22第二十二章 常见出血性疾病的实验诊断
临床检验技术师《基础知识》考前点题卷一

临床检验技术师《基础知识》考前点题卷一[单选题]1.小红细胞直径小于A.7.0μmB(江南博哥).6.0μmC.5.5μmD.5.0μmE.4.5μm参考答案:B参考解析:红细胞直径大于10μm者叫大红细胞;大于15μm者叫巨红细胞;小于6μm者称为小红细胞。
掌握“第二章红细胞检查3”知识点。
[单选题]2.羊水中的脂肪细胞出现率达到多少反映胎儿皮肤成熟A.>10%B.>20%C.>50%D.<10%E.<20%参考答案:B参考解析:羊水中的脂肪细胞出现率>20%示胎儿皮肤成熟;<10%示胎儿皮肤未成熟。
掌握“第十七章羊水检查2,3”知识点。
[单选题]3.白细胞计数各大方格间的计数结果相差不应超过A.5%B.7%C.9%D.10%E.15%参考答案:D参考解析:各大方格间细胞计数结果相差不超过10%,否则应重新充池。
掌握“第三章白细胞检查2”知识点。
[单选题]4.多数毛细胞白血病表现为一致和独特免疫表型的细胞是A.B细胞B.T细胞C.NK细胞D.粒细胞E.单核细胞参考答案:A参考解析:毛细胞白血病多数病例表现为一致和独特的B细胞的表型,即膜表面免疫球蛋白(SmIg)大部分阳性。
掌握“第十二章特殊类型白血病2”知识点。
[单选题]5.如果某种物质完全由肾小球滤过,然后又由肾小管完全重吸收,则该物质的清除率是A.0B.50%C.90%D.100%E.无法计算参考答案:A参考解析:肾清除率表示肾脏在单位时间内将多少量血浆中的某物质全部清除而由尿排出。
该物质被肾小管完全重吸收,从尿中无排出,故其清除率为0。
掌握“第十一章肾小球肾小管功能及早期肾损伤的检查3,4”知识点。
[单选题]6.用免疫荧光染料对细胞悬液进行染色,影响因素不包括A.温度B.pH值C.荧光染料的浓度D.固定剂E.增强剂参考答案:E参考解析:增强剂会加快染色速度,但对着色效果没有影响,其他选项的因素都会影响染色效果。
掌握“第八章荧光免疫技术的概述1”知识点。
常见出血性疾病的实验诊断习题_2

[单选,A2型题,A1/A2型题]声门上区的淋巴管主要汇入()。 A.喉前淋巴结 B.气管前淋巴结 C.气管旁淋巴结 D.颈深上淋巴结 E.颈深下淋巴结
[单选,A2型题,A1/A2型题]会厌软骨茎附着于()。 A.舌骨 B.甲状软骨板 C.甲状软骨切迹后下方 D.甲状软骨上角 E.环状软骨
[单选,A2型题,A1/A2型题]内痔初发的主要症状是() A.肛门疼痛 B.肛门坠胀 C.便秘 D.无痛性出血 E.便时肛内有肿物脱出
[单选,A2型题,A1/A2型题]肛痈的临床特点是() A.疼痛、便血、伴肿物脱于肛外 B.发热、便血 C.发病急骤、流脓、有肛漏形成 D.便秘、便血 E.发病急骤、疼痛剧烈、破溃后易形成肛漏
[单选,A2型题,A1/A2型题]内痔结扎术后第7~9天,嘱病人减少活动的主要目的是 () A.防止水肿发炎 B.避免大出血 C.防止感染 D.防止复发 E.防止尿潴留
[单选,A2型题,A1/A2型题]肛裂的主要症状是() A.便血、疼痛 B.便血、疼痛、脱垂 C.便秘、便血、疼痛 D.疼痛、便血、流脓 E.疼痛、便秘、异物感
[单选]血友病的实验室检查,错误的是() APTT延长 B.PT延长 C.因子活性(Ⅷ:C、Ⅸ:C.减低 D.因子抗原含量(FⅧ:Ag、FⅨ:Ag)可正常 E.排除试验可做BT、vWF:Ag检测及复钙交叉试验
[单选,A2型题,A1/A2型题]原发性纤溶亢进症时,实验室指标正常的是() A.PT B.APTT C.TT D.FDP E.D-二聚体
[单选]关于麻醉效果,利多卡因比普鲁卡因维持的时间() A.要短 B.一样长 C.长1倍 D.长2倍 E.长3倍
第二十二章 常见出血性疾病的实验诊断

?
一、出血性疾病的概念及分类 1.概念:出血性疾病是由于遗传性或获得性的因素,导致机体止血、凝血活性的减弱或抗凝血、纤溶活性的增强,引起自发性或轻微外伤后出血难止的一类疾病。本类疾病的诊断,除病史、 家族史和临床表现外,血栓与止血检查具有确诊的重要价值。 2.分类 (1)血管壁异常导致的出血性疾病:①遗传性血管性疾病:如遗传性毛细血管扩张症、有出血倾向的遗传性结缔组织病,包括马凡综合征、艾唐综合征等。②获得性血管壁结构、功能异常:本病统称为血管性紫癜,是一组较为复杂的皮肤、黏膜出血性疾病,包括过敏性紫癜、单纯性紫癜、药物性过敏性紫癜、感染性紫癜等。 (2)血小板数量与功能异常引起的出血性疾病:①血小板数量异常:如血小板减少性紫癜、血小板增多症。②血小板功能缺陷:遗传性血小板功能缺陷症、获得性血小板缺陷。 (3)凝血因子异常所致的出血性疾病:①遗传性凝血因子异常:如血友病等。②获得性凝血因子异常:如肝病、维生素K缺乏症。 (4)病理性抗凝物增多所致的出血性疾病:①获得性FⅧ抑制物;②肝素样抗凝物;③抗磷脂抗体综合征。 (5)纤溶活性增高所致的出血性疾病:①遗传性纤溶亢进;②获得性纤溶亢进。 (6)复合因素引起的出血性疾病。 二、血管壁异常导致的出血性疾病 1.过敏性紫癜,也称许兰-亨诺综合征。好发于儿童和青年人。20岁以前的发病率占80%以上,本症是一种变态反应性出血性疾病,主要是由于机体对某些致敏物质(过敏原)发生变态反应而引起全身性毛细血管壁的通透性和(或)脆性增加,导致以皮肤和黏膜出血为主要表现的临床综合征。 临床特征随病变部位不同而异。除发病前常有上呼吸道感染等前驱症状外,典型表现分为五型:①单纯紫癜型:好发于下肢、关节周围和臀部。紫癜常呈对称分布,分批出现,大小不等,暗紫红色,呈丘疹状或融合成片状,严重时形成大疱,中心有出血性坏死
出血性疾病的分类、临床表现和实验室诊断

第一一九章出血性疾病的分类、临床表现和实验室诊断关键词出血性疾病血小板凝血障碍纤溶筛选试验诊断试验第一节出血性疾病的分类人体正常止血机制包括血管、血小板、凝血系统和抗凝血系统以及纤溶系统的参与和协调。
止血机制的异常引起出血性疾病,也导致血栓性疾病。
这是止血机制两个相反方向异常产生的所谓“低凝”和“高凝”状态的不同临床疾病。
一些疾病或病理生理过程如弥漫性血管内凝血(DIC),骨髓增殖性疾病等既可以出现出血症状也可以同时发生血栓形成。
出血性疾病种类很多,根据参与止血机制的系统可分为血管因素的异常、血小板因素的异常、凝血系统的异常以及纤溶系统的异常。
依据功能异常和引起这种异常的发病机制分类最为理想。
血小板质和量的异常,以及先天性凝血因子异常常可根据这个原则分类。
但非血小板性紫癜这组疾病的发病机制了解有限,因其临床表现的相似性和基本是小血管的异常有时被称作血管性紫癜。
血管性血友病是血管内皮细胞和骨髓巨核细胞合成的vW因子异常所致、是与凝血因子缺乏不同的疾病,但仍分在凝血系统异常疾病中。
因子Ⅻ缺乏、血管舒缓素原缺乏和高分子量激肽原缺乏没有明显的出血倾向,但因凝血试验异常也归入凝血系统异常疾病类。
抗凝血系统的缺陷如:抗凝血酶Ⅲ、蛋白C、蛋白S、肝素辅因子Ⅱ,纤溶酶原激活剂(PA),纤溶酶原等的缺失,以及活化蛋白C抵抗均易引起血栓形成,将在有关章节叙述。
一、血管因素引起的出血性疾病(一)、透壁性压力梯度增加引起的紫癜急性:瓦尔索尔氏、咳嗽、呕吐、分娩、举重、吸入性紫癜。
慢性:静脉郁滞、高空。
(二)、微循环和支持组织机械完整性降低1、老年性紫癜2、单纯性紫癜(女性挫伤后青紫综合征)3、糖皮质激素过量:柯兴综合征、局部皮质激素4、维生素C缺乏5、结缔组织异常:爱一唐(Ehlers-Danlos)综合征、弹性假黄瘤6、淀粉样变化浸润血管7、Melas综合征(线粒体脑病、乳酸中毒和中风)(三)、血管损伤性紫癜1、物理性:损伤、难产、人为紫癜、昏迷大疱(Coma bullae)2、紫外线辐射:阳光灼伤性紫癜、日光性紫癜3、感染性:细菌、霉菌、病毒、立克次体、寄生虫4、栓塞性:感染性微生物、动脉粥样栓子(胆固醇晶体栓子)、脂肪栓子、肿瘤栓子、钙化防卫(皮肤血管钙化)5、变态反应及/或炎症:血清病、过敏性紫癜、自身红细胞过敏、DNA过敏、暴发性紫癜、药物性紫癜、色素紫癜性皮疹、接触性皮炎、坏疽性脓皮病,内脏炎症性疾病、婴儿急性出血性水肿、胶原脉管病、系统性脉管炎、超敏性脉管炎(包括药物性脉管炎)、感染性脉管炎、围新生物脉管炎6、血栓形成DIC,香豆素性皮肤坏死、肝素引起皮肤坏死、蛋白C或S缺乏、暴发性紫癜、阵发性睡眠性血红蛋白尿症、抗磷脂抗体综合征7、中毒:化学物质、节肢动物咬伤(四)、异常蛋白质血症引起的紫癜1、冷球蛋白血症2、巨球蛋白血症3、冷纤维蛋白原血症4、λ-轻链血管病(五)、毛细血管扩张的疾病1、遗传性出血性毛细血管扩张症2、“瘀点状”樱桃红血管瘤3、CREST综合征(钙化性筋膜炎、雷诺现象、指趾硬皮病、毛细血管扩张。
4—出血与血栓性疾病的实验诊断
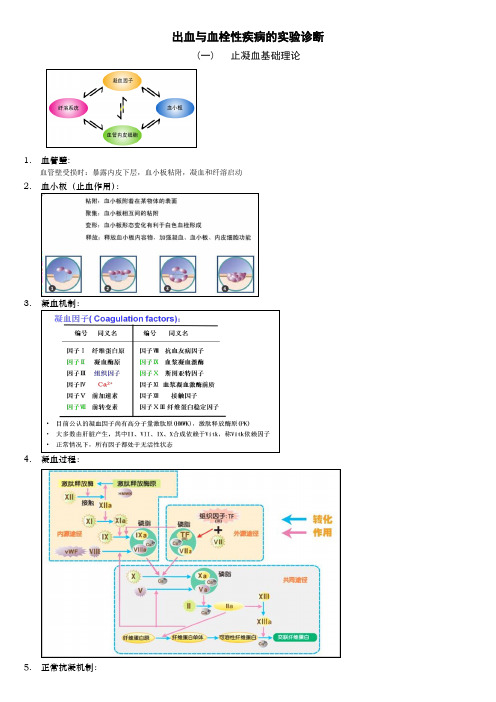
出血与血栓性疾病的实验诊断(一)止凝血基础理论1.血管壁:血管壁受损时:暴露内皮下层,血小板粘附,凝血和纤溶启动2.血小板(止血作用):3.凝血机制:4.凝血过程:5.正常抗凝机制:(1)体液抗凝机制:(2)纤溶系统(Fibrinolytic system)(二)实验室检验项目1.血管壁检测(1)筛检试验①出血时间(Bleeding Time,BT)测定原理:皮肤毛细血管刺破,出血自然停止所需的时间。
主要受血小板数量和功能影响,其次受血管壁完整性和收缩功能影响,凝血因子影响较小参考值:标准化出血时间测定器法(template bleeding time,TBT) 6.9±2.1min, 超过9min为异常(2)诊断试验血管性血友病因子抗原测定(vWF:Ag)血管性血友病因子活性测定(vWF:A)血浆凝血酶调节蛋白抗原测定(TM:Ag)血浆内皮素-1测定(ET-1)血浆6-酮-前列腺素F1 测定(PGI2的降解产物)2.血小板检测(1)筛检试验血小板计数(platelet count,PC) 参考值:(100-300) 109/L(2)诊断试验①血小板相关免疫球蛋白(platelet associated immunoglobulin, PAIg)PAIgG、PAIgM和PAIgA(ELISA法测定)临床意义: PAIg增高,见于ITP、同种免疫性血小板减少性紫癫及药物免疫性血小板减少性紫癫等。
②血小板粘附试验(platelet adhesion test, PAdT)临床意义:↓见于血管性血友病,巨PLT综合征,BM增生,抑制剂使用;↑见于高凝状态,血栓病③血小板聚集试验(platelet aggregation test, PAgT)④血小板激活的分子标志物β-血小板球蛋白(β-TG),血小板第4因子(PF4)测定P-选择素血浆血栓烷B2(TXB2)测定3.凝血功能检测(1)筛检试验①外源性凝血途径的综合检查血浆凝血酶原时间(Prothrombin Time, PT):PT延长常见于参与外源性凝血系统的凝血因子缺乏(遗传性因子Ⅶ缺乏、严重肝病、DIC等)。
常见出血性疾病的实验室诊断策略答案-华医网继续教育答案

常见出血性疾病的实验室诊断策略1.下列关于血友病,说法错误的是()参考答案:A2.A型血友病是哪种凝血因子缺乏所致()参考答案:A3.对凝血因子敏感性最高的激活剂是()参考答案:A4.对于儿童血友病患者,建议在首次接受凝血因子产品后的前20个暴露日每()个暴露日检测1次参考答案:A5.基于APTT检测的凝血因子是()参考答案:A6.APTT结果不明原因超出正常对照至少多久,患者有相关症状、体征或临床医生需要进一步了解APTT延长的原因时,可启动APTT纠正试验()参考答案:E7.vonWillebrand因子(vWF)是()的核心因子,vWF功能异常导致vWD。
参考答案:A8.vWF生物合成是在哪里()参考答案:C9.血小板减低是哪一型vWD的常见特征()参考答案:D10.专家组推荐无论是否有出血表现,VWF水平<()IU/ml即可确诊为1型VWD参考答案:A11.vWFpp/vWF:Ag比率在正常人群中为()参考答案:C12.vWD最常见的是哪个类型()参考答案:A13.免疫性血小板减少症以()为主要特点参考答案:A14.出血评分≥()分,诊断为重型ITP参考答案:D15.ITP诊断要点错误的是()参考答案:C16.成人原发免疫性血小板减少症的诊断中,外周血涂片检查的临床意义是()参考答案:B17.ITP一线治疗首选药物是()参考答案:A18.ITP患者出血症状评分≥()分应给予治疗参考答案:B19.积分>()分者,符合显性DIC诊断参考答案:D20.科学和标准化委员会(SSC)确定了脓毒症早期阶段DIC,称为()参考答案:A21.DIC的发病原因是()参考答案:E22.DIC的临床表现是()参考答案:E23.脓毒症相关DIC常出现()参考答案:A24.()是DIC的首选抗凝疗法参考答案:B25.MNPT的测定条件是()参考答案:D26.以下哪个不属于ISI的计算方法()参考答案:A27.PT延长的临床意义是()参考答案:C28.有关初级止血说法不正确的是()参考答案:C29.下列哪个不属于TT延长的临床意义()参考答案:B30.APTT是指()参考答案:B31.肝素诱导的血小板减少需要与一些疾病相鉴别,除外以下哪项()参考答案:D32.肝素诱导的血小板减少最常见的是哪个类型()参考答案:A33.肝素诱导的血小板减少常见发生于使用普通肝素治疗后的()参考答案:A34.根据4T评分系统,血小板相对降低30%~50%,评()参考答案:C35.根据4T评分系统,()分为高度怀疑参考答案:B36.肝素诱导的血小板减少,血小板减少定义为血小板计数至少下降()参考答案:B37.血友病的遗传特征是()参考答案:A38.关于血友病,说法错误的是()参考答案:A39.血友病患者最主要的治疗手段是()参考答案:C40.血友病A是缺乏哪种凝血因子()参考答案:B41.血友病B是缺乏哪种凝血因子()参考答案:C42.血友病A以()为特征参考答案:C43.实验室建立相应的校准程序文件,规定校准物,溯源性,校准方法,及校准周期等,应至少()个月进行一次校准参考答案:A44.出凝血实验分析,要严格按照说明书的要求保存试剂,必须加盖,不使用时置于()℃冰冻保存的试剂,不能反复冻融参考答案:A45.血栓和止血常见的失控原因不包括()参考答案:C46.出凝血检验的质控物至少检测()天,至少使用()个检测结果的均值作为质控图的中心线参考答案:B47.血细胞分析复检样本的数量每日在100份以下时,至少配备()人参考答案:B48.出凝血实验分析中质量控制,室内质控包括()参考答案:E49.动态平衡的维持不需要满足()参考答案:C50.创伤性凝血病早期使用凝血酶原复合物和静脉注射()mg维生素K1以紧急逆转维生素K依赖的口服抗凝药参考答案:A51.创伤性凝血病的出血表现主要包括()参考答案:E52.关于创伤性凝血病血栓预防,下列说法错误的是()参考答案:B53.创伤性凝血病的检测手段不包括()参考答案:C54.纤溶指数是MA后()min或()min血凝块消融速度参考答案:D55.随访6个月尿液检查仍异常者需要继续随访()参考答案:E56.尿液正常的HSP患儿随访时间为()参考答案:C57.以下关于过敏性紫癜的说法正确的是()参考答案:E58.下列不符合关节型过敏性紫癜临床表现的是()参考答案:C59.过敏性紫癜与血小板减少性紫癜的主要区别是()参考答案:C60.HSP诊断的必需条件是()参考答案:A61.关于家族性原发性血小板增多症的说法不正确的是()参考答案:C62.关于原发性血小板增多症说法错误的是()参考答案:D63.原发性血小板增多症血象检查不包括()参考答案:B64.原发性血小板增多症骨髓活检的说法错误的是()参考答案:D65.关于ET预后的说法不正确的是()参考答案:D66.原发性血小板增多症骨髓涂片的说法错误的是()参考答案:D67.新生儿弥散性血管内凝血,实验室检查可见()参考答案:C68.新生儿弥散性血管内凝血高凝期实验室检查描述错误的是()参考答案:E69.新生儿弥散性血管内凝血的基本症状为()参考答案:E70.新生儿弥散性血管内凝血最主要的临床表现是()参考答案:A71.新生儿弥散性血管内凝血,实验室检查描述错误的是()参考答案:B72.新生儿弥散性血管内凝血消耗性低凝期实验室检查描述错误的是()参考答案:A包含的内容,错误的是()参考答案:B74.急性DIC主要的临床表现是()参考答案:D75.对于没有出血的患者,只要血小板计数≥()/μL,我们便不会常规预防性输注血小板和凝血因子参考答案:D76.血管内凝血和纤溶事件的发生顺序,正确的是()参考答案:A77.非恶性血液病:每日计分1次,≥()时可诊断为DIC参考答案:E78.恶性肿瘤与慢性DIC相关的例外情况是()参考答案:E79.抗凝系统组成不包括()参考答案:C80.一期止血过程指()参考答案:A81.()是揭示动脉粥样硬化、血栓形成乃至于代谢综合征机制的重要途径参考答案:A82.血小板与vWF、内皮下胶原组织的结合称为()参考答案:A83.血小板与血小板的结合称为()参考答案:C84.血小板聚集的受体是膜糖蛋白()参考答案:A85.血凝学筛选实验中,用()监查口服抗凝治疗-香豆素/华法令等药物参考答案:B86.关于LA(狼疮抗凝物)测定的意义叙述错误的是()参考答案:D87.血凝学筛选实验中,((用于手术前测定内源性途径的凝血因子参考答案:A88.血凝学筛选实验中FIB的参考范围是()参考答案:B89.vWF功能通过()来测定参考答案:C90.()是D二聚体的标准检测方法参考答案:A91.是预防性输注的最佳时间是()参考答案:A92.关于获得性血友病A叙述正确的是()参考答案:C93.关于血友病的描述叙述有误的是()参考答案:C94.Utrecht方案的FVIII/IX剂量为()参考答案:B95.关于血友病预防性治疗叙述有误的是()参考答案:E96.血友病患者以下部位的出血率最高()参考答案:A。
临床检验技术师《专业实践能力》考前点题卷一

临床检验技术师《专业实践能力》考前点题卷一[单选题]1.瑞氏染色法的染色原理是A.物理(江南博哥)吸附B.物理吸附和化学亲和C.化学亲和D.化学结合E.物理性结合参考答案:B参考解析:细胞着色既有化学的亲和作用,又有物理吸附作用。
掌握“第一章血液样本采集和血涂片制备3,4”知识点。
[单选题]2.导致血清总蛋白增高的原因为A.营养不良B.消耗增加C.肝功能障碍D.脱水E.丢失过多参考答案:D参考解析:本题考查血清总蛋白增高的原因,脱水时,水减少,蛋白质被浓缩,浓度增高。
掌握“第四章血浆蛋白质检查3,4”知识点。
[单选题]3.对细菌斜面接种法用途的叙述正确的是A.用于杂菌多标本的培养B.用于杂菌少标本的培养C.主要用于牛乳标本细菌计数D.主要用于尿标本细菌计数E.主要用于单个菌落的纯培养或观察细菌某些特征参考答案:E参考解析:斜面接种法主要用于单个菌落的纯培养、保存菌种或观察细菌的某些特性。
掌握“第八章培养基1,3,4”知识点。
[单选题]4.“老核幼浆”可见于下列哪种疾病A.巨幼细胞贫血B.缺铁性贫血C.溶血性贫血D.阵发性睡眠性血红蛋白尿E.再生障碍性贫血参考答案:B参考解析:本题要点是缺铁性贫血的骨髓象特征。
缺铁性贫血是体内铁的储存不能满足正常红细胞生成的需要而发生的贫血,形态学表现为小细胞低色素性贫血。
骨髓象显示细胞增生活跃或明显活跃,尤其红系增生明显活跃(占30%以上),以中、晚、幼红为主。
各阶段幼红细胞体积较小,胞浆量少,且核固缩似晚幼红细胞,表明胞浆发育落后于核,是典型的核浆发育不平衡(老核幼浆)特点。
掌握“第七章贫血及其细胞学检验1”知识点。
[单选题]5.可用来鉴定人Tc细胞的单克隆抗体是A.CD2B.CD3C.CD4D.CD8E.CD16参考答案:D参考解析:CD8+T细胞主要指细胞毒性T细胞(Tc细胞),故可用CD8的单克隆抗体特异性的鉴定人Tc细胞。
掌握“第四章杂交瘤技术的基本原理和单克隆抗体制备3”知识点。
常见出血性疾病及检验

2.腹部症状
5.神经症状
3.关节症状
6.其他症状
第二节 常见出血性疾病的检验
一、过敏性紫癜
1.皮肤症状 皮肤紫癜为本病最常见的首发症状,大多在前驱症
状出现后2~3天出现,多位于下肢伸侧及臀部。 特点:对称性分布,分批出现,多呈紫红色,略高
于皮面,大小不等,可融合成片,紫癜可伴有局部或弥 漫性水肿、荨麻疹及多形性红斑,偶见瘙痒感。严重的 皮肤紫癜可融合成大疱,发生中心出血性坏死。
敏感性。 1.一般检测 2.凝血象检测 3.免疫学检测 4.皮肤或肾脏活检
第二节 常见出血性疾病的检验
一、过敏性紫癜
一般检测:
WBC常为正常或轻
RBC和Hb为正常或轻度
度升高,合并感染 血常规 降低,极少数出血严重
可增高
者可见重度贫血
PLT计数 多为正常
第二节 常见出血性疾病的检验
二、免疫性血小板减少症
1. 血小板无力症(G1anzmann病)
血小板膜糖蛋 白GPⅡb/IIIa 异常,同时伴 有GPIIb/IIIa基 因缺陷
血小板对胶原、ADP、 凝血酶、肾上腺素、花 生四烯酸等诱导的聚集 无反应
对瑞斯托霉素诱导的血 小板凝集反应良好
常染色体隐性遗传性疾病
第二节 常见出血性疾病的检验
四、遗传性血小板功能异常疾病
第二节 常见出血性疾病的检验
一、过敏性紫癜
(一)概述 过敏性紫癜(allergic purpura),又称为许兰-
亨诺综合征(Schonlein-Henoch syndrome),是一种 以皮肤、黏膜出血为主要临床表现的变态反应性出 血性疾病。
第二节 常见出血性疾病的检验
一、过敏性紫癜
(一)概述
诊断学出血与血栓性疾病的实验诊断课件

根据病因和发病机制,出血性疾病可分为遗传性出血性疾病和获得性出血性疾 病。遗传性出血性疾病通常由基因突变导致,而获得性出血性疾病则由其他疾 病或因素引起。
出血性疾病的病因与病理生理
病因
出血性疾病的病因多种多样,包括遗传因素、疾病、药物等 。例如,血友病、血管性血友病等遗传性出血性疾病是由基 因突变导致的;而获得性出血性疾病则可能由肝病、肾病、 内分泌疾病、免疫系统疾病等引起。
通过比较不同指标的异常表现,鉴别不同类型的血栓性疾 病,如深静脉血栓形成、肺栓塞等。
治疗效果评估与预后判断
治疗效果评估
通过定期进行实验室检查,监测病情变化和治疗效果, 调整治疗方案。
预后判断
根据实验室检查结果和患者的临床表现,评估疾病的预 后和复发风险,制定相应的预防措施。
05
实验诊断的局限性与发展 方向
04
实验诊断的临床应用Байду номын сангаас
出血性疾病的诊断与鉴别诊断
诊断
通过实验室检查,如血小板计数、凝血酶原 时间等指标,判断是否存在出血倾向或出血 性疾病。
鉴别诊断
通过比较不同指标的异常表现,鉴别不同类 型的出血性疾病,如血友病、血小板减少性
紫癜等。
血栓性疾病的诊断与鉴别诊断
要点一
诊断
要点二
鉴别诊断
通过实验室检查,如凝血酶时间、纤维蛋白原等指标,判 断是否存在血栓形成或血栓性疾病。
体征
体征主要包括贫血貌、皮肤黏膜瘀点瘀斑、肝脾肿大等。此外,根据出血部位和 严重程度的不同,还可能出现其他体征,如颅内压增高、休克等。
02
血栓性疾病概述
血栓性疾病的定义与分类
定义
血栓性疾病是指血液在血管内异 常凝集,形成血栓,导致血管狭 窄或闭塞,进而引发一系列临床 症状的疾病。
《出血性疾病的诊断》PPT课件_OK

– 凝血酶原时间(PT) – 活化的部分凝血活酶时间(APTT)
• 肝肾功能
21
• 血常规:白细胞11.37×109/L,血红蛋 白77g/L,血小板287×109/L
• 肝肾功能:正常 • PT不凝,APTT 62.5s,纤维蛋白原
(FbgC)275.5mg/dL • 血乙肝病毒标记物阴性,抗丙型肝炎
髋关节再次出现类似症状。
5
问诊
• 既往出血病史 • 慢性疾病史 • 药物毒物接触史 • 家族史
6
查体
• 皮肤出血的类型 – 淤点 – 紫癜 – 瘀斑
• 关节腔出血或血肿,是否存在关节畸形 • 皮肤或粘膜是否有毛细血管扩张 • 有无黄疸、贫血、淋巴结肿大或胸骨压痛等,出现这类体征通常提示某种全
身性疾病的伴随症状。
• 其它:DIC、循环抗凝物、抗磷脂综合征等
25
• 凝血因子异常
– 凝血因子缺乏? – 存在病理性抗凝物质?
1:1血浆纠正试验
26
• 1:1血浆纠正后复查
– PT 12.1s,APTT 29s • ANA、抗dsDNA抗体、ACL均阴性,LA正常 • 凝血因子活性
– 第二因子(FⅡ)8.1% – 第五因子(FⅤ)103% – 第七因子(FⅦ)5.1% – 第八因子(FⅧ)229.8% – 第九因子(FⅨ)6% – 第十因子(FⅩ)7.4% – 第十一因子(FⅪ)172% – 第十二因子(FⅫ)63%
出血性疾病的诊断
北京协和医院普通内科 黄晓明 陈嘉林
1
人的止血机制
• 血管 • 血小板 • 凝血因子 • Blood_clot.swf
2
出血性疾病
• 血管异常
– 先天性:遗传性出血性毛细血管扩张症 – 获得性:过敏性紫癜
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第二十二章常见出血性疾病的实验诊断一、出血性疾病的概念及分类1.概念:出血性疾病是由于遗传性或获得性的因素,导致机体止血、凝血活性的减弱或抗凝血、纤溶活性的增强,引起自发性或轻微外伤后出血难止的一类疾病。
本类疾病的诊断,除病史、家族史和临床表现外,血栓与止血检查具有确诊的重要价值。
2.分类(1)血管壁异常导致的出血性疾病:①遗传性血管性疾病:如遗传性毛细血管扩张症、有出血倾向的遗传性结缔组织病,包括马凡综合征、艾唐综合征等。
②获得性血管壁结构、功能异常:本病统称为血管性紫癜,是一组较为复杂的皮肤、黏膜出血性疾病,包括过敏性紫癜、单纯性紫癜、药物性过敏性紫癜、感染性紫癜等。
(2)血小板数量与功能异常引起的出血性疾病:①血小板数量异常:如血小板减少性紫癜、血小板增多症。
②血小板功能缺陷:遗传性血小板功能缺陷症、获得性血小板缺陷。
(3)凝血因子异常所致的出血性疾病:①遗传性凝血因子异常:如血友病等。
②获得性凝血因子异常:如肝病、维生素K缺乏症。
(4)病理性抗凝物增多所致的出血性疾病:①获得性FⅧ抑制物;②肝素样抗凝物;③抗磷脂抗体综合征。
(5)纤溶活性增高所致的出血性疾病:①遗传性纤溶亢进;②获得性纤溶亢进。
(6)复合因素引起的出血性疾病。
二、血管壁异常导致的出血性疾病1.过敏性紫癜,也称许兰-亨诺综合征。
好发于儿童和青年人。
20岁以前的发病率占80%以上,本症是一种变态反应性出血性疾病,主要是由于机体对某些致敏物质(过敏原)发生变态反应而引起全身性毛细血管壁的通透性和(或)脆性增加,导致以皮肤和黏膜出血为主要表现的临床综合征。
临床特征随病变部位不同而异。
除发病前常有上呼吸道感染等前驱症状外,典型表现分为五型:①单纯紫癜型:好发于下肢、关节周围和臀部。
紫癜常呈对称分布,分批出现,大小不等,暗紫红色,呈丘疹状或融合成片状,严重时形成大疱,中心有出血性坏死,可伴血管神经性水肿或荨麻疹;②关节型:多见于膝、肘、踝、腕等大关节,呈游走性红、肿、痛、热,可伴积液,但不留后遗症;③腹型:呈阵发性腹绞痛或持续性钝痛,有压痛但无肌紧张,可伴恶心、呕吐、腹泻、便血;④肾型:呈不同程度的蛋白尿、血尿和管形尿,表现为局灶性、节段性和增殖性肾小球肾炎,严重者可有高血压、少尿、浮肿和肾功能异常;⑤混合型:指上述两型或两型以上同时出现者。
实验室检查:本症缺乏特异性检验结果。
发作时可有自细胞数、中性或嗜酸性粒细胞增高。
骨髓象、血小板计数、血小板功能试验、凝血和纤溶试验均正常。
25%~50%的病人可见尿液改变、肾功能异常、束臂试验(CFT)阳性、血沉增快、血清IgA增高。
此外病变血管免疫荧光检查可见IgA与补体复合物的颗粒沉积,此对确诊较有价值。
2.遗传性出血性毛细血管扩张症,亦称 Rendu-Osier-Weber病。
它是一种由于毛细血管壁遗传性发育不全所引起的出血性疾病。
本病呈常染色体显性遗传。
主要病理缺陷是部分毛细血管壁的中层缺乏弹力纤维及(或)平滑肌层,因子病变部位的局部血管扩张、扭曲和破裂出血。
临床特征有:①成簇的毛细血管扩张:多见于上半身,如面部、唇部、口腔、鼻腔、胸部、上肢和手指等。
多为红色或暗红色,压之褪色,呈针头状、结节状、蜘蛛网状或瘤状的突起;②反复同一部位出血:幼年时多见鼻衄、龈血,成年后可有胃肠道、泌尿道、呼吸道反复出血,女性经量增多,手术、创伤和分娩后出血难止。
实验室检查:除部分患者的CFT阳性外,其他血栓与止血检验的结果多为正常。
出血严重者可有低色素小细胞性贫血。
病变部位组织病理学检查可见毛细血管壁缺乏弹力纤维及(或)平滑肌层,此是确诊的佐证。
3.其他血管性紫癜:包括单纯行紫癜、老年性紫癜、机械性紫癜、坏血病等。
这些紫癜都是由于血管壁的通透性、脆性增加或由于免疫球蛋白(IgG、IgM)、免疫复合物沉积于血管壁所致。
临床特征是皮肤反复发作性紫癜,有时伴有黏膜出现倾向。
实验室检查:除CFT阳性外,其他血栓与止血检验多数正常。
三、血小板病的实验诊断1.原发性(特发性)血小板减少性紫癜(ITP)是一种自身免疫性疾病。
临床上分为急性和慢性两型:①急性型典型病例见于3~7岁的婴幼儿,紫癜出现前1~3周常有上呼吸道感染史。
起病急骤,常伴发热、皮肤紫癜、黏膜出血和内脏(胃肠道、泌尿道)出血等,少数病例可发生颅内出血。
病程呈自限性,多数病例在半年内自愈。
②慢性型多数见于青壮年,以女性为多见。
常无诱发因素,起病缓慢,出血以皮肤、黏膜和经量过多为主,脾不肿大或稍肿大,病程长至1~数年,且有反复发作的倾向。
实验室检查:多次血小板计数明显减低;骨髓巨核细胞增生或正常,急性型患者以幼稚型增多为主,慢性型患者以颗粒型增多为主,但产生血小板的巨核细胞减少或缺如;MPV常增大,BT延长、血小板寿命缩短;血小板抗体检查,90%的患者PAIg和(或)PA-C3增高。
2.继发性血小板减少性紫癜是指有明确的病因或在某些原发病的基础上发生的血小板减少伴随临床出血症候群。
它不是一种独立性疾病而是原发病的一种临床表现。
(见表2-30-1)临床特点:有类似ITP的皮肤、黏膜和内脏出血的倾向。
实验室检查:除CRT阳性、PLT减少和BT延长外,可有血块收缩和凝血酶原消耗试验不佳。
依据不同病因有不同的检查结果。
3.血小板功能异常性疾病(1)血小板无力症亦称Glanzmann病。
系常染色体隐性遗传。
它的基本缺陷是血小板膜GPⅡb/Ⅲa数量减少或缺乏,也可出现GPⅡ b/Ⅲa基因的缺陷。
患者的血小板对ADP、胶原、肾上腺素、凝血酶、花生四烯酸等诱导剂无聚集反应,但对瑞斯托霉素有聚集反应。
(2)巨血小板综合征:亦称Bernard-Soulier综合征。
系常染色体隐性遗传。
它的基本缺陷是血小板膜GPⅠb/Ⅸ或GPV的数量减少或缺乏,也可有GPI b/Ⅸ或GPV基因的缺陷。
患者的血小板膜不能结合vWF,使其不能粘附于内皮下组织,并对瑞斯托霉素不发生聚集反应。
(3)贮藏池病:指血小板缺乏贮存颗粒或其内容物释放障碍。
包括致密颗粒缺陷症、α-颗粒缺陷症(亦称灰色血小板综合征)以及致密体与α-颗粒联合缺陷症。
系常染色体隐性遗传,有的呈常染色体显性遗传。
患者的血小板对ADP、胶原和凝血酶等诱导剂缺乏释放反应,故释放产物减少。
(4)血小板第3因子(PF3)缺乏症:亦称血小板病,系常染色体隐性遗传。
患者的血小板膜磷脂的结构或成分有缺陷。
故其表面缺乏凝血因子Va和Xa的受体,致使血小板不能有效地提供凝血催化表面,引起凝血机制异常,表现为白陶土凝血时间延长。
四、常见凝血因子异常所致出血性疾病的实验诊断1.血友病(1)概述:血友病是一组遗传性因子Ⅷ和Ⅸ基因缺陷、基因突变、基因缺失、基因插入等导致内源凝血途径激活凝血酶原酶的功能发生障碍所引起的出血性疾病。
包括血友病A或称血友病甲、因子Ⅷ缺乏症或AHG缺乏症;血友病B或称血友病乙。
血友病A、B均为性连锁隐性遗传病,基因分别位于Xq28、Xq27。
(2)临床特点:患者表现为自发性或轻微外伤后出血难止。
出血常发生于负重的大关节腔内(膝、踝、肘、腕、髋、肩关节)和负重的肌肉群内(肱三头肌、股四头肌、腓肠肌、腰大肌)。
尚可发生内脏出血或致命的颅内出血。
反复关节腔内出血是本病的出血特征之一,且常致关节腔纤维组织增生和粘连,造成关节畸形和残疾。
(3)实验室检查1)筛检试验:APTT延长,BT、PT、TT正常。
2)纠正试验:曾用STGT或TGT及其纠正试验。
目前也可用APTT纠正试验。
表5 不同纠正试剂中所含凝血因子------------------------------------------------------------纠正试剂FI FⅡFV FⅦFⅧFⅨFⅩFⅪFⅫ------------------------------------------------------------新鲜血浆+ + + + + + + + +硫酸钡吸附血浆+ - + - + - - + +硫酸钡吸附血清- - - - - - - + +新鲜血清- + - + - + + + +------------------------------------------------------------3)凝血因子促凝活性检测:因子活性(Ⅷ:C或Ⅸ:C)减低是常用的确诊试验,依此可将各因子缺乏症分为重型( <1%)、中型(2%~5%)、轻型(6%~25%)和亚临床型(26%~45%)。
4)凝血因子抗原含量检测:因子抗原含量(FⅧ:Ag和FⅨ:Ag)减低或正常。
若结合因子促凝活性检测的结果,可配对确定各因子的交叉反应物质(CRM)属于阳性或阴性。
5)排除试验:做BT和vWF:Ag检测以排除vWD;做复钙交叉试验以排除各因子的抑制物(尤其因子Ⅷ抑制物)。
6)携带者和产前诊断:根据临床需要尚可用基因探针、DNA印迹技术、限制性片段长度多态性作携带者诊断及产前诊断。
7)基因分析:深入研究尚需做遗传基因分析,以确定基因突变的类型,为研究病因、发病机制和基因治疗奠定理论基础。
2.血管性血友病(vWD)(1)概述:vWD是由于因子Ⅷ复合物中的血管性血友病因子基因的合成与表达缺陷而导致它的质和量异常所引起的出血性疾病。
vWD是仅次于血友病甲的另一种常见的出血性疾病。
(2)分型:根据遗传方式、临床表现和实验室检查的结果,大体可以将其分为三型:1)Ⅰ型:主要是由于vWF量的合成减少所致,而vWF的多聚体的结构基本正常。
本型患者为常染色体显性遗传。
2)Ⅱ型:主要是由于vWF的结构与功能缺陷所致,又分为ⅡA亚型、ⅡB亚型、ⅡM亚型和ⅡN亚型。
3)Ⅲ型:主要是由于vWF的抗原和活性均极度减低或缺如所致。
Ⅲ型患者为常染色体隐性遗传,临床出血严重。
(3)实验室检查:各型检验参见表2-30-2。
3.依赖维生素K凝血因子缺乏症(1)概述:由于缺乏维生素K所引起的因子Ⅶ、Ⅸ、Ⅹ和凝血酶原缺乏,称为依赖维生素K凝血因子缺乏症。
本症常有明确的病因,且呈多个因子联合缺乏,因此临床上除有原发病的表现外,尚有皮肤、粘膜和内脏的出血倾向。
临床常见的原因有吸收不良综合征、肠道灭菌综合征、新生儿出血症、口服抗凝药等。
(2)实验室检查:筛检试验APTT、PT延长和肝促凝血酶原激酶试验(HPT)减低。
然而因子Ⅶ:C、Ⅸ:C、X:C和Ⅱ:C水平减低有助于明确诊断。
蛋白C和蛋白S活性也降低。
4.肝脏疾病的凝血障碍(1)概述:出血是肝脏疾病的常见症状。
出血常表现为皮肤淤斑、黏膜出血(鼻出血、牙龈出血),月经过多,内脏出血(黑便、血尿等),且出血的严重程度与肝功能损害的程度成正相关。
肝病出血的原因甚为复杂,涉及一期止血、二期止血、纤溶亢进和血小板异常等各个方面,主要与以下几个方面有关:①凝血因子和抗凝蛋白合成减少,导致凝血和抗凝机制发生紊乱;②凝血因子和抗凝蛋白消耗增多;③异常抗凝物和血FDP增多;④血小板减少及其功能障碍。