进化树序列
生物信息学进化树

生物信息学进化树进化树是生物信息学中的一项重要工具,用于揭示生物物种之间的进化关系。
通过分析不同物种的基因组序列,可以推断它们之间的亲缘关系和进化历史。
进化树可以帮助我们了解生物的演化过程,揭示不同物种的共同祖先以及它们之间的分支关系。
在构建进化树的过程中,首先需要收集各个物种的基因组数据。
这些数据可以是DNA序列、蛋白质序列或其他形式的生物分子序列。
然后,通过比较这些序列之间的相似性和差异性,可以计算出它们之间的进化距离或相似性分数。
接下来,利用计算机算法可以根据这些进化距离或相似性分数构建进化树。
常见的算法包括最大简约法、邻接法和最大似然法。
这些算法会根据进化距离或相似性分数来确定物种之间的分支关系,从而构建出一棵树状图。
进化树的树枝代表物种的分支演化,而树叶代表当前的物种。
树枝的长度通常表示进化时间的长短,较长的树枝表示较早的分支,较短的树枝表示较晚的分支。
进化树的形态可以有很多种,例如二叉树、无根树和有根树等。
通过观察进化树,我们可以了解到不同物种之间的共同祖先以及它们之间的分支关系。
进化树的分支点代表物种的分裂事件,分支越早代表物种差异越大,分支越近代表物种差异越小。
进化树还可以显示出一些重要的进化事件,例如物种的起源、灭绝、迁移和适应等。
进化树在生物分类学、系统发育学和进化生物学等领域有着广泛的应用。
它可以帮助科学家研究物种的起源和演化过程,揭示生物多样性的来源和演变规律。
进化树还可以用于判断物种的分类和命名,帮助我们更好地理解和研究生物界的多样性。
近年来,随着高通量测序技术的发展,获得大规模的基因组数据变得越来越容易。
这使得构建进化树变得更加准确和可靠。
同时,生物信息学的快速发展也为进化树的构建提供了更多的工具和方法。
例如,基于分子标记的进化树、基于基因组的进化树和基于大数据的进化树等。
生物信息学进化树是一种重要的工具,可以帮助我们揭示生物物种之间的进化关系和演化历史。
通过构建进化树,我们可以了解到不同物种之间的共同祖先以及它们之间的分支关系。
构建进化树的步骤

构建进化树的步骤通常包括以下几个关键环节:
1. 数据收集:收集相关的生物序列数据,这些数据可以来自于公共数据库,如NCBI的GenBank,也可以通过实验获得。
序列数据包括DNA或蛋白质序列。
2. 序列alignment(序列比对):使用比对软件如Clustal Omega、MAFFT、MUSCLE等,将收集到的序列进行比对,以确保序列的同源性,并消除由于序列变异导致的噪音。
3. 序列拼接和校正:对测序得到的正向和反向序列进行拼接和校正,以获得完整的序列。
常用的拼接软件有Contig Express、Geneious 和Sequencher等。
4. 选择合适的模型:根据序列数据选择合适的进化模型。
可以使用软件如Modeltest来评估不同的进化模型,选择BIC(Bayesian Information Criterion)分数最低的模型。
5. 建树:选择合适的软件和建树方法来构建进化树。
常用的软件有MEGA、PhyML、MrBayes等,建树方法包括NJ(邻接法)、MP (最大简约法)、ML(最大似然法)等。
6. 建树检验:使用如Bootstrap方法等来检验所建树的稳定性和可靠性。
Bootstrap方法通过重复抽样来检验建树的节点支持度。
7. 绘制进化树:使用软件如TreeDraw、FigTree或在线工具来绘制进化树的图像,以便于分析和展示。
保守结构域序列构建进化树

保守结构域序列构建进化树是一个非常常见且重要的生物信息学分析步骤。
通过将同源蛋白中的保守序列区域聚合在一起,研究者可以对同一蛋白家族的多种蛋白质进行分析,并且使用这些保守结构域的序列信息进行进化树的构建,可以帮助我们理解蛋白质家族的进化关系和进化历程。
首先,我们需要收集一组同源蛋白的保守结构域序列。
这些序列通常来自于生物数据库中的已知蛋白质序列,通过比对和分析,我们可以找到这些序列中的保守区域。
这些保守区域通常代表了蛋白质的功能和结构的重要部分,因此,通过比较和分析这些序列,我们可以了解蛋白质家族的进化关系。
接下来,我们需要将这些序列导入到一个进化树构建软件中。
常用的软件包括MEGA、PHYLIP、Clustal等。
这些软件通常会使用一种叫做邻接法(Neighbor-joining)的算法来构建进化树。
邻接法是一种基于距离的算法,它通过比较序列之间的差异来构建树状图。
这种方法在处理大样本和复杂的进化关系时表现得尤为出色。
在构建进化树的过程中,我们需要对软件中的参数进行适当的设置。
例如,我们可能需要选择适当的距离度量方法、调整树的进化模型、考虑种间或种内的系统发生信息等。
这些参数的选择和调整可能会影响到进化树的精度和可靠性。
一旦进化树构建完成,我们可以利用一些可视化的工具进行观察和解读。
例如,我们可以使用专门的绘图软件(如TREE-PUZZLE或ITOL)将进化树绘制成漂亮的图形,或者使用一些专门的软件来分析树中的分支和节点,以了解蛋白质家族的进化关系和进化历程。
总之,保守结构域序列构建进化树是一个非常有用的生物信息学分析步骤。
通过比较和分析同源蛋白中的保守序列区域,我们可以了解蛋白质家族的进化关系和进化历程,这对于理解生物多样性和物种进化的机制具有重要意义。
生物信息学中的序列比对与进化树构建算法研究

生物信息学中的序列比对与进化树构建算法研究序列比对是生物信息学中重要的分析方法之一,通过比对不同生物种类的DNA、RNA或蛋白质序列,可以揭示它们之间的相似性和差异性,并为分析进化关系、功能预测等提供基础。
序列比对的基本思想是将两个或多个序列进行比对,并找出它们之间的相似性。
在序列比对中,常用的方法有全局比对、局部比对和多序列比对。
全局比对方法是将整个序列进行比对,一般采用Needleman-Wunsch算法或Smith-Waterman算法。
这些算法根据序列间的单个碱基或氨基酸之间的匹配、错配和缺失情况,计算出序列的相似度得分。
全局比对方法适用于较短的序列,优点是能够找到完全匹配的区域,但是对长序列不适用,计算复杂度较高。
局部比对方法主要用于比对较长的序列或存在较大插入缺失的序列。
常用的算法有BLAST和FASTA算法。
这些算法采用快速搜索的策略,先找出序列间的高度相似的片段,然后再进行比对和分析。
局部比对方法能够找到较长序列内的相似片段,但可能无法找到全局的最优比对。
多序列比对方法用于比对三个或更多序列,揭示它们之间的共同特征和区别。
常用的方法有多重序列比对和进化树构建。
多重序列比对旨在将多个序列按照匹配和错配的原则进行比对,以找到共同的序列区域。
进化树构建方法基于序列的相似性和进化关系,将多个序列构建成进化树,以揭示它们之间的进化关系。
在序列比对的过程中,常用的比对算法还包括Pairwise比对、局部比对、多重比对等方法。
这些方法都有自己的特点和适用范围,根据具体的研究目的和数据特点选择合适的方法进行序列比对。
进化树构建是生物信息学中的重要研究方向之一,用于揭示不同生物种类之间的进化关系。
进化树是一种图形化的表示方式,能够清晰地展示物种间的分支关系、共同祖先以及进化时间。
进化树的构建主要基于序列的相似性和进化关系。
在进化树构建中,常见的方法包括距离法、最大简约法和最大似然法。
距离法基于序列间的距离矩阵,通过测量序列间的差异程度来构建进化树。
序列谱进化树方法

活性中心序列谱及系统发育树的制作杨曼丽1 序列谱的制作1.1 搜集数据查找数据。
在CAZy数据库()中找到目标家族。
All显示的是该家族所有的序列条目,Structure显示的是结构已经被实验解析的条目,Characterized显示的是有功能标注的条目。
数据显示按Archaea、Bacteria和Eukaryota分类。
排列按字母顺序。
数据库中分别显示蛋白的名称、EC号、来源、GeneBank、Uniprot及PDB数据。
图1下载PDB文件。
将目标PDB文件下载。
在同种蛋白含有不同PDB文件的时候注意文件的选取。
可以通过上传时间、发表文献等找出最原始的结构,其他相关结构一般为该原始结构的突变结构。
下载Uniprot序列。
将目标Uniprot文件下载,保存为Fasta格式。
一般文件命名原则为:物种_EC号_Uniprot 号_PDB号(如果有的话)。
物种用A、B、E标注。
没有Uniprot号的用GenBank号代替。
为了方便后续的建树,可以先将每个序列文件抬头(一般为第一行”>”标识)名称改为文件所命之名,这样可以省去在建树后改leaf名称的麻烦1.2 活性中心架构的获取底物的选取。
用Pymol打开下载好的PDB文件,用present->ligands找出底物。
也可显示全序列,查找序列末端。
注意一般底物为BGC等糖环,而非ACT等小分子。
有的底物在结构解析时有丢失,因此需要详细研究整个家族的PDB,以便将一些底物进行拼接。
图2切点和方向的确定。
找好底物后将整条糖链横向放置,找出非还原端和还原端(一般非还原端在左,还原端在右)。
查找原始文献,找到切点位置,记录为0点。
0点往非还原端方向的糖环分别为-1、-2、-3、-4……,往还原端方向的糖环分别为+1、+2、+3、+4……。
活性中心氨基酸的筛选。
从一个糖环开始选取其邻近氨基酸。
以5埃为例。
选择第一个糖环的氧原子(如O6),Pymol的log会显示该原子的序号(如6006)。
序列比对,构建进化树教学提纲

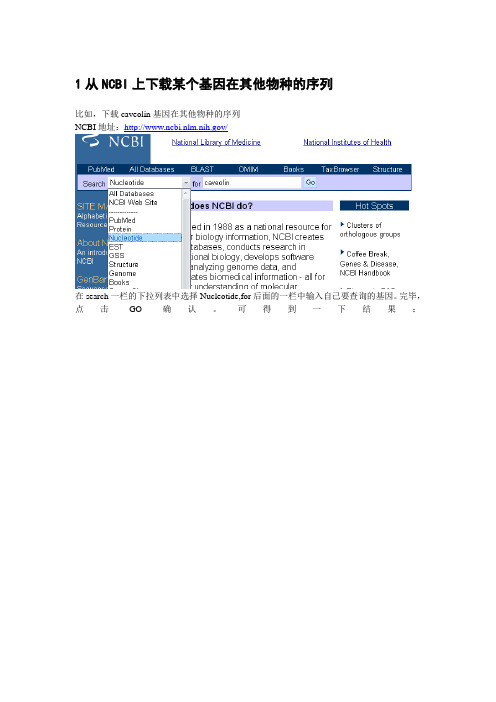
序列比对,构建进化树1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
生物信息学中的序列比对与进化树构建

生物信息学中的序列比对与进化树构建生物信息学是一门涉及生命科学和计算科学的交叉学科,其应用在分子生物学、生物医学、生态学、进化论、生物技术等诸多领域中。
序列比对和进化树构建是生物信息学的重要组成部分,是理解生物学进化的重要途径之一。
一、序列比对序列比对是将两个或多个蛋白质或核酸序列究竟有多少相同、多少不同进行比较的过程。
序列比对在生物学中极其重要,因为它可以帮助科学家确定两个生物物种之间的相似性,进而推断它们之间的亲缘关系以及共同祖先的时间。
序列比对中最基础和常用的方法是全局比对和局部比对。
全局比对试图比较两个序列的完整长度,一般用于比较相似性较高的序列,它最先被应用于分析DNA和蛋白质,是序列比对过程中最古老、最经典的算法方法。
而局部比对则更注重比较两个序列中的相似区域,忽略其中任何间隔,通常用于比较两个较短的序列或者两个相对较不相关的序列。
例如,在核酸序列比对中,这种算法更适用于获取多个剪接变异或者重复序列之间的相似性。
另外,序列比对有一个关键问题,就是如何准确的衡量两条序列的相似性和相异性。
在这方面有很多方法,例如编辑距离、盒子型、PAM矩阵、BLOSUM 矩阵等等,其中都采用了不同的评分标准。
二、进化树构建进化树(Phylogenetic Tree)是用来表示生物物种间亲缘关系的结构,也称演化树或家谱树。
进化树是通过对基于DNA和RNA等生物分子序列进行分析,推导出各物种之间共同祖先的关系构建起来的,同时它也综合了形态、系统和分子信息等其他生物学数据。
进化树的构建过程中涉及许多算法,其中最基础的是贪心算法。
贪心法从序列的最初状态开始,一步步选择最佳的演化路径,最终得到最优的进化树;而Neighborhood-joining (NJ)算法则是以序列之间的 Jukes-Cantor 模型距离或 Kimura 二参数模型距离为基础,使用最小进化步骤(Minimum Evolution,ME)标准构建进化树,是目前应用比较广泛的算法。
序列比对,构建进化树
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
3个基因构建进化树的方法

3个基因构建进化树的方法进化是生物学中一个重要的概念,它描述了生物种群随时间的演化过程。
进化树是一种用来表示不同物种之间演化关系的图表,它可以帮助我们理解生物的演化历史和亲缘关系。
构建进化树的方法有很多种,其中一种常用的方法是基于基因序列的比较。
本文将介绍基于3个基因的构建进化树的方法。
基因是生物体内用来传递遗传信息的分子,它们以DNA的形式存在于细胞中。
每个物种的基因组中都有很多基因,其中一些基因在不同物种之间保持高度保守,也就是说它们的序列变化很小。
这些保守的基因可以用来构建进化树。
在构建进化树的过程中,我们需要选择适合的基因进行比较。
一般来说,选择的基因应该满足以下几个条件:首先,基因在不同物种中的序列变化应该相对较小,这样才能准确地反映物种之间的演化关系;其次,基因在不同物种中应该有足够的变异,这样才能提供足够的信息来推断进化关系;最后,基因的比较应该能够得到可靠的结果,这就要求我们选择那些已经被广泛研究和验证的基因。
在基因选择完毕后,我们需要获取各个物种的基因序列。
这可以通过DNA测序技术来实现,现代的测序技术已经非常高效和准确,可以快速得到大量的基因序列数据。
在获取到基因序列后,我们需要对这些序列进行比对和分析,以便得到物种之间的差异。
比对可以使用一些开源的软件来完成,比如BLAST和ClustalW等。
通过比对,我们可以得到物种之间基因序列的异同点,这些差异点可以用来推断进化关系。
基于比对结果,我们可以使用一些计算模型来构建进化树。
常用的计算模型有距离法、最大简约法和最大似然法等。
这些方法都是基于不同的原理来进行计算的,它们可以根据基因序列的差异程度来计算物种之间的进化距离,并将这些距离用树状图的形式展示出来。
进化树的构建过程是一个迭代的过程,通过不断调整模型参数,我们可以得到更准确的进化树。
基于3个基因的构建进化树的方法可以提高进化树的准确性。
因为多个基因的比较能够提供更多的信息,可以避免单个基因的局限性。
进化树的构建和序列提交

f. 再将树复制到word上,然后进行修改。
二 如何提交序列
在NCBI提交序列主要有四种方式:Bankit, sequin,tabl2asn,Barcode submission tool.
我们实验室提交序列用的是Bankit, 因为它 是在网上直接提交便捷而且速度很快。
(2)打开treecon: a. 点击Distance estimation →start distance
estimation →载入刚刚保存的PHYLIP格式的 文件→ PHYLIP interleased → select all →ok → taken into account Yes → ok → Bootstrap samples 100 → ok b. 点击Infer tree topology →start inferring tree topology →Bootstrap analysis Yes →ok
c. 点击Root unrooted trees →start rooting unrooted trees →Bootstrap analysis Yes →ok →select root →ok
d. 点击Draw phylogenetic tree →file →open →(new) tree
1 16S序列的提交:
2 其他基因序列的提交:与16S相比,主要 是多了一步将序列翻译成蛋白,其他的填 写一样。http://www.expasy.ch/tools/dna.html
2 找比对的序列:一般都是把自己的序列在 NCBI上Blast, 然后找相近的序列进行比对。
DNA进化树揭示了物种间的亲缘关系

DNA进化树揭示了物种间的亲缘关系概述:自然界中存在着数量众多的物种,它们之间存在着复杂的亲缘关系。
为了揭示物种间的亲缘关系,科学家们使用了DNA进化树这一强大的工具。
通过比较物种的DNA序列,我们能够了解它们的共同祖先以及它们的演化历史。
DNA进化树的建立为生物学、生态学和进化理论等领域的研究提供了重要的依据和参考。
DNA进化树的基本原理:DNA进化树的基本原理是基于物种之间的DNA序列相似度来揭示它们的亲缘关系。
在自然选择和进化的过程中,物种的DNA序列会发生变异和累积差异。
这些差异反映了物种代际之间的演化关系和时间距离。
通过比较物种之间的DNA序列,科学家们可以推测它们的共同祖先和演化历史。
DNA进化树的建立过程:建立DNA进化树的过程包括收集物种样本、提取DNA、测定DNA序列、比较DNA序列、构建进化树和进行进一步的分析。
首先,科学家们收集不同物种的样本,可以是来自不同地区或不同环境的生物。
然后,他们从这些样本中提取DNA,并借助现代生物技术方法对DNA进行测序。
测定DNA序列后,科学家们将不同物种的DNA序列进行比较,计算它们之间的相似度。
根据这些相似度,科学家们使用计算机程序构建进化树,该树形象地表达了物种之间的亲缘关系。
最后,科学家们可以进行进一步的分析,如启示物种演化方式、挖掘共同祖先以及预测物种的演化趋势等。
DNA进化树揭示的亲缘关系:通过DNA进化树,我们可以深入了解物种之间的亲缘关系。
进化树以树状图的形式显示了物种的亲缘关系。
在进化树上,物种间较早分叉的节点代表了它们的共同祖先,而较近的分叉则代表了演化较新的物种。
树上的节点和分叉位置反映了物种之间的进化关系和时间距离。
除了揭示物种的亲缘关系外,DNA进化树还可以提供其他重要的信息。
例如,它可以揭示物种的地理分布和迁移路径,预测物种的进化趋势和适应能力。
此外,进化树还能帮助科学家们研究群落的结构和生态系统的功能,对于生态保护和物种保育也具有重要的意义。
基因进化树的构建

基因进化树的构建
基因进化树(Phylogenetic tree)是用来描述不同物种或个体之间基因演化关系的一种图形表示方法。
构建基因进化树可以帮助我们了解物种之间的亲缘关系和演化历史。
以下是构建基因进化树的一般步骤:
1.收集基因序列数据:首先,需要收集感兴趣物种或个体的基因序列数据。
这些基因序列可以是DNA序列、蛋白质序列或其他分子标记。
2.序列比对:将收集到的基因序列进行比对,找出相同的区域。
这可以通过使用比对算法(如ClustalW、MAFFT等)来完成。
比对后的序列将有助于确定物种或个体之间的相似性。
3.构建进化模型:选择适合你的数据的进化模型。
进化模型描述了基因在演化过程中的变化方式。
常见的进化模型包括Jukes-Cantor模型、Kimur a模型、GTR模型等。
选择适当的模型可以提高进化树的准确性。
4.构建进化树:使用构建进化树的方法,如最大似然法(Maximum Li kelihood)、贝叶斯推断(Bayesian Inference)或距离法(Distance-based m ethods)来构建进化树。
这些方法基于序列的相似性和进化模型来计算物种或个体之间的进化距离或相似性。
5.进化树评估和解释:评估构建的进化树的可靠性和准确性。
可以使用统计方法(如Bootstrap分析)来评估节点的支持度。
解释进化树的结果,包括物种或个体之间的亲缘关系和演化历史。
MEGA蛋白序列比对-保守序列分析-进化树

MEGA蛋⽩序列⽐对-保守序列分析-进化树
蛋⽩质序列进化(protein sequence phylogenetic},⼀种⽤于测定各种⽣物之间遗传关系的技术。
#百度百科#⼀般通过蛋⽩质的氨基酸序列进⾏⽐对后建树,⽅法过程如下:
⾸先由NCBI或其他查询基因途径获取要⽐对的⽬的蛋⽩氨基酸序列(⽹站上有很多此类说明)我的由于序列较多,就先把氨基酸序列复制到⽂本⽂件中
之后将序列⽂本⽂件扩展名改为.fas
之后打开MEGA软件进⾏序列⽐对,选择Align---Edit/Build/Alignment---Retrieve sequence from a file---选择⽂件---确定,输出结果默认以最右端蛋氨酸对齐,如图
在建树之前序列应该以保守序列⽐对模式进⾏,选择Alignment---Align by ClustalW,以输出以保守序列⽐对结果,如图
保存序列⽐对⽂件,默认格式为*.mas格式,并选择phylogeny---construct/Test UPGMA Tree进⾏建树,步骤如图
选择蛋⽩序列
之后就会输出树,如下
之后可以根据不同要求更改树形,选择下图按钮进⾏输出设置并输出环形树
之后可以保存到指定⽂件,同时也可以将树以pdf格式导出,选择image---Save as pdf file或者png file。
序列搜索_比对以及进化树的构建

Clustalx的输出结果
• .aln格式文件
– 这个文件是默认输出,可以转换成各种格式, 而且很多软件都支持这种格式。
• .dnd格式文件
– 引导树。就是根据两两序列相似值构建的一个 指导后面多重联配的启发树 – 不能做进化分析。进化分析要考虑的所有同源 位点的一个综合效应,因此应该用.aln格式文 件专门做进化分析。
• Blastn : 应该是出现较早的算法。比对的速度慢, 但允许更短序列的比对(如短到7个碱基的序列)。 • MEGABLAST : 主要用来鉴定一段新的核酸序列, 它并不注重比对各个碱基的不同和序列片断的同 源性,而只注重被比对序列是否是数据库未收录 的,是否为新的提交序列或基因。 速度快。同一 物种间的。 • Discontiguous MEGABLAST : 灵敏度 (sensitivity)更高,用于更精确的比对。主要用 于跨物种之间的同源比对。
• dnadist 计算核苷酸距离矩阵 • 把刚才的outfile改名,如dnadistinfile • 双击dnadist,输入dnadistinfile,回车
输入D,选择模型, 如改成kimura-2 输入M,然后输入 D,再输入1000, 和上面步骤要一致 即自举值 bootstrap=1000
• NCBI负责管理GenBank。 GenBank是
美国国立卫生研究院维护的基因序列数据库, 汇集并注释了所有公开的核酸序列。
• GenBank与日本DNA数据库(DNA Data Bank of Japan, DDBJ)以及欧洲生物信息研究所的欧洲 分子生物学实验室核苷酸数据库(European Molecular Biology Laboratory, EMBL),所有这 3个中心都可以独立地接受数据提交,而3个中心 之间则逐日交换信息,并制成相同的充分详细的 数据库向公众开放。因此他们是相等的。
进化树(Phylogenetic_tree)

修改M的参数为100 修改M的参数为100
修改后结果如图, 后运行, 修改后结果如图,选Y后运行,最后得到两个文件 后运行 最后得到两个文件neighbor,outfile 和neighbor,outtree
最后用CONSENSE读入neighbor,outtree, 最后用CONSENSE读入neighbor,outtree,运行后生成两个文 CONSENSE读入neighbor,outtree 件,outfile和outtree,打开outfile文件,即可查看结果。 outfile和outtree,打开outfile文件,即可查看结果。 outfile文件 如图: 如图:
运行后生成文件如下图
这个文件包含了与输入文件相同的100个 republicate,只不过每个republicate是以两 两序列的进化距离来表示。文件中的每个 republicate都省略了第一排的Mo3 Mo5 Mo6 Mo7 Mo8 Mo9 Mo12 Mo13。
以这个输出文件为输入文件,执行NEIGHBOR软件 以这个输出文件为输入文件,执行NEIGHBOR软件 NEIGHBOR
同样修改参数M 同样修改参数M
成功运行! 成功运行!
最后Dnapars ver3.61输出二个文件,分别命名为 输出二个文件, 最后 输出二个文件 分别命名为dnapars,outfile和dnapars,outtree 和
最后运行consense,导入dnapars,outtree 最后运行consense,导入dnapars,outtree consense
2
3
对进化树进行评估。主要采用 对进化树进行评估。主要采用Bootstraping法。 法
当前的任务是:
第一种方法: 第一种方法:最大简约法
一文读懂进化树(图文详解)

⼀⽂读懂进化树(图⽂详解)⽬录Content⼀、什么是进化树⼆、进化树的构成1. 根 (Root)2. 结点 (Node)3. 进化⽀ (Branch)4. 外群5. 进化分⽀长度6. 距离标尺7. Bootstrap value三、进化树评估1. Bootstrap检验2. 重复取样值3. Bootstrap value 阈值4. Bootstrap value 与分⽀四、⼏种进化树图1.经典树图(Traditional)Rectangle Tree2.圈图(Circle Tree)3.辐射树(Radiation Tree)什么是进化树系统发育进化树 (Phylogenetic tree):⼀般也叫系统进化树,进化树。
它可以利⽤树状分⽀图形来表⽰各物种或基因间的亲缘关系。
建进化树的过程,⽤术语讲:分⽀系统发育分析 (Molecular phylogenetic analysis):是⽤来研究物种或序列进化和系统分类的⼀种⽅法。
⼀般研究对象是碱基序列或氨基酸序列,通过数理统计算法来计算⽣物间进化关系。
最后,根据计算结果,可视化为系统进化树。
进化树的构成我们模拟⼀个项⽬,使⽤⼈和⿏的各两个基因做进化树,结果如下:可以看到上⾯有⼀堆标注,下⾯来看看它们代表什么意义:1. 根 (所有分⽀的共同祖先叫做根根据有⽆根可分为:有根树:上⾯的图就是有根树,可以从树中找到共同的祖先。
⽆根树:顾名思义,没有根,也就找不到共同的祖先。
⽐如后边会提到的 Straight Tree2. 结点 (每个结点代表⼀个分类单元,物种上可以是属,种群等,基因上可以是基因家族,同源物等。
这⾥需要注意,有的⼈会把 node 翻译为节点,但是节点与结点有着不⼀样的含义:节点:通常被认为是⼀个实体,⽐如互联⽹上的每台计算机,蛋⽩互作⽹络的每个蛋⽩质。
结点:只是⼀个交叉点,指交汇点,并不代表⼀个实体或事物但是,也有另外⼀种解释:这种解释将 node 分为外部节点与内部节点:外部节点⼜叫叶节点,也就是最外层的⼈基因1,⼈基因2等,代表参与分析的序列样本内部节点,也就是我们使⽤蓝⾊标注的位置,代表假定祖先。
多序列比对进化树合并的方法

多序列比对进化树合并的方法多序列比对是生物信息学中的一个重要步骤,它用于研究不同物种或个体之间的基因或蛋白质序列的差异和相似性。
多序列比对可以帮助我们理解物种的进化关系,寻找保守区域和功能位点,以及预测蛋白质的结构和功能。
在多序列比对中,常用的算法包括Pairwise算法和多序列比对算法。
Pairwise算法是将两个序列进行比对,通过计算相似性得分来评估它们的相似性。
而多序列比对算法则是将多个序列进行比对,通过比对得分来评估它们的相似性和差异性。
在多序列比对的基础上,我们可以构建进化树来研究物种的进化关系。
进化树是描述物种或序列之间进化关系的一种图形化表示。
进化树可以帮助我们推断物种的分支顺序和时间,进而研究物种的起源和演化。
在多序列比对进化树合并的方法中,有两种常用的方法,分别是距离法和最大似然法。
距离法是根据序列之间的距离矩阵来构建进化树,常用的距离法包括邻接法、UPGMA法和Neighbor-Joining 法。
最大似然法则是基于统计模型来计算进化树的似然度,常用的最大似然法包括最大似然方法和贝叶斯方法。
在距离法中,邻接法是最简单的方法之一,它根据序列之间的距离来构建进化树。
邻接法的基本思想是将距离最近的序列合并为一个节点,然后再继续合并其他序列,直到构建出一棵完整的进化树。
UPGMA法是一种基于平均距离的方法,它通过计算序列之间的平均距离来构建进化树。
Neighbor-Joining法是一种基于最小进化距离的方法,它通过计算序列之间的最小进化距离来构建进化树。
最大似然法是一种基于统计模型的方法,它通过最大化序列数据出现的概率来计算进化树的似然度。
最大似然方法使用了复杂的数学模型和算法,可以更准确地估计进化树的拓扑结构和分支长度。
贝叶斯方法则是在最大似然方法的基础上引入了贝叶斯统计学的思想,通过计算后验概率来估计进化树的拓扑结构和分支长度。
除了距离法和最大似然法,还有其他一些进化树构建方法,如最小进化法、最大平均法和最小冲突法。
系统进化树的构建

进化支
结点
猩 猩
根
一个单位
分支 长度
狒 狒
距离标尺
外 群
系统发育进化树示例
系统发育树重建分析步骤
多序列比对(自动比对,手工校正)
选择建树方法
建立进化树
进化树评估
系统发育树重建的基本方法
• 1. 距离法 (distance)
适用序列有较高相似性时
• 2. 最大简约法 (maximum parsimony, MP)
•实例讲解
文件下载完之后,这里我们采用事先准备好的序列。 将Fasta 文件直接用 ClustalX 1.83打开
•实例讲解
在进行多序列比对之前我们需要对软件进行一些设置 1.选择Alignment标签 2.选择Output format options PHYLIP软件:PHYLIP MEGA软件:FASTA
99 BANNAch 68 100 88
BJ9575 YN6 YN0556 LN0684
100
LN0688
81 LN0689
JKT6969
100 94
JKT6423 JKT7043 LNVNE9712
0.05
如果结点的Bootstrap Value >70我们认为 这个分支是可靠的
优化图标
优化选项栏
适用序列有很高相似性时
• 3. 最大似然法 (maximum likelihood, ML)
– 可用于任何相关序列集合
1. 基于序列距离特征 2+3基于序列离散特征
• 计算速度:
– 距离法 >最大简约法 >最大似然法
系统发育树重建分析过程
直系同源序列 合理的外群
点阵法
进化树——精选推荐

进化树进化树英⽂名叫“Phyligenetic tree”。
对于⼀个完整的进化树分析需要以下⼏个步骤:⑴要对所分析的多序列⽬标进⾏排列(To align sequences)。
做ALIGNMENT的软件很多,最经常使⽤的有CLUSTALX和CLUSTALW,前者是在WINDOWS下的⽽后者是在DOS下的。
⑵要构建⼀个进化树(To reconstrut phyligenetic tree)。
构建进化树的算法主要分为两类:独⽴元素法和距离依靠法●独⽴元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:⼀个序列上可能包含很多的酶切位点,⽽每个酶切位点的存在与否是由⼏个碱基的状态决定的,也就是说⼀个序列碱基的状态决定着它的酶切位点状态,当多个序列进⾏进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。
●距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。
进化树枝条的长度代表着进化距离。
独⽴元素法包括最⼤简约性法和最⼤可能性法;距离依靠法包括除权配对法(UPGMAM)和邻位加⼊法(Neighbor-joining)。
⑶对进化树进⾏评估。
主要采⽤Bootstraping法。
进化树的构建是⼀个统计学问题。
我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果我们采⽤了⼀个适当的⽅法,那么所构建的进化树就会接近真实的“进化树”。
模拟的进化树需要⼀种数学⽅法来对其进⾏评估。
不同的算法有不同的适⽤⽬标。
⼀般来说,最⼤简约性法适⽤于符合以下条件的多序列:i 所要⽐较的序列的碱基差别⼩,ii 对于序列上的每⼀个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数⽬较多(⼤于⼏千个碱基);⽤最⼤可能性法分析序列则不需以上的诸多条件,但是此种⽅法计算极其耗时。
如果分析的序列较多,有可能要花上⼏天的时间才能计算完毕。
UPGMAM (Unweighted pair group method with arithmetic mean)假设在进化过程中所有核苷酸/氨基酸都有相同的变异率,也就是存在着⼀个分⼦钟。