高等有机化学——反应机理(4)
《高等有机化学—反应和机理》(Bernard Miller)笔记

●Woodward-Hoffmann规则一:4n电子的热电环化反应,如果按照顺旋方式进行是允许的;4n+2电子的热电环化反应,如果按照对旋的方式进行时允许的。
●顺旋:多烯烃的末端碳原子或环烯烃的饱和碳原子,以相同方向(同为顺时针或同为逆时针)旋转成键或断键,这种方式称为顺旋。
顺旋:多烯烃的末端碳原子或环烯烃的饱和碳原子,以不相同方向旋转成键或断键,这种方式称为对旋。
●最高已占分子轨道(HOMO)在4n+2的体系中是对称的;最低未占分子轨道(LUMO)在4n+2的体系中是反对称的。
●前线轨道理论:忽略较低的能级轨道,只考虑HOMO。
前线轨道理论能简单、形象化,但是理论上不完善,在理论上应该有更精确的处理方法。
在电环化反应中,对旋是允许的,顺旋是禁阻的。
●轨道对称性守恒:反应物中的每个轨道的对称性,在反应后对称性保持不变。
●用相关图法处理电环化反应遵循轨道对称性守恒。
●相关图法处理4n+2体系的热环化反应(对旋):以1,3,5-己三烯为例:(1)形成6个分子轨道(2)用能量最低的形成键,和的对称性相同,都是镜面对称的。
(3)是由6个原子轨道组成,键是2个原子轨道组成,故转化为时,可以想象其中有4个原子轨道的系数降低为0。
(4) 1,3,5-己三烯的,不能转化为1,3环己二烯的,因为前者的的对称性是镜面反对称,后者的的对称性是镜面对称,对称性不匹配。
故1,3,5-己三烯的事转化为1,3环己二烯的,同理1,3,5-己三烯的事转化为1,3环己二烯的(5)能量分配很合理,故反应是允许的。
用相关图法处理4n体系的热环化反应(对旋):以1,3-丁二烯为例:(1)用能量最低的形成键(2)用1,3-丁二烯的形成环丁烯的;用1,3-丁二烯的形成环丁烯的。
理由同4n+2体系,因为对称性不守恒。
(3) 1,3-丁二烯的上有2个电子,而要形成的环丁烯的电子在上。
但是1,3-丁二烯要转化为环丁烯的,如果发生这样的转化,就会形成能量很高的环丁烯的激发态。
高级有机化学反应机理的讲解

高级有机化学反应机理的讲解有机化学是研究碳元素及其化合物的学科,是化学中的一个重要分支。
在有机化学中,反应机理是理解和解释化学反应过程的关键。
本文将讲解一些高级有机化学反应的机理,帮助读者更好地理解有机反应的本质。
一、亲电取代反应机理亲电取代反应是有机化学中最基本的反应类型之一。
它是指一个亲电试剂攻击一个亲核试剂中的亲电位点,形成一个新的化学键。
亲电取代反应的机理可以分为三步:亲电试剂的攻击、亲电位点的断裂和新键的形成。
以酯水解为例,酯分子中的羰基碳是亲电位点,而水分子中的氧原子是亲核试剂。
在反应中,水分子的氧原子攻击酯分子的羰基碳,形成一个中间体,然后中间体断裂,生成醇和羧酸。
二、亲核取代反应机理亲核取代反应是另一种常见的有机化学反应类型。
它是指一个亲核试剂攻击一个亲电试剂中的亲核位点,形成一个新的化学键。
亲核取代反应的机理也可以分为三步:亲核试剂的攻击、亲核位点的断裂和新键的形成。
以卤代烷的亲核取代反应为例,卤代烷分子中的卤素是亲电位点,而亲核试剂(如氢氧根离子)是亲核试剂。
在反应中,亲核试剂的氧原子攻击卤代烷分子中的卤素,形成一个中间体,然后中间体断裂,生成醇和卤化物。
三、加成反应机理加成反应是有机化学中常见的反应类型之一。
它是指两个或多个分子中的两个亲电位点结合,形成一个新的化学键。
加成反应的机理可以分为两步:亲电位点的攻击和新键的形成。
以烯烃的加成反应为例,烯烃分子中的双键是亲电位点,而亲核试剂(如溴气)是亲核试剂。
在反应中,亲核试剂的溴原子攻击烯烃分子中的双键,形成一个中间体,然后中间体断裂,生成溴代烷。
四、消除反应机理消除反应是有机化学中常见的反应类型之一。
它是指一个分子中的两个官能团结合,形成一个双键或三键,同时释放一个小分子。
消除反应的机理可以分为两步:亲电位点的断裂和新键的形成。
以醇的脱水为例,醇分子中的羟基和氢原子是亲电位点,而酸催化剂是亲核试剂。
在反应中,酸催化剂攻击醇分子中的羟基和氢原子,形成一个中间体,然后中间体断裂,生成烯烃和水。
高等有机化学反应和机理

高等有机化学反应和机理
高等有机化学反应和机理是有机化学的重要分支之一,它涉及到有机化合物的合成、转化以及反应机理的研究。
在高等有机化学反应中,原子、分子或离子之间的相互作用导致了反应产物的生成,而反应机理则是研究这些化学反应发生的步骤和反应物、中间体以及产物之间的相互转化关系。
高等有机化学反应的种类繁多,包括加成反应、消除反应、置换反应、重排反应等。
这些反应都具有一定的特点和规律,可以通过实验研究和理论计算来揭示其反应机理和反应路径。
在高等有机化学反应中,反应条件和催化剂选择对反应产率和化学选择性具有重要影响。
因此,研究反应条件和催化剂的优化也是高等有机化学反应和机理研究的重要内容之一。
总之,高等有机化学反应和机理是有机化学领域中的重要研究方向,对于促进有机合成化学、药物研发等领域的发展都具有重要意义。
- 1 -。
南开大学高等有机化学课件第四章有机反应机理的研究和描述

Ea ln k ln A RT
R: 气体常数, A: 频率因数,
在不同温度下测速率常数, 可计算出 Ea: Arrhenius活化能
Ea ΔH RT ΔS Ea log k 10 .753 log T 4.576 4.576T
4.3.1 简单速率表达式的积分形式
正常情况下动力学数据用微分方程的积分形式来处理:
如简单的一级反应和二级反应:
1 C0 一级级反 : k ln( ) t C 1 b0(a) 二级反应 : k (a0 - b0)ln t a0(b)
a, b, c: 时间t时浓度
a0, b0, c0: 起始浓度
一些反应速率方程积分形式的推导:
4.2 动力学数据(Kinetic Data)
动力学数据使我们能更详细地洞察反应机理。用跟踪反 应物消失和产物出现的方法可以测定某一个反应的速度。波 谱技术提供了一个迅速又连续地监测浓度变化的方法,因而 往往被用来测量反应进行的程度。总之,任何与一种反应物 或产物的浓度有关而且能被测量的性质,都可利用来测定反 应速度。 动力学研究的目的是为了在反应物和催化剂的浓度以及 反应速度之间建立定量关系。
k1[A][B] [C] k -1
d[D] k1 k2[C] k2 [A][B] kobs.[A][B] dt k -1
大多数反应不止一步, 可以参考一些重要的多步反应例子来得出动 力学表达式, 例如在决速步之前可以有一个快速平衡:
ROH + H+
+ ROH2
快 k1 k -1
_
ROH2 RBr +H2O
计算出一个反应的自由能变化,就使反应平衡位置的计算有了 可能,也就指出了某一化学过程的可实现性。 有兴趣的反应大多数发生在溶液中,任何这种反应的焓、熵和 自由能都与溶剂介质有关。 但是,热力学数据并不能说明是否存在一个能量上有利的潜在 反应途径,即反应速度上的情报。因此,深入了解反应机理以及 有机反应进行是中间所经各步的速度和能量要求是极为重要的。
高等有机化学第四部分4-6金属有机化合物

O
四、反应还被用来合成高分子(特殊 的高分子,如导电子分子)
X Ar MgCl
Ni (Ar)n
Br Ar Br+ (HO)2B Ar B(OH)2 Pd
(Ar) (Ar')
*从机理看反应应用了 a)O.A. b)烷基化 c)R.E.
R'M'
RX
M(O) O.A
R MX
M'X R
M R'
R R' +M(o)
实验二:
CH3 CH = CH CH3
+
CO3 CO = CO CO3
CH3 CH = CO CO3
"W"
CH3 CH = CH CO3
CH3 CO = CO CH3
CH3 CH = CO CH3
只有这种产物所以证明反应中没有氢或烷基的转移。
最早认为机理:
R M ⅡⅡ
M
R
R
RR
R
R
到1972年.E.O.Fishec发现:
1、
(CO)5CR = C
Ar OCH3
+ CH2 = C
CO3Et H
Ar CH2 = C OMe
2、
Ar (CO)5W = C Ar +
CH2 = C
OCH3 Ar
(CO)5W = C
R.E.
O ?Ⅱ ?Ⅱ ?Ⅱ
X_ Pd
OH
R
X
O
Pd R
HO ?Ⅱ ?Ⅱ H
O
Ph R CH CH2 + CO
O
Ph
R CH CH2 CO
O
β_ ⅡⅡ
O CH2 Rh CHR
高等有机化学第四章有机反应中间体解析

正电荷分散程度大
共轭体系的数目越多,正碳离子越稳定:
CH2 CH 3C > CH2 CH 2CH > CH2 CHCH2
当共轭体系上连有取代基时,供电子基团使正碳离子 稳定性增加;吸电子基团使其稳定性减弱:
CH3
CH2 >
CH2 > O2N
CH2
环丙甲基正离子比苄基正离子还稳定:
3C >
2CH > CH2 >
含有带负电荷的三价碳原子的原子团。 是最早被确认的活性中间体
1、碳负离子的结构
两种构型: 未共用电子对占据p轨道
未共用电子对占据sp3杂化轨道
有利构型!
桥头碳负离子 角锥结构可以快速翻转,不具有手性
三元环碳负离子难于翻转 得到构型保持的氘代产物
当碳负离子与相邻的不饱和体系共轭时,平面结 构变为有利结构
CH2
环丙甲基正离子的结构:
C
其结果是使正电荷分散
CH2
空的 p 轨道与弯曲轨道的交盖
随着环丙基的数目增多,
CH2
CH2
正碳离子稳定性提高。
直接与杂原子相连的碳正离子结构:
氧上未共有电子对所 占 p 轨道 与中心碳原子上的空的 p轨道 侧面交盖,未共有电子对离域, 正电荷分散。
CH3 O CH2
CH3O CH2
HC CH
NaNH3 液 NH3
HC CNa
NH3
Ph3C H
NaNH3 液 NH3
Ph3CNa
NH3
CH3COCH2COOEt NaOEt CH3COCHCOOEt
常用的碱 ■ 有机锂试剂:n-BuLi, PhLi, MeLi ■ KOBut ■ LDA
高等有机化学第十九章 自由基取代反应

7
第二节 自由基取代反应影响因素 在链锁反应中,决定形成什么产品的步骤时常是 提取。自由基几乎从来也不提取四阶的或三价的原 子,也很少提取两价的原子。自由基提取的几乎全是 一价的原子, 因而从有机化合物提取的是氢或者是 卤素。
19.2.1 取代基的影响
a.取代基诱导效应的影响: 伯氢、仲氢和叔氢被提取的相对活性
第十九章 自由基取代反应
自由基取代反应是指自由基试剂与底物发生的 取代反应:
C X +
Y
C Y +
X
自由基反应机理包括四种类型: (1)自由基形成和转变:
1
(2)自由基袭击作用物(双分子反应) 自由基可以和稳定的有机分子作用,形成另 外一个自由基,后者自由基可以作为加成、聚合、 或取代反应中的引发剂。这种反应经常是链锁反应。 (3) 自由基消失过程
6
19.1.3 非经典自由基取代反应机理 例1,在光催化的卤代反应里,一般得到的是许多产品 的混合物。但是,溴代一个含有溴原子的碳链则位置 选择性很高。溴代烷烃的溴代反应,84∼94%取代在分 子中原有的溴的邻位碳原子上。
例2, Skell及同事报导在0∼72°C之间溴代光学活性1溴-2-甲基丁烷(α27=2.86°),得到了光学活性1,2-二溴2-甲基丁烷(α27=-2.33°)。再有,在DBr存在的情况下 进行这个反应时,则回收的1-溴-2-甲基丁烷中在2-位 置被重氢化,并且保持构型不变。又如,在顺式4-溴 权丁基环己的溴代反应里,取代发生在3-位置上。而 且是反式的:
在本章中仅就自由基取代反应进行讨沦。至于自 由基对不饱和化合物的加成,以及重排反应等将在有 关章节再行介绍。
第一节 自由基取代反应机理 19.1.1 脂肪族自由基取代反应机理 按自由基进行的脂肪族取代反应,在原则上可能 按两种途径进行: a.
高等有机化学-反应机理

4.1.1 Classes of Pericyclic Reactions
Electrocyclic reactions (ring openings or ring
4.2.3 Stereoselectivity
4.3 Cycloadditions
4.3.1 Typical Reactions
4.3.1.1 The Diels–Alder Reaction
Normal electron-demand Diels–Alder reactions
Most Diels–Alder reactions occur with what is called normal electron-demand, in which an electron-rich (nucleophilic) diene reacts with an electronpoor (electrophilic) dienophile.
用前线轨道理论解释反应活性
炔烃的D-A反应活性低
Inverse electron-demand Diels–Alder reaction
Very electron poor dienes can undergo Diels–Alder reactions with electronrich dienophiles in the inverse electron-demand Diels–Alder reaction. The dominant interaction in the TS of inverse electron-demand Diels–Alder reactions is between the LUMOdiene and the HOMOdienophile.
高等有机化学作业答案

非芳香性:(1)、(6)反芳香性:(2)、(10)
作业5
请简述三种"酸碱理论”的要点。
一、经典的酸碱定义是指溶解在水中能放出质子的物质为酸,能产生氢氧离子的物质为碱。
重要性及优点:是建立在电离理论之上,大众最熟知的经典酸碱理论。建立了对酸碱强度的定量描述;适用于pH计算、电离度计算、缓冲溶液计算、溶解度计算。
缺点:不能说明Ac-,F-,(CO3)2-,NH3也是碱;不能说明NH3与HCl是酸碱反应;错误的认为水中有"NH4OH”这种物质;不能说明非水质子溶剂(液NH3,液HF)和非质子溶剂(液SO3,液N2O4,液BrF3)中的酸碱反应;不能说明无溶剂体系的酸碱反应(固体BaO和液态或气态SO3反应生成固体BaSO4)
(3)酯的种类有:小分子链状酯、环酯、聚酯、内酯、硝酸酯、酚酯。
7.水解反应
(1)能发生水解反应的物质:卤代烃、酯、油脂、二糖、多糖、蛋白质等
(2)从本质上看,水解反应属于取代反应。
(3)注意有机物的断键部位,如乙酸乙酯水解时是与羰基相连的C-O键断裂。(蛋白质水解,则是肽键断裂)
比较下列各组化合物的酸性强弱,并予以解释。
比较下列各组化合物的碱性强弱,并从结构上予以解释
1、乙胺比苯胺强这是由于s亚层比p亚层距原子核近,受到核的吸引力较强,因而电负性较强。或者说s轨道的能量低于p轨道,杂化轨道的s成分愈大,其能量愈低,所形成的碳负离子就愈稳定。
2、对羟基苯胺比对硝基苯胺强
3、六氢吡啶比苯胺强
4、吡啶比吡咯强
作业4
请区别"立体选择性反应”和"立体专一性反应”。立体选择性是指一个反应物可能产生几个立体异构,而其中某个异构体占绝对优势,即产率高。立体专一性应该是选择性相当高,或者是这个条件下只有一个构型的反应物能发生反应而其对映体不能。
西南大学高等有机化学复习资料

西南⼤学⾼等有机化学复习资料当前位置:第⼀章电⼦效应和空间效应1、下列羰基化合物分别与亚硫酸氢钠溶液加成,哪⼀个反应速度快?哪⼀个最慢?为什么?(1)CH3COCH2CH3(2)HCHO(3)CH3CH2CHOHCHO反应速度最快,CH3COCH2CH3反应速度最慢。
羰基化合物的亲核加成反应,其速度主要由空间效应决定。
同时,与羰基相连的取代基的性质也将影响其加成速度。
烷基是供电⼦基,将减少碳原⼦上的正电荷,不利于亲核加成反应。
甲醛中与羰基相连的是两个氢原⼦,空间位阻最⼩,因此加成反应速度最快。
丁酮中供电的甲基和⼄基与羰基相连,空间位阻⼤,因此加成反应速度最慢。
2、⽐较下列各组化合物的酸性强弱,并予以解释。
(1)HOCH2CH2COOH和CH3CH(OH)COOH(2)对硝基苯甲酸和对羟基苯甲酸(3)a.ClCH2COOH b.CH3COOH c.FCH2COOH d.CH2ClCH2COOH e.CH3CHClCOOH(4)CH3COCH2COCH3和CH3COCH2CO2C2H5(1)-羟基丙酸的酸性⽐-羟基丙酸的强;羟基有-I效应,-I效应常随距离的增长⽽作⽤迅速减弱。
(2)对羟基苯甲酸的酸性⽐对硝基苯甲酸弱;NO2是强吸电⼦基,其诱导效应和共轭效应⽅向⼀致,OH有-I和+C效应,⼀般取代基诱导效应和共轭效应⽅向不⼀致时,往往以共轭效应为主,OH基总的来看是供电基。
(3)c>a>e>d>b。
(4)后者酸性较弱。
3、试解释亲核加成反应中,ArCH2COR的反应活性为何⽐ArCOR⾼。
Ar直接与>C=O相连时,羰基和芳环共轭使羰基碳的正电荷离域分散,使羰基碳正电性减少,活性降低。
在ArCH2COR中,Ar 与羰基⽆共轭效应,只有吸电⼦诱导作⽤,增加羰基碳正电性。
故加速亲核反应进⾏。
当前位置:第⼆章⽴体化学1、具有n个C=C双键的化合物,总共应存在多少个Z-E异构体,并举例说明。
高等有机化学 第四章 亲电加成反应(2010)

implies formation of a complex between one
molecule of the reagent and the reactant and also
is expected to result in anti addition.
11
(2)双分子历程 ① 碳正离子历程
H
+
+
13
CH3 C C H H
DCl CH3COOD
D
+C
D Cl C C CH3 H H
C CH3 H
H
通常不具有立体选择性
CH CH3 3 + CH3 + H2O CH3 H
+
CH3 CH3 顺式 OH HO + H CH3
H
OH
OH CH3 H CH3
反式
CH3 H
14
有时有重排产物出现
鎓离子存在的直接证据?
Biadamantylidene bronomium
19
(3)三分子历程(AdE3)
某些非共轭烯烃与HX加成按AdE3历程进行。
立体化学通常为反式加成
20
complex
21
HBr + H3C C C H H H CH3 H3C C C CH3 H
22
2. 烯烃亲电加成反应的立体化学
5
(2) Formation of carbocation ion pair from
alkene and electrophile.
6
Mechanism(2)also involves a carbocation intermediate, but it is generated in the presence of an anion and exists initially as an ion pair. Depending on the mutual reactivity of the two ions, they might or might not become free of one another before combining to give product.
沈阳药科大学高等有机化学课件(胡春版)——第三章 有机反应总论

三、反应机理的研究方法 3. 同位素标记
在机理研究中常常用同位素效应和同位数标记来 确定反应历程。
三、反应机理的研究方法 3. 同位素标记
(1)同位素效应
最常用的是用氘来代替氕,当反应底物中的 一个原子被它的同位素取代后,对它的化学反应 性没有影响,但反应速度有显著的影响。同位素 效应分一级同位素效应和二级同位素效应。 一级同位素效应:在决定速度步骤中与同位 素直接相连的键发生断裂的反应中所观察到的效 应, 其值通常在KH/KD 为2或更高。 二级同位素效应:在反应中与同位素直接相 连的键不发生变化,而是分子中其它化学键变化 所观察到的效应,其值通常在KH/KD = 0.7-1.5范 围内。
N
CN
-
-
O
+
O C-
O N
O C N H
N
hydrolysis ( NH
3
)
H OH O H
+
O H C O
-
N
O C
N O
O hydrolysis COOH + NO 2
-
an acyl nit rit e
Rosenblum Observations (1960)
Observation I: Inst ead of nit rite, molecular nit rogen was a by-product of t he von Richt er reaction. Apparent ly, in t he 1871 von Richt er paper, nitrit e had never been demonstrated to be a by-product. It had been deduced based on stoichiometry considerations. In t he revised Bunnet t mechanism, amm onia and nit rit e are stipulat ed by-products of the von Richter reaction. Ammonia and nit rit e can react t o form ammonium nitrit e which, upon heat ing, decom poses t o give molecular nitrogen and wat er. NH 3 t o an on-going von Richt er reaction, Upon addit ion of t he only molecular nit rogen obt ained contained no nitrogen-15! If t he revised Bunnet t mechanism were correct , the added NH 3 and the expelled NO 2 (from t he last st ep) should have produced N
有机反应机理和反应类型

有机反应机理和反应类型有机反应机理是研究有机化合物在反应过程中发生的变化的一种方法。
它揭示了反应底物与产物之间的化学变化,以及反应中可能涉及的中间体和过渡态。
有机反应类型则是根据反应中的特定特征和机制将反应分类的方法。
一、酯化反应酯化反应是一种有机反应,通过酸催化或酶催化,醇与酸酐之间的酯结合,生成酯化合物。
该反应的机理包括酸催化步骤、裂解步骤和酯化步骤。
酸催化步骤中,酸负责质子化醇,并使酸酐发生裂解,生成酸和酰氧离子。
裂解步骤中,酸酐的酰氧离子与醇的质子化醇发生求核取代反应,形成酯和酸。
酯化步骤中,酸催化下,酸与醇发生质子化和水解反应,生成酯。
二、亲电取代反应亲电取代反应是一种有机反应,通过亲电试剂与有机物中的亲核试剂之间的相互作用,进行化学变化。
该反应包括亲电试剂的进攻和亲核试剂的离开,生成产物。
亲电取代反应的机理可以分为两步:亲电试剂进攻和亲核试剂离开。
在第一步中,亲电试剂通过与反应物的亲电中心之间的相互作用,形成中间体。
在第二步中,亲核试剂攻击中间体,将原来的反应物的基团替换为新的基团。
三、自由基反应自由基反应是一种有机反应,通过自由基与有机物中的亲核试剂之间的相互作用,进行化学变化。
该反应的机理包括自由基的产生、自由基的进攻和自由基的消除。
在产生自由基的步骤中,常使用氧化剂或光照射来打断反应物的化学键,产生自由基。
在自由基进攻的步骤中,自由基通过与反应物中的亲电中心之间的相互作用,形成中间体。
在自由基消除的步骤中,反应产物中的两个自由基相互结合,生成较稳定的产物。
四、环加成反应环加成反应是一种有机反应,通过酸催化或碱催化,烯丙基复合物与具有亲核性的试剂之间的反应,生成环化合物。
该反应的机理包括烯丙基离子的形成、环中间体的形成和中间体的断裂。
在烯丙基离子的形成步骤中,烯丙基复合物通过酸催化或碱催化,生成带正电荷的烯丙基离子。
在环中间体的形成步骤中,烯丙基离子与具有亲核性的试剂发生求核取代反应,生成环中间体。
有机反应机理知识点归纳

有机反应机理知识点归纳
有机反应机理是有机化学中非常重要的一部分,它描述了有机分子之间发生化学反应的详细过程。
下面是一些常见的有机反应机理知识点归纳:
1. 反应类型:
- 加成反应:两个单体结合形成一个新的化合物。
- 消去反应:一个大分子分解成两个或更多小分子。
- 变位反应:分子内原子或基团的位置重新排列。
- 取代反应:一个原子或基团被另一个原子或基团取代。
2. 反应机理的步骤:
- 初始步骤:包括反应物的活化和生成中间体。
- 中间体的转化:中间体经历一系列的转化步骤,最终形成产物。
- 生成产物:最终产物生成并结束反应。
3. 催化剂的作用:
- 催化剂可以加速反应速率,降低活化能。
- 酶是生物体内常见的催化剂。
4. 反应速率与反应底物浓度的关系:
- 当反应底物浓度增加时,反应速率也会增加。
- 反应速率与浓度之间的关系可以通过速率方程式表示。
5. 质子转移反应:
- 质子可以从一个分子转移到另一个分子,形成质子化和去质子化产物。
- 质子转移反应在有机化学中非常常见。
6. π电子的参与:
- π电子可以作为电子云,参与化学反应中的电子迁移。
以上是有机反应机理的一些常见知识点归纳,希望对您有所帮助。
高等有机化学各章习题及答案 (2)
m-Cl
ρ值
1.24
1.24
1.08
1.06
1.01
0.94
注:ρ值愈正,说明积聚的负电荷越多。
从ρ值的变化趋势说明当环 A 上的取代基不同时,过渡态是怎样变化的?
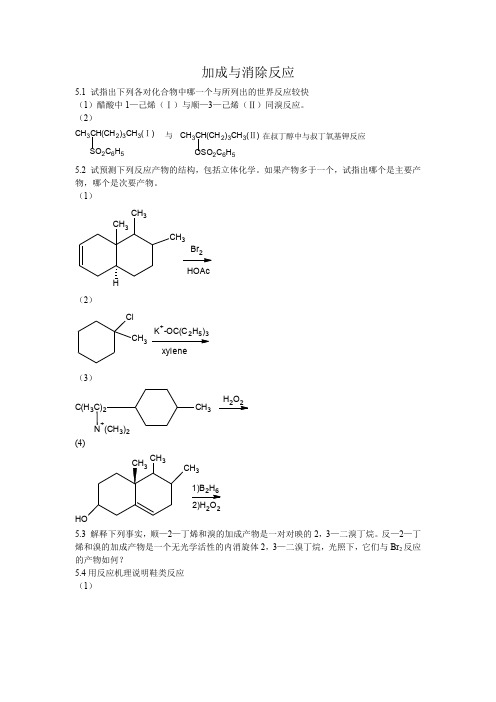
答案: 5.1
(1)(Ⅱ)>(Ⅰ)
H
C2H5
H5C2
+ Br2 H
(Ⅱ)
H C
C2H5 C
H5C2
+
Br
H
H
H
C4H7
H2C
+ Br2
CC
C4H7
H Br+ H
(Ⅰ)
烯烃与溴的加成反应中速率决定步骤是中间体溴鎓离子的生成,很明显溴鎓中间体上碳 所连的推电子基越多,正电荷越易被分散,中间体越稳定,从而越降低了反应活化能,使反 应加快。 (2)(Ⅱ)>(Ⅰ)
因为它们的离去基团是—OSO2C6H5,—SO2C6H5。离去能力显然是前者大于后者,这 是由于离去后的碎片稳定性是:C6H5SO3->C6H5SO2—。 5.2 (1)
O
O
O
Cl
Cl
+ Cl-
H
-Cl-
(2)
Br2
O
O
Br
Br -OH
COOO-
Br
Br
-OH
O
O(±)
Br
COOO
H
Br
Br OH -OH
O
O-
COO- H3+O O
COOH O
这是一个组合型的机理题,它涉及:①溴的亲电加成;②内酯的水解反应;③分子内 的 SN2;④E2 消除反应;⑤酸碱反应。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
完整版课件ppt
54
Aldol reaction的立体化学
完整版课件ppt
55
Aldol reaction的立体化学
完整版课件ppt
56
完整版课件ppt
57
练习
(安息香缩合反应)
完整版课件ppt
58
Retro-aldol 反应
完整版课件ppt
59
Knoevenagel 缩合反应
完整版课件ppt
-NR2的PKb为35,根据规则此基团的离去能力弱,但是它真正的离去基团是HNR2,
PKb为10.所以此化合物具有强的亲电性。完整版课件ppt
21
独特的C=C键(变色龙)
C=C键既可以是亲核性的也可以是亲电性的,它的性质取决于所连的官 能团的性质。一般来说,C=C键连有亲核性官能团如RO–,R2N–,– CH2MgBr时,这个烯烃或芳香化合物是亲核性的;C=C键连有亲电性官 能团如–COR, –CO2R,–CN,–NO2,–CH2X时,这个烯烃或芳香化 合物是亲电性的。
完整版课件ppt
76
格式试剂对酰胺的加成反应
完整版课件ppt
77
α,β-不饱和羰基化合物的烯基碳上的取代反应
α,β-不饱和羰基化合物上的β碳上带有离去基团时,此碳 上可以发生加成——消除反应。
(反应性与酰氯相当)
完整版课件ppt
78
带有强吸电子基团的芳基环上的取代反应
当苯环上带有强吸电子基团时,芳基碳上也可以发生加 成——消除反应。
完整版课件ppt
48
羰基上的加成反应
(CeCl3能促进羰基上的加成反应)
完整版课件ppt
49
羰基加成反应的立体化学
完整版课件ppt
50
羰基化合物与含氮亲核试剂的加成反应
醛、酮与可以与胺发生加成反应,也可以和胺的衍生物(羟氨、肼、苯肼)发生 反应。与一级胺反应,氮上还有氢,加成物失去一份子水,变为亚胺称为西佛碱。 亚胺在稀酸中水解,可得回原来的羰基化合物及其胺,因此可以用来保护羰基化 合物。反应过程如下:
8
区域选择性
完整版课件ppt
9
亲核性
大多数情况下,碱性增加则亲核性增加。 以下是一些特例:
ⅰ.在同族元素中,周期高的原子亲核性大,碱性则降低。
亲核性:I- > Br- > Cl- > F-; RS- > RO-
碱性:I- < Br- < Cl- < F-; RS- < RO-
完整版课件ppt
10
32
杂原子的SN2反应机理
完整版课件ppt
33
SN2反应机理中的特例
完整版课件ppt
34
SN2反应机理中的立体化学
完整版课件ppt
35
β消除反应中的E2反应机理
完整版课件ppt
36
E2反应机理的立体化学
完整版课件ppt
37
E2反应机理的立体化学
完整版课件ppt
38
C(sp2)-X的E2消除反应机理
完整版课件ppt
51
二级胺与羰基化合物反应
二级胺与羰基化合物反应,可以生成烯胺。反应历程如下:
完整版课件ppt
52
羟醛缩合反应(Aldol reaction)
一分子醛或酮,在碱的作用下,形成碳负离子,此碳负离子与另一分子醛 或酮的羰基发生亲核加成反应。 反应历程如下:
完整版课件ppt
53
羟醛缩合反应(Aldol reaction)
完整版课件ppt
16
路易斯酸亲电试剂
ⅰ. 路易斯酸式亲电性化合物含有价电子数不到8的原子, 具有能量低的空轨道,通常是P轨道。
完整版课件ppt
17
π键亲电试剂
ⅱ. π键亲电试剂的亲电原子满足八偶体结构,但是π键与一 个能接受孤对电子的原子或官能团相连。 π键亲电试剂通常 含有C=O,C=N,C≡N。
完整版课件ppt
46
亲核加成反应
羰基化合物上的加成反应
羰基化合物有两个主要的共振结构式,R2C=O
R2C+-O-, 从第二个共振结构式
中,可以看出碳原子带有正电性,很容易发生羰基上亲核加成反应。
羰基化合物的α位上的氢有一定的弱酸性,在碱性条件下可被夺去,形成碳负离子。 O=CR--CR2 -O-CR=CR2 碳负离子是一个很好的亲核试剂。
消除-加成反应机理:没有α氢的羰基化合物不能进行此反应机理。
完整版课件ppt
72
加成-消除反应的催化剂
(I- 也有类似作用)
完整版课件ppt
73
Claisen 缩合反应
酯在碱的催化下缩合为β-酮酯。完整版课件ppt
75
格式试剂、金属负氢试剂对酯基的加成反应
酸性条件和碱性条件:pKa值
完整版课件ppt
24
酸性条件和碱性条件:pKa值
碱性条件下的反应机理:
完整版课件ppt
25
酸性条件和碱性条件:pKa值
酸性条件下的反应机理:
完整版课件ppt
26
酸性条件和碱性条件:pKa值
完整版课件ppt
27
第三章 碱性条件下的极性反应
完整版课件ppt
28
C(sp3)-Xσ键的取代反应
离去,形成新的π键。 )
完整版课件ppt
43
β消除反应中的E1cb反应机理
完整版课件ppt
44
消除反应或取代反应的预测
在C(sp3)-X化合物的反应中,以消除反应或取代反应进行主要取决于 两个最主要因素:第一,亲核试剂的亲核性的强弱和含有孤对电子的化 合物的碱性的强弱。第二,反应底物是否Me、Bn或是伯碳,仲碳,叔 碳卤代烷烃。
完整版课件ppt
13
非亲核性碱
完整版课件ppt
14
Meier’s Rule
O
O-
+
完整版课件ppt
15
亲电试剂和离去基团
(1)亲电试剂
亲电试剂含有能够形成新的化合键的能量较低的空电子轨道。 亲电试剂可以是中性的,也可以是电正性的。
亲电试剂可以分为三种类型:
路易斯酸亲电试剂;
π键亲电试剂;
σ键亲电试剂。
完整版课件ppt
1
路径依赖
➢ 马屁股决定航天飞机助推器的宽度; ➢ 道格拉斯.若思的伟大发现(1993年诺贝尔经济学奖); ➢ 自我强化和锁定效应; ➢ 可怕的沉没成本; ➢ 习惯——缠在你身上的铁链; ➢ 播种行为,收获习惯;播种习惯,收获性格;播种性格,
收获命运。
➢ 男怕入错行,女怕嫁错郎; ➢ 做正确的事比正确的做事更重要; ➢ 知错就改,善莫大焉。
完整版课件ppt
79
烯基和芳基碳上的取代反应
消除——加成反应机理:
完整版课件ppt
80
金属插入反应
卤代烯烃和卤代芳烃可以与金属(Zn、Mg、Li)发生 插入反应,从C-X生成C-Metal。反应活性顺序: I>Br>>Cl
反应实例:
完整版课件ppt
81
金属插入反应
反应机理:
完整版课件ppt
82
60
Knoevenagel 缩合反应
(E1cb消除)
完整版课件ppt
61
共轭加成反应
完整版课件ppt
62
Michael 加成反应
完整版课件ppt
63
Robinson annulation
完整版课件ppt
64
Robinson annulation
第一步( Michael 加成反应)
完整版课件ppt
亲核性
ⅱ. 当亲核性原子的位阻变大时,亲核性大大下降,而碱性 稍有增加。
亲核性:t-BuO- << EtO-
碱性:t-BuO- > EtO-;
完整版课件ppt
11
iii 负电荷的离域使碱性大大下降;相对而言,亲核性则只 是部分下降
完整版课件ppt
12
亲核性
iv. 非质子极性溶剂可以溶解阴离子,因此化合物的碱性和 亲核性都会增加,但亲核性增加得更多。
完整版课件ppt
39
练习
完整版课件ppt
40
β消除反应中的E2’ 反应机理
完整版课件ppt
41
E2消除反应机理
完整版课件ppt
42
β消除反应中的E1cb反应机理
(H原子的酸性很强,而且离去基团的离去性很弱时发生)
E1cb反应机理
(特点:两步完成,第一步是碱进攻酸性的H原子,形成碳负离子。第二步是离去基团
完整版课件ppt
45
亲核性和碱性
1. 亲核性强,碱性弱的基团, 如:Br-, I-, R2S, RS, R3P, CN-, 丙二酸酯负离子 等基团。
2. 亲核性强,碱性强的基团,如:RO-, R2N-, RC≡C-, Cl-。 3. 亲核性弱,碱性强的基团,如:t-BuO, i-Pr2NLi (LDA), (Me3Si)2NK (KHMDS), i-Pr2NEt,DBU, DBN, TMG.
C=C,C≡C键与具有亲电性的原子相连时具有亲电性。
完整版课件ppt
18
σ键亲电试剂
ⅲ. 含有σ键的亲电化合物,具有E-X结构。E为亲电性原子, 满足八偶体结构,因其与离去基团X相连,导致E具有亲电 性。
亲电原子为C原子
亲电原子为杂原子
完整版课件ppt
19
离去基团
完整版课件ppt
20
离去基团
注意:离去基团的PKb一般是可以反映离去基团的离去能力的,但个别是例外的,如: RCONR2可在强碱水溶液中水解。
是加成-消除反应机理,不是SN2反应机理。