荧光定量PCR实验指南(一)
荧光定量PCR原理及实验步骤精选全文
可编辑修改精选全文完整版
荧光定量PCR原理及实验步骤
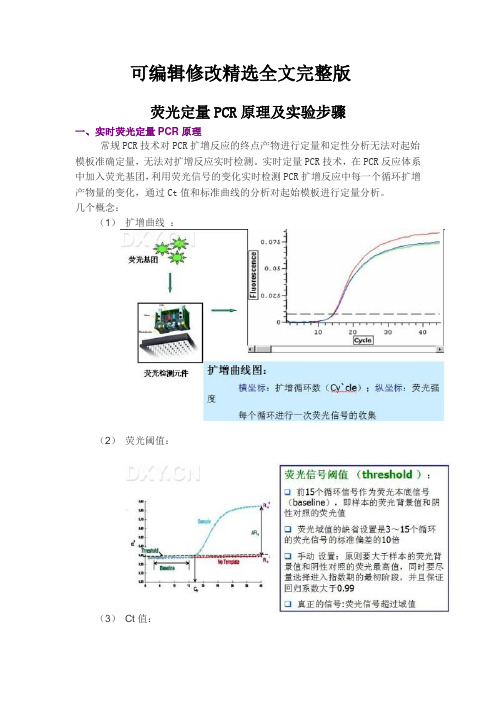
一、实时荧光定量PCR原理
常规PCR技术对PCR扩增反应的终点产物进行定量和定性分析无法对起始模板准确定量,无法对扩增反应实时检测。
实时定量PCR技术,在PCR反应体系中加入荧光基团,利用荧光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,通过Ct值和标准曲线的分析对起始模板进行定量分析。
几个概念:
(1)扩增曲线:
(2)荧光阈值:
(3)Ct值:
(4)标准曲线
SYBR Green工作原理:
1、SYBR Green 能结合到双链DNA的小沟部位
2、SYBR Green 只有和双链DNA结合后才发荧光
3、变性时,DNA双链分开,无荧光
4、复性和延伸时,形成双链DNA,SYBR Green 发荧光,在此阶段采集荧光
信号。
二、实验步骤
1. 实验前先在大型仪器共享平台上预约多元荧光定量PCR仪。
1、将所需引物和SYBgreen(避光)拿出,解冻。
计算好所有引物和SYBgreen
的用量。
2、反应体系(25μL)如下:
H2O 11μL
SYBgreen 12.5Μl
上游引物0.25μL
下游引物0.25μL
cDNA 1μL
可先将H2O 和SYBgreen按照所需量配好后,分装,再根据需要加引物和模板。
4、加完所有试剂后,盖上盖子,混匀,离心。
上机。
荧光定量PCR实验报告

荧光定量PCR实验报告摘要:荧光定量PCR(quantitative polymerase chain reaction,qPCR)是一种通过扩增和定量DNA的方法。
本实验的目的是利用荧光定量PCR技术,对给定的DNA样品进行定量检测,并比较不同浓度的模板DNA的扩增情况。
实验结果表明,荧光定量PCR是一种快速、准确、高灵敏度的定量PCR方法。
引言:荧光定量PCR技术是PCR技术的一种发展,并且在分子生物学中得到广泛应用。
它可以实时监测PCR扩增反应中DNA的扩增情况,因此被广泛用于基因表达检测、基因拷贝数检测、病原体检测等领域。
在这个实验中,我们将利用荧光定量PCR技术检测给定DNA样品的浓度并比较不同浓度的模板DNA的扩增情况。
材料与方法:1.实验材料:(1)实验电脑:配备实时荧光定量PCR软件的计算机。
(2)实验试剂:-PCR反应混合液:包含DNA模板、引物、荧光染料、酶等。
- 模板DNA:浓度为10 ng/μL的DNA样品。
-引物:用于扩增目标DNA的引物。
-荧光染料:用于实时检测PCR扩增过程中的DNA量。
(3)实验仪器:实时荧光定量PCR仪。
2.实验步骤:(1)制备PCR反应混合液,包括DNA模板、引物、荧光染料和酶。
(2)将PCR反应混合液分装到PCR试管中。
(3)将不同浓度的模板DNA分别加入PCR试管中。
(4)使用实时荧光定量PCR仪将PCR试管放入仪器中,进行PCR扩增反应。
(5)实时监测PCR扩增过程中的荧光信号,并记录荧光信号的变化。
结果与讨论:本实验中,我们选取了不同浓度的模板DNA样品并进行PCR扩增反应。
实时荧光定量PCR仪可以实时监测PCR反应中的荧光信号,并根据荧光信号的强度来定量PCR扩增产物的量。
实验结果显示,荧光信号随着PCR反应的进行逐渐增强,并且荧光信号的强度与模板DNA的浓度呈正相关关系。
通过测量荧光信号的强度,我们可以计算出不同浓度的模板DNA的扩增产物的量,进一步定量DNA样本的浓度。
荧光定量PCR实验操作流程

荧光定量PCR实验操作流程1、实验器材多样品研磨珠均质仪台式高速冷冻型微量离心机荧光定量PCR仪超净工作台分光光度计离心管TIP头2、主要实验试剂及耗材RNA提取液三氯甲烷异丙醇无水乙醇引物75%乙醇:HyPure TMMolecular Biology Grade Water配制离心管、TIP头均购湿热灭菌40min,干燥。
二、荧光定量PCR实验步骤1、总RNA抽提(枪头和离心管均经过湿热灭菌,无RNA酶)1)取匀浆器,加入1ml的Trizol Reagent,置冰上预冷。
2)取100mg组织,加入到匀浆器中。
3)充分研磨直至无可见组织块。
4)12000rpm离心10min取上清。
5)加入250 μl三氯甲烷,颠倒离心管15s,充分混匀,静置3min。
6)4℃下12000rpm离心10min。
7)将上清转移到一新的离心管中,加入0.8倍体积的异丙醇,颠倒混匀。
8)-20℃放置15min。
9)4℃下12000rpm离心10min,管底的白色沉淀即为RNA。
10)吸除液体,加入75%乙醇1.5ml洗涤沉淀。
11)4℃下12000rpm离心5min。
12)将液体吸除干净,将离心管置于超净台上吹3min。
13)加入15μl无RNA酶的水溶解RNA。
14)55℃孵育5min。
15)使用UV1800检测RNA浓度及纯度:仪器空白调零后取2.5μl 待测RNA溶液于检测基座上,放下样品臂,使用电脑上的软件开始吸光值检测。
16)将浓度过高的RNA进行适当比例的稀释,使其终浓度为200ng/μl.左右。
2、反转录(枪头和PCR均经过湿热灭菌,无RNA酶)1)取一PCR管,加入含2μg RNA的溶液。
2)加入1μl 逆转录引物。
3)用无核糖核酸酶的去离子水补足至12μl。
4)于PCR仪上65℃保温5min,迅速置冰上冷却。
5)依次加入4μl 5×buffer,2μl 10mM dNTPs,1μl RNA inhibitor和1μl 反转录酶,用枪抽吸混匀。
荧光定量pcr实验步骤

荧光定量pcr实验步骤荧光定量PCR实验步骤引言:荧光定量PCR(qPCR)是一种广泛应用于生物学研究和临床诊断的技术,可用于准确、快速地定量检测DNA的含量。
本文将介绍荧光定量PCR实验的步骤,以及注意事项和数据分析方法。
一、实验准备1. 准备所需试剂和仪器:包括PCR反应体系的各种试剂(如引物、探针、酶等)和实时荧光定量PCR仪。
2. 根据实验设计,制定合适的实验方案。
确定需要扩增的目标序列,设计引物和探针。
二、样品处理1. 提取待测样品中的DNA,确保提取得到高质量的DNA。
可以使用商业DNA提取试剂盒进行提取,按照厂家说明进行操作。
2. 测定DNA的纯度和浓度,确保测量到的DNA适用于PCR扩增反应。
使用比色法或分光光度计检测DNA的纯度和浓度。
3. 对提取得到的DNA进行稀释,以便在PCR反应中使用。
确保稀释后的DNA浓度恰当,以避免PCR反应的干扰。
三、荧光定量PCR反应体系的准备1. 根据实验设计和目标序列的长度,计算出所需的试剂和反应体系的配比。
2. 根据计算结果,将引物、探针和模板DNA按照适当的比例加入PCR反应管中。
注意保持反应管的清洁和无菌。
3. 加入合适的PCR反应缓冲液、酶和核酸酶抑制剂等试剂。
根据实验设计的需要,可以在反应体系中添加适当的试剂,如酶切酶、胶束等。
四、PCR扩增反应1. 将PCR反应管放入实时荧光定量PCR仪中,设置好PCR反应的程序和参数。
通常包括预热、变性、退火和延伸等步骤。
2. 启动PCR反应,开始扩增。
在反应过程中,实时监测PCR产物的荧光信号强度,并记录下来。
五、数据分析与结果解读1. 在实时荧光定量PCR仪中,可以实时获得PCR反应体系中荧光信号的强度和变化趋势。
根据实验设计的需要,可以选择合适的荧光信号通道进行监测。
2. 根据荧光信号和PCR反应的周期数,可以绘制荧光增幅曲线。
通过观察曲线的形态和特征,可以初步判断PCR反应的特异性和效果。
荧光定量PCR-实验步骤

荧光定量PCR检测MMP-2 mRNA的表达一、提取细胞总RNA(一)材料准备1.注意事项(1)RNA酶广泛存在于人体表面和环境中,戴口罩,勤换手套,避免皮肤与实验器具接触;专门的RNA操作区,保持清洁,定期消毒离心机,移液器均应专用。
(2)注意冰上操作,低温离心,实验所需试剂均应-4℃冰箱中预冷处理。
(3)所有实验器皿,材料都要经过去RNA酶处理,所需的氯仿、异丙醇、乙醇等都确保没有被RNA酶污染。
如果发现有污染,立即更换。
(4)操作人员保证操作的准确性,避免人为产生误差和偏倚。
2.实验器具处理:(1)塑料制品:包括枪头、EP管等,尽量使用一次性塑料制品。
若已表明为RNase-free 制品,且没有开封使用过,不需要再处理。
去除RNA酶处理步骤为:玻璃烧杯中加三蒸水和DEPC使其终浓度为0.1%(DEPC二乙基焦碳酸酷为剧毒物,通风橱中小心使用),将待处理的塑料制品逐个浸泡入DEPC水中,保证塑料制品的所有部分都浸泡到溶液中,在通风橱中室温下处理过夜,取出后尽量控干其中液体,高压后烘干,置洁净处备用。
(2)玻璃制品:泡酸过夜,冲洗干净后0.1%DEPC-H20浸泡过夜,烤干备用。
(二)试剂配制1.DEPC水:吸取1ml DEPC放在1000ml容量瓶中加双蒸水定容至1000ml,配制成0.1%DEPC水,充分搅拌混匀后分装,备用。
2.75%乙醇:用无水乙醇加DEPC水(高压灭菌处理后的DEPC水)按比例配制,-4℃冰箱保存备用。
3.异丙醇:DEPC水处理过的棕色瓶中保存。
4.氯仿:DEPC水处理过的棕色瓶中保存。
(三)实验操作步骤1.标本收集根据实验设计,给予不同浓度的药物作用于人卵巢癌SKOV3细胞24h,观察MMP-2 mRNA表达的变化。
按实验要求收集样本,分别提取细胞的总RNA。
2.Trizol法提取细胞总RNA(1)吸除6孔板中的旧培养基,使用PBS清洗2次。
(2)吸除PBS,每孔加入0.9ml RNA裂解液(Trizol),轻轻平晃板身,使裂解液覆盖底面与细胞充分接触并完全裂解细胞。
荧光定量PCR实验指南设计(一)

荧光定量PCR实验指南〔一〕一、根本步骤:1、目的基因〔DNA和mRNA〕的查找和比对;2、引物、探针的设计;3、引物探针的合成;4、反响体系的配制;5、反响条件的设定;6、反响体系和条件的优化;7、荧光曲线和数据分析;8、标准品的制备;二、技术关键:1、目的基因〔DNA和mRNA〕的查找和比对;从/网点的genbank中下载所需要的序列。
下载的方式有两种:一为打开某个序列后,直接点击“save〞,保存格式为“.txt〞文件。
保存的名称中要包括序列的物种、序列的亚型、序列的注册号。
然后,再打开DNAstar软件中的Editseq软件,点击“file〞菜单中的“import〞,打开后点击“save〞,保存为“.seq〞文件。
另一种直接用DNAstar软件中的Editseq软件,点击“file〞菜单中的“open entrez sequence〞,导入后保存为“.seq〞文件,保存的名称中要包括序列的物种、序列的亚型、序列的注册号。
然后要对所有的序列进展排序。
用DNAstar软件中的Seqman软件,点击“sequence〞菜单中的“add〞,选择要比拟的“.seq〞的所有文件,点击“add〞或“add all〞,然后点击“Done〞导入要比拟的序列,再点击“assemble〞进展比拟。
横线的上列为一致性序列,所有红色的碱基是不同的序列,一致的序列用黑色碱基表示。
有时要设定比拟序列的开始与结尾。
有时因为参数设置的原因,可能分为几组(contig),假如想全部放在一组中进展比拟,就调整“project〞菜单下的“parameter〞,在“assembling〞的“minimum math percentage〞默认设置为80,可调低即可。
再选择几个组,点击“contig〞菜单下的“reassemble contig〞即可。
选择上下的原如此是在保证所分析的序列在一个“contig〞的前提下,尽量提高“minimum math percentage〞的值。
biorad-荧光定量PCR应用指南完整压缩

目录目录1.荧光定量PCR概述21.1 荧光定量PCR的主要概念21.1.1 什么是荧光定量PCR?21.1.2 荧光定量PCR工作原理31.1.3 优化的qPCR实验特点42.实验开始82.1 总体注意事项82.2 qPCR实验设计注意事项92.2.1 单重实验或多重实验?92.2.2 方法选择92.2.2.1 DNA结合染料102.2.2.2 基于荧光标记的引物或探针的化学机理122.3 SYBR Green I反应设计及优化192.3.1 引物和扩增子的设计192.3.2 实验体系的确定及优化202.3.2.1 退火温度的优化202.3.2.2 用标准曲线进行反应的评估222.4 TaqMan探针反应设计及优化232.4.1 引物和探针设计232.4.2 实验体系的确定及优化243. 多重实验注意事项283.1 多重实验的引物及探针设计283.2 多重实验的报告基团和淬灭基团选择293.3 多重实验前单个实验的优化293.4 多重实验的验证293.5 多重实验的优化30目录4. 荧光定量PCR数据分析344.1 绝对定量354.1.1 什么时候使用绝对定量?354.1.2 使用标准曲线进行绝对定量354.2 相对定量374.2.1 什么时候使用相对定量374.2.2 用质量单位作为标准进行相对定量384.2.3 用参照基因作为标准进行相对定量404.2.3.1 2–∆∆C T(Livak) 方法41法424.2.3.2 用参照基因的∆CT4.2.3.3 Pfaffl 方法435. 基因表达分析465.1 实验设计465.2 RNA 分离475.2.1 样品收集475.2.2 RNA 提取485.2.3 检测核酸的质量和数量485.3 cDNA 模板的制备(反转录)495.4 qPCR 实验方法选择505.5 应用举例515.5.1 多重分析的反应组分515.5.2 循环条件515.6 基因表达数据分析526. 基因型/等位基因分析546.1 实验设计556.2 使用TaqMan方法设计引物和探针566.3 DNA提取和样品处理576.4 TaqMan探针实验的反应组分586.5 实验优化596.6 TaqMan探针实验的循环条件596.7 等位基因分析的数据处理596.7.1 等位基因分析的结果验证596.7.2 基因型识别72目录7. 转基因生物(GMO) 检测767.1 实验设计777.2 DNA提取及样品制备777.3 利用SYBR Green I方法对转基因大豆进行定量PCR分析787.3.1 反应成分787.3.2 循环条件797.3.3 数据分析797.4 利用TaqMan探针对转基因大豆进行多重定量PCR分析817.4.1 反应成分827.4.2 循环条件827.4.3 数据分析828. 产品指南858.1 荧光定量PCR检测系统868.2 样品制备试剂868.3 样品测定868.4 反转录878.5 定量PCR试剂888.6 常规PCR试剂888.7 耗材898.3 软件892 23 4荧光定量PCR概述21. 荧光定量PCR概述核酸扩增和检测技术是当今生物学研究的重要手段之一。
荧光定量PCR应用指南

荧光定量PCR应用指南荧光定量PCR(Fluorescent quantitative PCR)是依靠荧光信号的强度来定量PCR产物的一种PCR技术。
该技术通过在PCR反应体系中添加特异性荧光探针,利用荧光信号的强度来反映PCR产物的数量。
荧光定量PCR在生物医学研究、基因表达分析、致病微生物检测等领域有着广泛的应用。
下面将介绍荧光定量PCR的原理、实验步骤和一些注意事项。
一、原理:荧光定量PCR最常用的荧光探针是TaqMan探针,TaqMan探针是由两个DNA引物和一个荧光标记的探针组成。
当PCR反应进行到延伸阶段时,引物和探针结合到靶序列上形成荧光信号。
荧光信号的强度与PCR产物的数量成正比,通过检测荧光信号的强度可以定量PCR产物的数量。
二、实验步骤:1.设计引物和探针:选择特异性的引物和探针对目标序列进行扩增。
引物和探针的设计需要遵循一些原则,如避免引物和探针之间的自互补性、探针需选择合适的荧光染料等。
2.准备PCR反应体系:根据PCR反应需要,准备PCR反应体系。
一般包括模板DNA、引物、探针、聚合酶、缓冲液等。
3.荧光定量PCR反应:将PCR反应体系加入到荧光定量PCR仪中,进行PCR反应。
PCR反应的条件需要根据目标序列的特性进行优化,包括温度、延伸时间等。
4.数据分析:通过荧光定量PCR仪记录荧光信号的强度,根据荧光信号的强度可以反应PCR产物的数量。
通过数据分析软件对荧光信号进行定量计算,获得PCR产物的数量。
三、注意事项:1.引物和探针的设计需要严格控制,确保其特异性,避免与非靶序列结合。
2.PCR反应的条件需要进行优化,不同的目标序列可能需要不同的温度、延伸时间等。
3.控制实验中的阳性对照和阴性对照,确保实验结果的可靠性。
4.严格遵守无菌操作,避免PCR反应体系受到外源性污染。
5.选择合适的荧光定量PCR仪进行实验,确保获得准确的荧光信号强度数据。
6.实验过程中需严格按照操作规程进行,避免操作失误对实验结果产生影响。
荧光定量PCR实验指南

荧光定量PCR实验指南荧光定量PCR(qPCR)是一种能够对DNA或RNA的特定序列进行定量分析的技术。
它结合了PCR技术和荧光检测技术,能够检测出非常低浓度的靶分子。
本文将为您提供一份荧光定量PCR实验的指南,以帮助您完成这一实验。
实验前的准备工作:1.设计引物和探针:引物和探针是荧光定量PCR实验的核心组成部分,因此需要确保它们与目标序列特异性结合。
同时,还需选择合适的荧光染料和探针的浓度。
2.准备实验材料:包括引物、探针、荧光染料、模板DNA或RNA、逆转录酶酶、dNTP、聚合酶、缓冲液、MgCl2等。
3.准备质量控制样品:根据需要制备相应的阳性对照样品,并准备阴性对照样品。
实验步骤:1.反应体系的准备:根据PCR试剂盒使用说明或自行优化,将引物、探针、荧光染料、逆转录酶酶、dNTP、聚合酶、缓冲液、MgCl2等加入PCR管(注意按顺序添加,避免污染)。
2.加入模板:将模板DNA或RNA加入PCR管中,注意保持反应体系的稳定。
3.PCR条件设定:根据目标序列的特性和PCR试剂盒的要求设定PCR反应的温度、时间和周期数。
4.PCR反应:将PCR管密封,然后置于热循环仪中进行PCR反应。
5.数据分析:使用荧光定量PCR软件分析PCR反应产生的荧光信号,得到目标序列的定量结果。
注意事项:1.严格控制实验室的污染,使用无核酸污染的试剂和器材,避免引物和探针的降解。
2.对于荧光定量PCR的标准曲线方法,可以使用标准品系列的不同浓度来制作标准曲线,并根据荧光信号的强度进行定量。
3.在实验过程中,建议进行阳性和阴性对照样品的实验,以确保实验结果的准确性。
4.合理选择PCR管的容量,适量调整反应体系的配比,以保证实验的稳定性和可重复性。
总结:荧光定量PCR是一种非常强大的技术,能够在基因表达调控、感染病原体检测、突变检测等多个领域发挥重要作用。
在进行荧光定量PCR实验时,我们需要设计特异性的引物和探针,并合理选择荧光染料和探针的浓度。
荧光定量PCR技术的使用教程

荧光定量PCR技术的使用教程PCR(聚合酶链式反应)技术是分子生物学中常用的实验手段之一,它可以扩增起始序列的DNA片段。
荧光定量PCR技术是PCR技术的一种改进方法,它结合了PCR扩增和荧光检测技术,可以对扩增产物进行精确的定量分析。
本文将详细介绍荧光定量PCR技术的使用步骤和相关注意事项。
第一步:准备实验材料与试剂在进行荧光定量PCR实验前,首先需要准备好以下实验材料和试剂:1. DNA模板:可以是DNA提取物、血浆、组织样本等。
2. 引物(Primer):引物对应于目标序列的两端,用于扩增DNA片段。
应选择合适的引物,包括正向引物和反向引物。
3. 标记荧光的探针(Probe):探针可以是DNA分子、RNA分子或合成的寡核苷酸,用于检测PCR扩增产物。
4. dNTPs混合液:包含dATP、dCTP、dGTP和dUTP。
5. 磷酸缓冲液:用于维持PCR反应的酸碱平衡。
6. DNA聚合酶:用于酶切和DNA复制。
7. MgCl2:用于在PCR反应体系中提供镁离子。
8. 蒸馏水:用于稀释试剂和制备反应体系。
第二步:制备PCR反应体系1. 在无菌试管中依次加入如下试剂,制备PCR反应体系:- 1 μL DNA模板- 1 μL 正向引物- 1 μL 反向引物- 0.5 μL 荧光探针- 10 μL 2× PCR反应缓冲液- 0.5 μL 10 mM dNTPs混合液- 0.2 μL DNA聚合酶- 1 μL 25 mM MgCl2- 35.8 μL 蒸馏水注意:根据实验需求和试剂浓度的不同,反应体系中有时需要调整试剂的用量。
2. 轻轻混匀试管中的液体,避免形成气泡。
第三步:进行PCR扩增反应1. 将PCR反应体系分装至PCR管或96孔板中。
2. 将PCR管或96孔板放入PCR仪中,并设置合适的PCR程序。
- 初始变性:95°C,5分钟- 循环变性:95°C,30秒- 退火温度:根据引物的Tm值进行调整,通常为50-65°C,持续30秒- 延伸:72°C,持续1分钟- 终止延伸:72°C,10分钟3. 根据需要,可以设置PCR程序的循环次数。
荧光定量PCR实验技术干货

37
相对定量分析--管家基因筛选
• 管家基因
– 维持细胞基本代谢活动所必须的基因,
– 如:GAPDH、Actin、18S rRNA等
• 筛选方法
– 根据文献提供 – 通过具体实验筛选
实验值
6 5 4
Pβ-Actin PGAPDH
3 2 1
Oβ-2-microglobulin PHPRT
荧光定量PCR原理--Ct值的重现性
纵 轴 : 荧 光 信 号 量
横轴:PCR反映循环数
➢ Ct值的特点: • 相同模板进行96次扩增,终点处产物量不恒定; • Ct值极具重现性
19
荧光定量PCR原理--Ct值定量的数学原理 理想的PCR反应 Xn=X0 * 2n
n:扩增循环数 X0 :起始模板数量 Xn:第n次循环后扩增产物数量
• 方法:检测毕赤酵母的A基因 • 标准品:质粒标准品浓度为5个浓度;3个重复;设阴性空白对照 • 实验步骤:
提取A DNA; 设计特异引物; 设计TaqMan探针并标记探针; 荧光定量扩增; 结果分析:获取毕赤酵母的A的精确copy数。
31
绝对定量实验例--毕赤酵母A基因的绝对定量
32
绝对定量实验例--毕赤酵母A基因的绝对定量
➢ 根据样品Ct值,就可以计算出样品中所含的模板量
Sample
2 5
27
绝对定量分析几要素
• 一个目的基因 ——即需要确定其量值的核酸序列。 • 一组标准样本 ——用来生成标准曲线。可以是含有目的基因的质粒
、PCR产物、基因组DNA等。 • 重复反应孔 –——建议每个样本使用三个或更多重复反应,以确保统
荧光定量PCR手册

荧光定量PCR手册一、什么是荧光定量 PCR荧光定量 PCR(Quantitative Realtime PCR,qPCR)是一种在 DNA 扩增反应中,通过荧光信号的实时监测来定量分析特定核酸序列的技术。
简单来说,它能告诉我们样本中特定基因的数量有多少。
这一技术在生物学、医学、农学等众多领域都发挥着重要作用。
比如,在医学诊断中,能检测病毒或细菌的感染量;在基因研究中,可分析基因的表达水平。
二、荧光定量 PCR 的原理荧光定量 PCR 的原理基于传统的 PCR 技术,但增加了荧光检测的环节。
在 PCR 反应中,DNA 会被不断复制。
我们会在反应体系中加入一种特殊的荧光探针或荧光染料。
这些荧光物质的荧光强度会随着 PCR 反应的进行而发生变化。
当 PCR 反应开始时,荧光信号很弱。
随着反应的进行,DNA 不断扩增,荧光信号逐渐增强。
通过检测荧光信号的强度变化,我们就能实时了解 PCR 反应的进程,并计算出初始样本中目标 DNA 的含量。
三、荧光定量 PCR 的实验流程(一)样本准备首先,需要从待检测的生物样本(如血液、组织、细胞等)中提取出 DNA 或 RNA。
提取的质量和纯度对后续实验结果至关重要。
(二)引物和探针设计设计合适的引物和探针是关键步骤。
引物是一小段能与目标 DNA 序列特异性结合的寡核苷酸,它们决定了 PCR 反应扩增的区域。
探针则用于检测扩增产物,通常带有荧光标记。
(三)PCR 反应体系配置将提取的核酸模板、引物、探针、酶、缓冲液、dNTPs 等试剂按照一定的比例混合,配置成 PCR 反应体系。
(四)PCR 反应将配置好的反应体系放入荧光定量 PCR 仪中,设置好反应程序,进行 PCR 反应。
(五)数据分析PCR 反应结束后,仪器会给出荧光信号的变化曲线。
通过对这些曲线进行分析,可以得出目标基因的初始含量。
四、荧光定量 PCR 的关键要素(一)引物和探针的设计引物和探针的特异性、长度、GC 含量等都会影响 PCR 反应的效率和特异性。
荧光定量pcr 实验报告

荧光定量pcr 实验报告荧光定量PCR 实验报告引言:荧光定量PCR(Polymerase Chain Reaction)是一种非常重要的分子生物学技术,它能够在短时间内扩增特定DNA片段,并通过荧光信号实时监测扩增过程。
本实验旨在利用荧光定量PCR技术,检测目标基因在不同组织中的表达水平。
材料与方法:1. 提取RNA:从待测组织中提取总RNA,采用RNA提取试剂盒按照说明书操作,获得高质量的RNA样品。
2. RNA逆转录:使用逆转录试剂盒将RNA逆转录为cDNA,反应条件为37℃反应30分钟,然后95℃反应5分钟,最后冷却至4℃。
3. 荧光定量PCR反应体系准备:将PCR反应体系按照说明书中的推荐比例配制,包括模板cDNA、引物、荧光探针和PCR Master Mix。
4. 荧光定量PCR反应条件设置:根据引物和荧光探针的特性,设置PCR反应条件,包括初始变性、循环扩增和荧光信号检测等步骤。
5. 荧光定量PCR实验操作:将反应体系加入PCR管或板中,放入荧光定量PCR 仪中,进行扩增和信号检测。
记录荧光信号变化曲线。
结果与讨论:通过荧光定量PCR实验,我们成功检测到目标基因在不同组织中的表达水平。
在实验中,我们选择了心脏、肝脏和肺脏作为待测组织,以GAPDH作为内参基因。
在荧光定量PCR实验中,我们观察到了荧光信号的实时变化曲线,通过计算Ct值(Cycle threshold),可以获得目标基因的相对表达量。
实验结果显示,在心脏组织中,目标基因的表达量较高,Ct值较低;而在肝脏和肺脏组织中,目标基因的表达量较低,Ct值较高。
这与我们的预期相符,心脏是目标基因的主要表达器官,而肝脏和肺脏则是次要表达器官。
这些结果为我们进一步研究目标基因的功能和调控机制提供了重要线索。
荧光定量PCR技术具有高灵敏度、高特异性和高准确性的优点,能够快速、准确地检测目标基因的表达水平。
通过该技术,我们可以对基因的表达调控机制进行深入研究,为疾病的诊断和治疗提供重要依据。
荧光定量pcr 实验报告

荧光定量pcr 实验报告荧光定量PCR 实验报告引言:荧光定量PCR(Polymerase Chain Reaction)是一种常用的分子生物学技术,用于扩增和定量特定DNA序列。
它通过在PCR反应中引入荧光探针,可以实时监测PCR扩增过程中的DNA产物累积情况。
本实验旨在利用荧光定量PCR技术,检测目标基因在不同样本中的表达水平。
材料与方法:1. 样本准备:从不同组织中提取总RNA,并利用逆转录酶将RNA转录成cDNA。
2. PCR反应体系的配制:根据厂家提供的荧光定量PCR试剂盒说明书,配制PCR反应体系。
3. 荧光定量PCR扩增条件:预热酶解反应,然后进行PCR扩增,最后进行荧光信号检测。
4. 数据分析:利用荧光定量PCR仪器提供的软件对实验数据进行分析,计算目标基因的表达量。
结果与讨论:通过荧光定量PCR实验,我们成功检测到目标基因在不同样本中的表达水平。
实验结果显示,在肝脏组织中,目标基因的表达量最高,而在肺组织中表达量最低。
这与已有的研究结果一致,说明我们的实验结果是可靠的。
荧光定量PCR技术的优点在于其高灵敏度和高特异性。
相比传统的定量PCR技术,荧光定量PCR可以实时监测PCR反应过程中的荧光信号,从而准确测量目标基因的表达量。
此外,荧光定量PCR还可以同时检测多个基因的表达水平,提高实验效率。
然而,荧光定量PCR也存在一些限制。
首先,实验过程中需要使用荧光探针,这增加了实验的复杂性和成本。
此外,荧光定量PCR对样本的质量要求较高,如果样本中存在较多的抑制物质,可能会导致PCR扩增效率降低。
在今后的研究中,我们可以进一步探索荧光定量PCR技术的应用。
例如,可以利用这一技术研究基因表达的调控机制,或者检测特定基因在疾病发展过程中的变化。
此外,我们还可以结合其他分子生物学技术,如RNA测序和蛋白质组学,深入研究基因的功能和调控网络。
结论:荧光定量PCR是一种可靠、高效的分子生物学技术,用于检测目标基因的表达水平。
荧光定量pcr实验步骤

荧光定量pcr实验步骤荧光定量PCR实验步骤荧光定量PCR(Quantitative PCR,qPCR)是一种用于测量特定DNA序列数量的技术。
它可以快速、准确地定量检测目标DNA的含量,广泛应用于基因表达分析、病原体检测、遗传变异分析等领域。
下面将介绍荧光定量PCR实验的步骤。
一、实验前准备在进行荧光定量PCR实验之前,需要做好实验前的准备工作。
1. 设计引物和探针:根据目标DNA序列设计引物和探针,确保其特异性和互补性。
2. 准备模板DNA:从样品中提取目标DNA,并进行纯化和定量。
3. 制备PCR反应体系:根据PCR反应的需要,准备好PCR反应体系,包括引物、探针、模板DNA、Taq DNA聚合酶、缓冲液和dNTP等。
4. 验证引物和探针的特异性:使用目标DNA和非目标DNA进行聚合酶链式反应,通过凝胶电泳验证引物和探针的特异性。
二、荧光定量PCR实验步骤1. 反应体系配置:按照实验设计,配置好PCR反应体系。
将引物、探针、模板DNA、Taq DNA聚合酶、缓冲液、dNTP等加入反应管中,然后加入适量的去离子水。
2. PCR反应条件设定:根据引物和探针的特性,设定PCR反应的温度和时间参数。
一般来说,PCR反应包括预变性、变性、退火和延伸四个阶段,其中变性温度为95℃,变性时间为30秒,退火温度为60℃,退火时间为30秒,延伸温度为72℃,延伸时间根据目标片段的长度而定。
3. PCR反应体系装入仪器:将装有PCR反应体系的反应管放入荧光定量PCR仪器中。
4. 荧光定量PCR实验运行:启动荧光定量PCR仪器,按照预设的PCR反应条件进行PCR反应。
仪器会根据设定的温度和时间参数进行PCR反应,并实时检测荧光信号。
5. 数据分析与结果解读:荧光定量PCR仪器会自动记录PCR反应过程中的荧光信号,根据荧光信号的变化可以计算出目标DNA的数量。
通过对比不同样品的荧光信号差异,可以定量分析目标DNA 的含量。
实时荧光定量 PCR 仪快速操作指南说明书

实时荧光定量PCR仪快速操作指南1.连接电源从包装箱里取出仪器放置在实验台上,拿出电源线和电源适配器,将电源线一端与电源适配器相连,另一端连接至交流220V电源插座(AC85~264V)电源适配器的输出端连接到仪器电源插孔。
接口插入仪器电源插孔,注意插孔方向箭头方向朝上。
打开仪器背面的电源开关开启仪器,仪器自检成功,自动进入到软件界面。
2.样品准备◼准备试剂Q1600实时荧光定量PCR仪使用0.2ml透明离心试管根据试剂要求选用10-100μl合适的加液量。
◼离心操作配置完成试剂样本的试管在放入仪器之前,要用离心机进行离心操作,确保试剂液体处于试管底部,且液体内部不含有气泡。
◼放置样品将反应管按照顺序放入加热孔内。
3.设置程序,运行实验软件操作基本步骤:◼新建实验新建实验,选择运行槽◼基本设置设置实验名称,实验类型,检测项目,选择使用的染料。
◼样本设置设置样品名称,样本ID号,选择样本类型◼程序设置设置需要的反应程序◼保存文件单击保存,保存实验文件◼运行单击运行,运行实验。
4.结果分析◼实验结束,软件自动计算Ct值,根据判定规则自动出结果。
◼如需进行熔解分析和基因分型,在软件分析类型里选择相应的分析功能。
5.结果导出◼导出实验结果单击导出,导出实验数据。
◼结果打印连接配置的热敏打印机,选中要打印的样品孔,单击打印,打印实验结果。
6.关闭仪器◼实验结束,取出反应管◼长按仪器前面板关机按键,关闭仪器◼长时间不使用仪器,请关闭仪器背部左下角电源开关。
荧光定量PCR实验及数据分析

荧光定量PCR实验及数据分析荧光定量PCR(Fluorescence Quantitative PCR,qPCR)是一种常用于检测和定量分析DNA或RNA浓度的技术。
该技术利用荧光探针与靶分子结合产生荧光信号,通过荧光信号的强度可以确定靶分子的数量。
本文将介绍荧光定量PCR实验及数据分析过程。
实验步骤:1.样品制备:根据研究需要选择DNA或RNA样品,提取并纯化目标DNA或RNA,并将其浓度测定。
2.酶切反应:如果需要对目标DNA或RNA进行酶切,可在此步骤中将其酶切为较小的片段。
3.扩增反应体系的准备:根据实验设计和厂家提供的建议,配置扩增反应所需的试剂。
4.PCR扩增:将目标DNA或RNA与引物和荧光探针一起添加到PCR反应管中,并进行PCR扩增。
根据实验设计,设置反应的温度梯度和时间。
5.实时荧光检测:在PCR扩增的过程中,使用实时PCR仪不断监测PCR反应管中的荧光信号,记录其强度变化。
6.构建标准曲线:选取一系列已知浓度的标准样品进行PCR扩增,并记录每个标准样品的荧光信号强度。
根据标准曲线绘制荧光信号强度和目标分子浓度的对应关系。
7.分析样品数据:将样品的荧光信号强度与标准曲线进行比较,可以计算样品中目标分子的浓度。
根据实验目的,可以对样品数据进行统计分析,如计算平均值、标准差等。
数据分析:1.标准曲线分析:使用标准曲线中的已知浓度和相应的荧光信号强度,通过拟合曲线或插值方法,可以计算出待测样品中目标分子的浓度。
2.相对定量分析:若需要比较不同样品之间目标分子的相对表达水平,可选取一个内参基因作为参照,通过计算目标分子基因和内参基因相对表达量的比值,进行比较分析。
3.统计分析:根据实验设计和样品数量,可以使用合适的统计方法对数据进行分析。
常见的统计方法包括t检验、方差分析等。
总结:荧光定量PCR技术在生物学研究中具有重要的应用价值,能够快速、准确地定量测定DNA或RNA的浓度。
在进行实验时,需要注意实验步骤的正确操作,并合理选择实验设计和数据分析方法,以确保结果的可靠性和准确性。
荧光定量PCR实验

荧光定量PCR实验Real-time PCR(SYBR GreenⅠ)实验实时荧光定量PCR技术于1996年由美国Applied Biosystems公司推出,由于该技术不仅实现了PCR从定性到定量的飞跃,而且与常规PCR相比,它具有特异性更强、有效解决PCR污染问题、自动化程度高等特点,目前已得到广泛应用。
实时荧光定量PCR仪包括热循环仪、检测系统、计算机及软件分析系统。
一.实验目的定量待测样品中靶DNA含量的多少及其动态变化。
二.实验原理所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
每个管内的荧光信号达到设定的域值时所经历的循环数(Ct值)与该模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,Ct值越小。
利用已知起始拷贝数的标准品可作出标准曲线,其中纵坐标代表起始拷贝数的对数,横坐标代表Ct值。
因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。
定量的方法一般常用的有三种:SYBR green I,TaqMan探针和Molecular Beacon。
图1.Ct值的确定SYBR荧光染料法:在PCR反应体系中加入过量的SYBR荧光染料,SYBR荧光染料特异性地掺入DNA双链后发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。
但是在有非特异性扩增的情况下,不能采用SYBR green Ⅰ定量,因为非特异的扩增片段也会贡献荧光增量。
TaqMan荧光探针法:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个猝灭荧光基团。
探针完整时,报告荧光基团发射的荧光信号被猝灭基团吸收。
PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和猝灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的积累与PCR产物形成完全同步,TaqMan探针特异性地结合在靶序列上,只有真正发生靶序列扩增的反应才会有荧光增量的产生。
荧光定量pcr操作方法

荧光定量pcr操作方法荧光定量PCR(qPCR)是一种广泛应用于生物学实验室的分子生物学技术,用于定量分析DNA和RNA样本中的特定序列。
本文将详细介绍荧光定量PCR的操作方法。
1. 材料准备荧光定量PCR所需的常见材料包括:PCR试剂盒、模板DNA或RNA样本、引物、探针、荧光染料、DNA聚合酶、缓冲液、dNTPs和PCR管。
2. 引物和探针设计引物和探针的设计是荧光定量PCR的关键步骤。
引物应特异性结合目标序列,并具有相近的Tm(退火温度),以确保同步扩增。
探针通常是一种荧光标记的引物,与扩增产物的靶标序列相完全互补。
可以使用在线工具(如NCBI Primer-BLAST)来设计引物和探针。
3. PCR体系准备根据PCR试剂盒的说明书,准备包含缓冲液、dNTPs、引物、探针、聚合酶和水的PCR体系。
确保在无菌条件下操作,避免污染。
4. DNA模板和RNA样本的制备荧光定量PCR可以用于DNA和RNA的定量分析。
DNA样本可通过提取体液、血样、组织样本等制备。
RNA样本则需要在制备前将RNA转录为cDNA,可通过反转录反应来完成。
5. PCR反应条件设定根据所用PCR试剂盒的要求和具体实验需求,设定PCR反应条件,包括退火温度、扩增周期数等。
合适的PCR程序是保证扩增反应成功和准确性的重要因素。
6. PCR反应体系配制根据上述准备的PCR体系和模板DNA或RNA样本的需要量,将所需试剂添加到PCR管中。
轻轻混合液体,避免气泡的产生。
7. 荧光定量PCR反应将PCR管放入PCR仪中,启动PCR程序。
PCR程序通常包括初始变性步骤,退火和延伸步骤,以及一些可选的温度变化步骤。
8. 数据分析与结果解读在PCR仪运行期间,荧光定量PCR会持续记录发光信号。
PCR结束后,可以利用相关软件对数据进行分析,如绝对定量法、相对定量法和标准曲线法。
这些分析方法可用于计算样本中目标序列的浓度。
需要注意的是,在进行荧光定量PCR实验时要注意以下几点:- 严格按照操作规程进行操作,确保实验的准确性和可重复性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
荧光定量PCR实验指南(一)一、基本步骤:1、目的基因(DNA和mRNA)的查找和比对;2、引物、探针的设计;3、引物探针的合成;4、反应体系的配制;5、反应条件的设定;6、反应体系和条件的优化;7、荧光曲线和数据分析;8、标准品的制备;二、技术关键:1、目的基因(DNA和mRNA)的查找和比对;从/网点的genbank中下载所需要的序列。
下载的方式有两种:一为打开某个序列后,直接点击“save”,保存格式为“.txt”文件。
保存的名称中要包括序列的物种、序列的亚型、序列的注册号。
然后,再打开DNAstar软件中的Editseq 软件,点击“file”菜单中的“import”,打开后点击“save”,保存为“.seq”文件。
另一种直接用DNAstar软件中的Editseq软件,点击“file”菜单中的“open entrez sequence”,导入后保存为“.seq”文件,保存的名称中要包括序列的物种、序列的亚型、序列的注册号。
然后要对所有的序列进行排序。
用DNAstar软件中的Seqman软件,点击“sequence”菜单中的“add”,选择要比较的“.seq”的所有文件,点击“add”或“add all”,然后点击“Done”导入要比较的序列,再点击“assemble”进行比较。
横线的上列为一致性序列,所有红色的碱基是不同的序列,一致的序列用黑色碱基表示。
有时要设定比较序列的开始与结尾。
有时因为参数设置的原因,可能分为几组(contig),若想全部放在一组中进行比较,就调整“project”菜单下的“parameter”,在“assembling”内的“minimum math percentage”默认设置为80,可调低即可。
再选择几个组,点击“contig”菜单下的“reassemble contig”即可。
选择高低的原则是在保证所分析的序列在一个“contig”内的前提下,尽量提高“minimum math percentage”的值。
有时因此个别序列原因,会出现重复序列,碱基的缺失或插入,要对“contig”的序列的排列进行修改,确保排列是每个序列的真实且排列同源性最好的排列。
然后,点击“save”保存即可。
分析时,主要是观察是否全部为一致性的黑色或红色,对于弥散性的红色是不可用的。
2、引物和探针设计2.1引物设计细心地进行引物设计是PCR中最重要的一步。
理想的引物对只同目的序列两侧的单一序列而非其他序列退火。
设计糟糕的引物可能会同扩增其他的非目的序列。
下面的指导描述了一个可以增加特异性的引物所具有的令人满意的特点:序列选取应在基因的保守区段;扩增片段长度根据技术的不同有所分别:sybr green I技术对片段长度没有特殊要求;Taqman探针技术要求片段长度在50bp-150bp;避免引物自身或与引物之间形成4个或4个以上连续配对;避免引物自身形成环状发卡结构;典型的引物18到24个核苷长。
引物需要足够长,保证序列独特性,并降低序列存在于非目的序列位点的可能性。
但是长度大于24核苷的引物并不意味着更高的特异性。
较长的序列可能会与错误配对序列杂交,降低了特异性,而且比短序列杂交慢,从而降低了产量。
Tm值在55-65℃,GC含量在40%-60%;引物之间的TM相差避免超过2℃;引物的3’端避免使用碱基A;引物的3’端避免出现3个或3个以上连续相同的碱基。
为避免基因组的扩增,引物设计最好能跨两个外显子。
2.2 T aqman 探针设计一般设计原则:探针位置尽可能地靠近上游引物;探针长度通常在25-35bp,Tm值在65-70℃,通常比引物TM高5-10℃,GC含量在40%-70%。
探针的5’端应避免使用碱基G。
整条探针中,碱基C的含量要明显高于G的含量。
为确保引物探针的特异性,最好将设计好的序列在blast中核实一次,如果发现有非特异性互补区,建议重新设计引物探针。
(/BLAST)2.3 T aqman MGB 探针设计介绍MGB探针的优点:MGB探针较短(14-20bp),更容易找到所有排序序列的较短片段的保守区。
短片段探针(14-20bp)加上MGB后,Tm值将提高10℃,更容易达到荧光探针Tm值的要求。
MGB探针的设计原则探针的5’端避免出现G,即使探针水解为单个碱基,与报告基团相相连的G碱基仍可淬灭基团的荧光信号。
用primerexpress软件评价Tm值,Tm值应为65-67℃。
尽量缩短Taqman MGB探针,但探针长度不少于13bp。
尽量避免出现重复的碱基,尤其是G碱基,应避免出现4个或4个以上的G重复出现。
原则上MGB探针只要有一个碱基突变,MGB探针就会检测到(MGB探针将不会与目的片段杂交,不产生荧光信号)。
因此,在进行SNP检测时,为了检测到突变子,即Taqman MGB不与目的片段杂交,不产生荧光信号,探针目的片段产生荧光信号检测将探针的突变位点尽量放在中间1/3的地方。
注意:为了满足上述要求的4个条件,探针的突变位点可向3’端移动,但突变位点至少在离3’端2个碱基的前方(即必须确保探针的后两个碱基是绝对的保守),以进行SNP检测。
反过来,若要进行同类检测,找的是保守片段区,探针中不应有突变位点。
若探针即便是只有13个bp,探针仍不完全保守。
有几个突变,突变位点也应靠近探针的5’端,这样,即便是突变,探针也可与目的片段杂交,产生荧光信号。
另一种方法是设计简并探针,也可达到即使是突变,仍可检测到突变。
2.4 实时多重PCR探针的选择:多重实时PCR的多种含意有两种:一为选择保守的探针和引物,利用不同的染料标记探针,在检测时可根据荧光的颜色来判定不同的产物。
另一种为选择保守的引物,扩增不同长度的目的片段,反应中加入SYBRN染料,最后根据不同目的片段的Tm值来判定不同的物品。
多重实时PCR的荧光探针应为同一类型:如同时为Taqman 探针、或同时为MGB探针、或同时为Beacon 探针。
在多重PCR中,多重PCR的各个引物之间相互干扰和各个探针之间相互干扰分析:设计好各对引物和探针后,重新在用DNAstar软件中的Primerselect软件,打开保守在同一文件中的多重PCR的引物文件,然后两两分别选中所设计的多重引物或两两分别选中所设计的多重探针后,在“report”菜单下“primer pair dimers”,分析上下游引物的dimers。
弹出的窗口中就告诉此对引物有多少个dimer,并对此对引物用dG值进行评价(通常给出最差的dG值,理论上是dG值越大越好)。
3、影响PCR及荧光PCR 的其他因素引物的设计和选择符合荧光PCR的探针并进行设计对于实时荧光PCR尤其重要。
可以说,不合理的设计意味着绝对的失败。
但是,好的设计并不等于好的实验结果,影响PCR和荧光PCR的因素非常多,下面择其重要进行介绍。
3.1 引物退火温度引物的一个重要参数是熔解温度(Tm)。
这是当50%的引物和互补序列表现为双链DNA 分子时的温度。
Tm对于设定PCR退火温度是必需的。
在理想状态下,退火温度足够低,以保证引物同目的序列有效退火,同时还要足够高,以减少非特异性结合。
合理的退火温度从55℃到70℃。
退火温度一般设定比引物的Tm低5℃。
设定Tm有几种公式。
确定引物Tm最可信的方法是近邻分析法。
大部分计算机程序使用近邻分析法——从序列一级结构和相邻碱基的特性预测引物的杂交稳定性。
所有oligo软件会自动计算引物的Tm值。
在设置退火温度时可以如下进行:以低于估算的Tm5℃作为起始的退火温度,以2℃为增量,逐步提高退火温度。
较高的退火温度会减少引物二聚体和非特异性产物的形成。
为获得最佳结果,两个引物应具有近似的Tm值。
引物对的Tm差异如果超过5℃,就会由于在循环中使用较低的退火温度而表现出明显的错误起始。
如果两个引物Tm不同,将退火温度设定为比最低的Tm低5℃。
或者为了提高特异性,可以在根据较高Tm设计的退火温度先进行5个循环,然后再根据较低Tm设计的退火温度进行剩余的循环。
这使得在较为严谨的条件下可以获得目的模板的部分拷贝。
3.2 引物浓度引物的浓度会影响特异性。
最佳的引物浓度一般在0.1到0.5μM。
较高的引物浓度会导致非特异性产物扩增。
为了确定引物浓度,可以在260nm(OD260)测量光密度值。
然后使用光吸收值和微摩消光系数的倒数(nmol/OD),通过Beers法则(公式1)计算引物浓度。
微摩消光系数可以使用公式2计算。
与大分子双链DNA可以使用平均消光系数不同,确定引物的精确浓度必须使用计算的消光系数。
这是因为引物较短,碱基组成差异很大。
在oligo软件上可以计算出引物的的消光系数(OD/μmol)和消光系数的倒数(μmol/OD)。
一般商业合成的引物以O.D.值表示量的多少,在一般情况下,20个碱基长的引物,1个O.D.加100ul水后引物浓度为50pmol/ul(50μM)。
也可以用OLIGO软件,根据引物的的消光系数(OD/μmol),计算出一定的工作浓度下,引物的加水量。
浓度(μM)=A260(OD/ml)×稀释系数×消光系数的倒数(nmol/OD)举例:计算某寡核苷酸(溶于1ml水中),取其中的10μl稀释100倍(加入990μl)水中。
在A260处测吸光度为0.2。
计算消光系数的倒数为4.8 nmol/OD,代入得:浓度=0.2(OD/ml)×100×4.8(nmol/OD)=96nmol/ml=96μM3.3 引物、探针的纯度和稳定性定制引物的标准纯度对于大多数PCR应用是足够的。
部分应用需要纯化,以除去在合成过程中的任何非全长序列。
这些截断序列的产生是因为DNA合成化学的效率不是100%。
这是个循环过程,在每个碱基加入时使用重复化学反应,使DNA从3'到5'合成。
在任何一个循环都可能失败。
较长的引物,尤其是大于50个碱基,截断序列的比例很大,可能需要纯化。
引物产量受合成化学的效率及纯化方法的影响。
定制引物以干粉形式运输。
最好在TE重溶引物,使其最终浓度为100μM。
也可以用双蒸水溶解。
引物的稳定性依赖于储存条件。
应将干粉和溶解的引物储存在-20℃。
以大于10μM浓度溶于TE的引物在-20℃可以稳定保存6个月,但在室温(15℃到30℃)仅能保存不到1周。
干粉引物可以在-20℃保存至少1年,在室温(15℃到30℃)最多可以保存2个月。
探针即寡核苷酸进行荧光基团的标记,标记本身有效率的区别。
探针标记以后一般应该纯化,纯度高、标记效率高的探针不仅荧光值高,且保存时间可以高达一年以上。
不同的生物技术公司探针标记效率和纯度有很大的区别。
由于反复冻融易导致探针降解,在确认探针质量好的情况下,最好稀释成2uM(10×),作为工作浓度分装多支,避光保存。