确定洗脱模式。
色谱柱是一根容量为10ml的小针管,内径10mm,高接近7cm,上样量0.1g,溶于1ml丙酮,拌入0.5g硅藻土,风干,柱内硅胶分段洗脱后用TCL分析确定色斑位置。