羰基化合物的α-烷基化和催化烷基化反应
第四章 手性药物的制备技术2

3.手性配体的来源及其与过渡金属的络合
不对称合成中使用的大量手性配体主要来自手性 库中的天然原料,典型的例子是酒石酸及其酯类和 金鸡纳生物碱,酒石酸在非均相镍催化的不对称氢 化和均相钛催化的不对称环氧化等反应中充当手性 配体,金鸡纳生物碱作为手性配体用于非均相钯催 化的不对称氢化和均相锇催化的烯烃的不对称二羟 基化。金鸡纳生物碱本身作为不对称催化剂,用于 一系列碱催化反应中。大部分手性二瞵配体是以相 对便宜的天然化合物原料合成的。
在不对称催化合成中,手性配体有两方面的作用, 一是加速反应,二是手性识别和对映体控制。
在不对称催化合成反应中,手性配体与过渡金属 的络合加快了反应速度,并提高了反应的立体选择性, 这种现象被称为配体促进的催化。换句话说,当过渡 金属配合物催化活性远远高于过渡金属本身时,才能 看到反应的高度立体选择性。
5.生物碱类 生物碱类分子量大、价格高。常用的金鸡纳生物
碱类仅作为拆分剂用于某些外消旋酸的拆分,结构见 图4-7。关于金鸡纳生物碱类作为不对称催化剂的手性 配体或不对称催化反应的碱性催化剂的研究很多,有 一定的应用前景。
三、手性药物合成实例
直接结晶法简单经济,但适用范围有限。 非对映体结晶法较通用,但需要大量的拆分剂和 溶剂,操作繁琐,还有非目标对映体的消旋化、拆分 剂回收套用等工序。 动力学拆分中非目标异构体的自发消旋化提高了 收率,并可通过调节转化率控制产物光学纯度,可与 不对称合成相媲美。 催化不对称合成所用的手性催化剂结构明确、种 类繁多;反应条件温和,生产效率高,已成为合成手 性药物的重要方法。
▪
树立质量法制观念、提高全员质量意 识。20.12.2020.12.20Sunday, December 20, 2020
▪
羰基化合物的α-烷基化和催化烷基化反应
2.4 配位型的环内手性传递 配位型的手性烯醇体系,金属离子在固定原有的手性和烯醇部分之 间的立体化学关系至关重要。 2.4.1β-羟基酸 在烯醇化过程中这些底物可以形成E烯醇或者Z烯醇,对这两种烯醇 体系的任何一种而言,配位作用在确定烯醇对映面的选择中都可能 是决定性的。如图2.12所示,由于Re面进攻Z-或E-烯酵,结果都形 成了主要的组分。
从一些易得的环氧基硅烷基醚开始,合成α,α—二取代α—氨基酸衍 生物:
具有张力的氨基醇衍生的酮酯或酰胺与格氏试剂反应,制备对映体 纯叔-α -羟基酸 :
2.6 双内酰亚胺体系 甘氨酸和其它氨基酸经过二酮哌嗪,进行O—甲基化得到六元杂环产 物,水解以后以高对映体过量获得了α-甲基氨基酸。
2.7 用于羰基化合物的α-烷基化的手性辅剂一览表
三乙基铝化合物可从氢化铝和乙烯很经济地以工业规模制备,因此 这类化合物成功的烷基化反应肯定会开辟一个活跃的新研究领域。
一些新配体:
2.10 ZnR2对酮的催化不对称加成: 叔醇不对称中心的对映选择性形成 许多生物活性的天然产物含有季碳原子,通过碳亲核试剂对酮的加 成来不对称合成叔醇近来越来越受到关注(也参见2.5节)。 DAIB(142)催化二苯基锌对酮的对映选择性加成:
樟脑磺酰胺-钛醇盐衍生物催化二甲基锌和二乙基锌对潜手性酮的对 映选择性加成:
2.11 不对称氰醇化反应 光学活性的磺肟145和Ti(OPri)4生成手性钛试剂催化三甲基甲硅烷基 氰化物对醛的不对称加成。
假定的反应机理:
139(X=CN)催化三甲基甲硅烷基氰化物与醛的反应。手性的二氰 基络合物在原位生成,不对称氰硅烷化给出的e.e.值达到75%。
在同环桥键羟基氨基二茂铁(—)—130存在下,11种芳族和脂族醛与 Et2Zn进行烷基化反应。生成的醇具有67%一97%的e.e.值。这种 二茂铁催化剂成功地用于使芳族醛和直链或支链脂族醛的烷基化, 产生的仲醇e.e.值高达97%。即使用β—支链的脂族醛,得到的光 学产率也比用131所报道的要高。
第5章第四节 利用前手性原料制备手性药物

要点:
不对称合成概念 手性源合成 手性合成子与手性辅剂 一个成功的不对称合成反应的标准
不对称合成分为对映体选择性合成和非对映异构体选择性 合成两类 对映体选择性合成指潜手性底物在反应中有选择的生 成一种对映异构体; 非对映异构体选择性合成指手性底物在生成一个新的 不对称中心时,选择性生成一种非对映异构体。例如潜手 性烯烃和手性烯烃的环氧化反应,前者需手性催化剂的不 对称诱导作用,而后者本身结构中的不对称中心具有不对 称诱导作用,不需另加手性催化剂。 不对称合成反应成为合成手性药物的重要途径,与经 典的拆分技术和酶催化的不对称合成反应等技术一道广泛 地用于工业生产。
萘普生合成
二、手性源的组成和应用 手性源中相对便宜的手性原料有糖类、羟基酸 类、氨基酸类、萜类和生物碱类。
手性药物合成实例 地尔硫卓(Diltiazem) 地尔硫卓属于钙离子拮抗剂,适用于缺血 性心脏病、运动性心绞痛及陈旧性心肌梗塞引 起的心绞痛。
随着酶催化动力学拆分技术的发展,发现粘 质沙雷氏菌中的酯酶可选择性地水解(2S,3R)环氧化物,有效地拆分Darzen反应的一对产物, 得到(2R,3S)-环氧化物。在这条路线中,拆分 一步提前至第二步,且反应总步骤缩短为6步, 逐渐取代了旧路线。
4.不对称催化氢化等还原反应 手性二膦铑催化剂的发现与应用,改变丁烯烃、 烯胺、羰基不对称催化氢化的发展进程,并使 不对称催化氢化成为制备各种光学纯化合物的 最有效最方便的方法之一。
例如:抗高血压药物L-多巴中的烯胺还原 采用DIPAMP手性铑催化剂立体选择性可达95%。
5. Sharpless环氧化反应 20世纪80年代初发现的烯丙基伯醇的 Sharpless环氧化反应,已经成为催化不对称 合成中的经典反应。Sharpless环氧化利用可 溶性的四异丙氧基钛(DET)和酒石酸二乙酯 (DIPT)或酒石酸二异丙酯(TBHP)为催化 剂,叔丁基过氧化物为氧化剂,得到立体选择 性大于95%和化学收率70%-90%的环氧化物。
羰基阿尔法位烷基化

羰基阿尔法位烷基化
羰基阿尔法位烷基化(β-Ketoenol α-Alkylation)是一种化学反应,由羰基酯与叔丁基烷基或三甲基烷基反应构建羰基阿尔法烷基产物。
羰基阿尔法位烷基化反应主要用于有机合成中,是合成羰基烯、烷、醇、酮和天冬酰脲等化合物的重要化学路线。
本反应是加成反应,生成的烯化合物易于形成对称的酮、醇等合成中间体。
羰基阿尔法位烷基化反应的催化剂主要有两类,一种是活性羰基酸催化剂(如金,铂,等),另一种是以碘化物为活性位点的热稳定催化剂。
在羰基阿尔法位烷基化反应中,活性羰基酸催化剂和热稳定催化剂的机理是一样的,都是羰基酯的活化,使叔丁基烷基或三甲基烷基可以与它发生反应。
羰基酯的活化会改变它的结构,促使叔丁基烷基或三甲基烷基发生加成反应,形成羰基阿尔法位烷基。
然而,这种反应受到温度和酸催化剂水溶液的影响,当反应体系受任何一种影响时,反应产物易发生水解而产生羰基醛,因此温度和pH值需要调节以得到所需的反应效果。
羰基阿尔法位烷基化反应是重要的合成路线,其中活性羰基酸催化剂和热稳定催化剂都很有效,可以合成多种化合物、中间体和介电材料。
此外,在复杂反应体系中,可以进行多步反应,形成更复杂的有机物。
高等有机化学第6章羰基化合物的反应精品PPT课件

R
H
C2H 5
+
H
OH
Ph
OH
H
C2H 5
H
R
Ph
Ph
H
C 2H 5
HO
H
R
主要产物
Ph
H
C 2H 5
H
OH
R
次要产物35oC源自R主次CH3
2.5 : 1
C6H5
>4 : 1
(CH3)2CH 5 : 1
(CH3)3C
49 : 1
-70oC
R (CH3)3C CH3
主
次
499 : 1
5.6 : 1
Cram规则二:当醛或酮的α-碳原子上连有羟基或氨基 等可以和羰基氧原子形成氢键的基团时,试剂将从含氢 键环的空间阻碍较小的一边对羰基进行加成。
Cram规则一:如果醛或酮的α-碳原子上连有三个大
小不同的基团(L、M、S分别代表大、中、小三个
基团)时,其优势构象为羰基键处在两个较小基团
之间的构象,试剂优先从空间阻碍较小的一边进攻
羰基。
O
次要产物
Nu-
M
S
M O S 主要产物
LR LR
Ph
O
H
H
C 2 H 5
H
O
P hH
C 2H 5 1 RMgX 2 H2O
——羰基C=O的吸电子作用使得a-H具有明显的酸性,在碱
性条件下可以离解,生成烯醇负离子,从而成为亲核试剂, 进攻羰基碳或卤代烃,发生亲核加成反应、亲核取代反应。
O CC H
BH2
O CC
O CC
烯 醇 负 离 子
烯醇负离子由于羰基的共轭作用得以稳定
第二章烷基化

OM R''X R' R' n-C4H9 CH2=CHCH3
O R'' R' R''X MeI MeI CH2=CHCH2Br
O R'' R' anti:syn 88:12 75:25 89:11
B. 五员环
O LDA O Br O Only product O
Ph LDA N O MeI Me
R'X Me2SO4 Me2SO4 MeI n-C3H7
OBut Li N O H R
e.e% 84(S) 98(S) 97(S) 97(S)
OBut Li N O S Me OMe O
E.手性腙烷基化
OMe OMe
N NH2 SAMP
N NH2 RAMP
N N Me Me
OMe LDA n-C4H7I Me H Me N N
常用的碱
(Me2CH)2NLi N Li N Li LTMP (Me3Si)2NLi
LDA
LICA
LHDS
手性的传递方式
A. 环内手性传递
OM EI+ R H O EI H H major R O H EI H R
B. 环外传递
OLi H Me Ph N N Me PhCH2Br -60 H Me Ph N O CH2Ph N Me >95 H Me Ph N O Me N CH2Ph 5
LiAlH4 RO H OH 肾上腺能阻断剂 NH2
B.醛的手性加成
O R2M HX*
-RH
R' RMX*
R'' R2M
R' R
R'' OMX*
烷基化反应

烷基化反应有机化合物分子中连在碳、氧和氮上的氢原子被烷基所取代的反应。
碳原子上的烷基化①羰基的a碳上氢的烷基化。
羰基的a碳上的氢呈弱酸性,羰基的a碳原子在强碱(如氨基钠、氢化钠)的作用下,能与卤代烷发生烷基化反应,生成a碳烷基化产物:酮和酯的直接烷基化会发生自身缩合;也会发生多烷基化反应。
要获得a-碳单烷基化产物,可用四氢吡咯、吗啉等仲胺制成烯胺,再与活泼的卤代烷(碘甲烷、卤代苄等)反应,生成取代的烯胺,经水解即得烷基化的羰基化合物:②活泼亚甲基的烷基化。
处于两个活性基团之间的亚甲基比较活泼,在醇钠作用下容易烷基化。
活性基团可以是硝基、羰基、酯基或氰基等。
例如取代的丙二酸酯合成法和乙酰乙酸酯合成法:H2C(COOC2H5)2+C2H5O-Na+CH(COOC2H5)2-Na++C2H5OHCH(COOC2H5)2-Na+RXRCH(COOC2H5)2+NaXCH3COCH2COOC2H5+C2H5O-Na+(CH3COCHCOOC2H5)-Na++C2H5OH式中R为烷基;X为卤素。
取代的丙二酸酯、乙酰乙酸酯水解后容易脱羧、分解成取代乙酸或酮,此反应广泛用于有机合成。
这些烷基化反应都是在无水条件下进行的。
③相转移催化的烷基化。
利用相转移催化剂使处于两个互不相溶的液相系统中的反应物进行反应。
无需在无水条件下操作,可以用浓氢氧化钠水溶液代替无水醇钠。
反应条件温和,操作简便。
常用的催化剂有四级铵盐(Q+X-),如(n-C4H9)4N+HSO4-、四级磷盐[(C2H5)3P+CH2C6H5]Cl-或冠醚等。
反应物于界面处与碱作用,生成负碳离子。
后者与四级铵盐正离子形成离子对,转移到有机相中,与卤代烷进行烷基化反应。
例如:氧原子上的烷基化醇钠与卤代烷反应生成醚,这是合成不对称醚的重要方法:RONa+R′XROR′+NaX酚的酸性较醇强,采用氢氧化钠可生成芳香氧负离子,然后进行烷基化。
例如:硫酸二酯类也是常用的烷基化试剂,其活性比卤代烷高,反应条件温和,一般只有一个烷基参加反应。
第十一讲 羰基化合物a-取代反应和缩合反应 PPT课件

三、Aldol缩合(羟醛缩合)
3、分子内Aldol缩合(分子内羟醛缩合)—五、六元环
2020年6月17日
温州医学院药学院
三、Aldol缩合(羟醛缩合)
3、分子内Aldol缩合(分子内羟醛缩合)—五、六元环
2020年6月17日
温州医学院药学院
三、Aldol缩合(羟醛缩合)
3、分子内Aldol缩合(分子内羟醛缩合)—五、六元环
亲核试剂
底物——亲电
2020年6月17日
-羟基醛 -羟基酮 温州医学院药学院
三、Aldol缩合(羟醛缩合) (P286)
1、简单Aldol缩合(羟醛缩合) —— 反应机理
2020年6月17日
-羟基醛 温州医学院药学院
三、Aldol缩合(羟醛缩合)
1、简单Aldol缩合(羟醛缩合) —-羟基醛(酮)的脱水
2、丙二酸二乙酯(小结)
2020年6月17日
温州医学院药学院
近期复习预习
■ 复习巩固第 13、14章化学反应部分,完成 相应习题(见学习指导与习题集)
■ 预习第15章化学反应部分
2020年6月17日
温州医学院药学院
三、Aldol缩合(羟醛缩合)
3、Aldol缩合(羟醛缩合)——应用示例(合成2)
用不多于3个碳的有机原料
HCHO 浓-OH
1. NaClO 2. H+
稀-OH 稀-OH
2020年6月17日
温州医学院药学院
四、酮的烷基化和酰基化反应(P415)
1、酮的烷基化——通过烯醇负离子和烯胺两种方法
E = R-,RCO-
2020年6月17日
温州医学院药学院
1、乙酰乙酸乙酯
(2)反应——与溴的反应(可使溴水褪色)
有机化学基础知识点整理烷基化反应与烷化反应机制

有机化学基础知识点整理烷基化反应与烷化反应机制【有机化学基础知识点整理】烷基化反应与烷化反应机制烷基化反应是有机化学中一类重要的反应,主要涉及底物中的氢原子被烷基基团取代的过程。
烷化反应机制则是指在此类反应中所形成的反应中间体和过渡态的详细变化过程。
本文将整理烷基化反应的几个基础知识点,并探讨其中常见的烷化反应机制。
一、烷基化反应的基本概念烷基化反应是有机化学中最为常见和广泛应用的一类反应,其中最常见的就是烷基磺酸盐的合成。
其原理是通过引入一个“烷基基团”,将底物中的氢原子取代。
烷基化反应可以形成新的碳-碳键或碳-氧键,产生新的有机化合物。
二、烷基化反应的分类烷基化反应根据反应过程的类型和反应底物的不同,可以分为以下几类:1.质子接受型烷基化反应:底物中的质子被烷基离子或烷基金属试剂取代。
2.质子捐赠型烷基化反应:底物中的烷基基团将质子给予另一分子,形成新的碳-碳键。
3.亲电取代型烷基化反应:底物中的亲电试剂与烷基基团发生亲电取代反应。
4.自由基烷基化反应:底物中的烷基离子与自由基试剂发生反应,形成新的碳-碳键。
三、常见的烷化反应机制1.质子接受型烷基化反应机制:在质子接受型烷基化反应中,底物中的质子被烷基离子或烷基金属试剂取代。
这类反应通常采用强碱作为反应试剂,如氢氧化钠(NaOH)。
反应机理如下:底物 + 烷基离子/烷基金属试剂→ 产物 + 相应的离子/金属盐该反应机制属于亲核取代反应,质子首先被碱中的氢氧根离子去质子化,然后进一步与烷基离子或金属离子发生亲核取代反应,最终形成新的碳-碳键。
2.自由基烷基化反应机制:在自由基烷基化反应中,底物中的烷基离子与自由基试剂发生反应,形成新的碳-碳键。
这类反应通常通过热、光或自由基引发剂来产生自由基,如过氧化氢(H2O2)或过氧化苯甲酰(Benzoyl peroxide)。
反应机理如下:底物 + 自由基试剂→ 产物该反应机制属于自由基取代反应,首先由自由基引发剂产生自由基,然后自由基与底物反应,形成新的碳-碳键。
[理学]第六章 羰基化合物的反应
![[理学]第六章 羰基化合物的反应](https://img.taocdn.com/s3/m/f28edb13f111f18583d05a59.png)
sp3杂化 四面体
产物中基团拥挤程度增大。
R 越大,妨碍Nu:从背后进攻C原子。
6.2 羰基加成反应及产物
a. 与水加成
O
H+或OH-
OH R' + H2O R C OH R'
R
C
除甲醛、多卤代醛外,其它醛的水合反应平衡偏向 左边。
(2)与ROH的加成
H+
OCH2CH3 CH3CH OH
CH3CH=O + CH3CH2OH
H
O
C2H5
PhH
Ph H H C2H5 O
R H H Ph C2H5 OH
+
H
O
C2H5
1 RMgX 2 H2O
PhH
OH H H Ph C2H5 R
35oC R CH3 C6H5 (CH3)2CH (CH3)3C -70oC R (CH3)3C CH3
主 2.5 > 4 5 49
: : : :
H O O
L
R
S
Nu-
C2H5 H C6H5
O C C CH3
C 2 H5 H C6H5
LiAlH4 乙醚
OH H CH3
H2 O
C
C
+
C2H5 H C6H5
C
C
H OH CH3
75%
25%
当羰基和一个手性中心连接时,反应符合 克莱姆规则一。
6.5 碳负离子
O R C CH3
-
OH
O R C
CH2
第六章 羰基化合物的反应
6.1 羰基化合物的反应机理 6.2 羰基加成反应及产物 6.3 加成-消除反应 6.4 羰基化合物的反应活性和加成的立体选择性 6.5 碳负离子 6.6 各种重要的缩合反应 6.7 羰基与叶子立德的反应 6.8 羧酸衍生物的亲核加成 6.9 亲核性碳 6.10 分子内催化作用
不对称反应及其应用

⒉不对称性
2.2.2 DIP规则——以顺序规则为基础
(1)有较高原子序数的原子排在有较低原子序数原子的前面。对 同位素原子,有较高质量的同位素排在有较低质量的同位素 的前面
(2)如果两个或多个相同的原子直接连接在不对称原子上,按照 相同的顺序对侧链原子进行比较,如果在侧链中没有杂原子 ,则烷基的顺序是叔基>仲基>伯基。当两个基团有不同的取 代基时,先比较在每个基团仲具有最高原子序数的取代基, 依据这些取代基的顺序来觉得这些基团的循序,含有优先取 代基的基团有最高的优先权,对于含有杂原子的基团,可以 应用类似的规则
映体引起的可能的副作用。
第7页/共59页
⒉不对称性
? 不对称性的创立条件 ? 具有手性的化合物的命名
第8页/共59页
⒉不对称性
2.1 创立条件 (通过以下任何一个条件而创立)
(1)化合物带有不对称碳原子(然而,不对称碳原子的 存在对于光学活性既非必要条件也非充分条件) (2)化合物带有其他四价共价键联的不对称原子,四个 价键指向四面体的四个角,但四个基团不相同,它们 是:Si,Ge,N(在季胺盐或N-氧化物中),Mn,Cu,Bi和 Zn——形成四面体配位 (3)化合物带有三价的不对称原子,原子带有角锥键, 与三个不同的基团相连,未共享电子对类似于第四基 团——倒伞效应:(a)三元环;(b)三元杂环,杂 原子含未共用电子对;(c)桥头键
若xyzw为不同基团,且 x>y>z>w,则从C到w方向观 察,x→y → z为顺时针方 z 向,则为R构型,反之为S 构型
x
Cw y
第13页/共59页
(2)轴手性
沿轴向看,比较靠近观
察者的一对配体在优先顺序
中排在头两ቤተ መጻሕፍቲ ባይዱ,另一对配体
叔丁醇钾做羰基α位烷基化

叔丁醇钾做羰基α位烷基化
叔丁醇钾可以用来进行羰基α位烷基化反应。
这个反应是通过在叔丁醇钾存在下与醛或酮反应,将烷基基团连接到羰基化合物的α位。
反应机理一般涉及到以下几个步骤:
1. 叔丁醇钾通过失去一个甲基离子生成 tert-BuO-。
2. tert-BuO-攻击羰基化合物的α位,形成一个负离子中间体。
3. 中间体经过质子转移生成碳碳键。
4. 质子化生成产物。
这个反应通常在室温下进行,由于叔丁醇钾具有较高的碱性,可以加速反应的进行。
此外,反应选择性较高,可以通过调整反应条件和底物结构来实现所需产物的选择。
化学制药工艺学

1、药物合成工艺路线设计方法:类型反应法分子对称法追溯求源法模拟类推法2、类型反应法:指利用常见的典型有机化学与合成方法进行合成路线设计的方法。
分子对称法:具有分子对称性的化合物往往由两个相同的分子经化学合成反应制得,或可以在同一步反应中将分子的相同部分同时构建起来。
追溯求源法(倒推法、逆向合成分析):从药物分子的化学结构出发,将其化学合成过程一步一步逆向推导进行寻缘的思考方法。
模拟类推法:从初步的设想开始,通过文献调研,改进他人尚不完善的概念和方法来进行药物工艺路线设计。
3、平顶型反应:反应条件易于控制,可减轻操作人员的劳动强度。
P39 图2-1尖顶型反应:反应条件苛刻,条件稍有变化收率就会下降;与安全生产技术、三废防治、设备条件等密切相关。
4、一勺烩(一锅合成):在合成步骤改变中,若一个反应所用的溶剂和产生的副产物对下一步反应影响不大时,可将两步或几步反应按顺序,不经分离,在同一反应罐中进行,习称“一勺烩”5、常见的设备材质:铁、铸铁、搪玻璃、陶瓷、不锈钢6、①可逆反应:特点:正反应速率随时间逐渐减少,逆反应速率随时间逐渐增大,直到两个反应速率相等,反应物和生成物浓度不再随时间而发生变化。
可以用移动方法来破坏平衡,以利于正反应的进行,即设法改变某一物料的浓度来控制反应速率。
平行反应(竞争性反应):级数相同的平行反应,其反应速率之比为一定常数,与反应物浓度及时间无关。
即不论反应时间多长,各生成物的比例是一定的。
可通过改变温度、溶剂、催化剂等来调节生成物的比例。
②工业生产的合适配料比确定:A凡属可逆反应,可采取增加反应物之一的浓度(即增加其配料比),或从反应系统中不断除去生成物之一的办法,以提高反应速率和增加产物的收率。
B当反应生成物的生成量取决于反应液中某一反应物的浓度时,则增加其配料比。
C倘若反应中,有一反应物不稳定,则可增加其用量,以保证有足够量的反应物参与反应。
D当参与主、副反应的反应物浓度不尽相同时,利用这一差异,增加某一反应物的用量,以增加主反应的竞争能力。
羰基的加成反应在有机合成中的应用

一、概述羰基的加成反应是有机化学中一种重要的反应类型,具有广泛的应用价值。
本文将探讨羰基的加成反应在有机合成中的应用。
二、羰基的加成反应基本原理1. 羰基的结构特点羰基是含有碳氧双键的有机化合物官能团,一般表示为“C=O”。
羰基通常分为醛、酮和羧酸三种类型,它们具有较强的电性,是有机合成中常见的反应物和产物。
2. 羰基的加成反应羰基的加成反应是指具有亲核试剂(如胺、醇等)与羰基发生亲核加成反应,形成加成产物的过程。
这种反应通常在碱性或酸性条件下进行,产物可以是醇、醛、酮、羧酸等化合物。
三、羰基的加成反应在有机合成中的应用1. 羰基的还原羰基的加成反应可用于醛酮的还原反应,常见的还原试剂有金属氢化物(如氢化钠、氢化铝锂等)和还原醇(如醇、胺等)等。
借助该反应,可以将醛酮还原为相应的醇,扩大有机合成的应用范围。
2. 羰基的羟化反应在羰基的加成反应中,羟胺(氨和水的混合物)可以与醛酮发生羟化反应,形成羟醇。
这种反应被广泛应用于药物合成和其他有机合成领域,具有重要的化学和生物活性。
3. 羰基的羟胺加成在温和的酸性条件下,羟胺可以与羰基形成加成产物。
该反应常用于合成β-羟基酮或β-羟基醛的过程中,产物可以进一步转化为药物分子或生物活性分子。
4. 羰基的羟胺甲酰化在适当的反应条件下,羰基与羟胺发生甲酰化反应,生成羰基甲酰胺。
这种反应在药物合成和有机合成中具有重要的应用价值,可以构建含氨基酰胺结构的化合物。
5. 羰基的醇加成在碱性条件下,醇可以与羰基形成加成产物。
这种反应常用于合成醛醇或酮醇的过程中,产物在有机合成中具有重要的应用价值。
6. 羰基的氧化反应在适当的氧化条件下,羰基可以与氧化剂发生氧化反应,形成羧酸。
这种反应在生物活性分子或有机合成中具有重要的应用价值。
7. 羰基的胺加成在适当的酸性条件下,胺可以与羰基形成加成产物。
这种反应在合成酰胺类化合物中具有重要的应用价值,是有机合成中一种有效的方法。
四、结论羰基的加成反应在有机合成中具有广泛的应用价值,可以用于合成各种类型的有机化合物,包括醇、醛、酮、羧酸等。
有机物α位β位γ位

有机物α位β位γ位有机物是指由碳、氢、氧、氮、硫等元素组成的化合物,它们广泛存在于自然界中,包括生物体内和地球表面的各种环境中。
有机物的结构复杂多样,其中的一些特定位置对于它们的性质和用途具有重要影响。
本文将着重介绍有机物中的α位、β位和γ位,以及它们在有机化学中的重要性。
α位是有机物中最重要的位置之一,它指的是与官能团相连的碳原子上的那个碳原子。
这个位置的化学性质通常比较稳定,因为它与官能团之间有较强的σ键作用力。
许多有机反应中,α位是反应的主要位置,因为它的反应性较高。
例如,酮类化合物在酸性条件下会发生α位羟基化反应,生成α-羟基酮。
这个反应可以通过羰基化合物的质子化来促进。
此外,α位还可以进行α-取代反应,例如通过酸催化或金属催化剂促进的α-烷基化和α-芳基化反应。
β位是与α位相邻的碳原子位置,也是有机化学中非常重要的位置之一。
β位的性质通常比α位更加活泼,因为它与α位之间的碳-碳单键比较弱,容易发生断裂。
在许多有机反应中,β位是反应的次要位置。
例如,α-烯酸酯在酸性条件下可以进行β-消除反应,生成烯烃。
这个反应可以通过β位的质子化来促进。
此外,β位还可以进行β-取代反应,例如通过金属催化剂促进的β-烷基化和β-芳基化反应。
γ位是与β位相邻的碳原子位置,它的化学性质比α位和β位更加不稳定。
在许多有机反应中,γ位往往是反应的次次要位置。
例如,酮类化合物在酸性条件下可以发生γ-位羟基化反应,生成γ-羟基酮。
这个反应可以通过酮类化合物的质子化来促进。
此外,γ位还可以进行γ-取代反应,例如通过金属催化剂促进的γ-烷基化和γ-芳基化反应。
有机物中的α位、β位和γ位在有机化学中具有重要的意义。
它们可以通过特定的反应进行取代或转化,从而改变有机物的性质和用途。
例如,α-烯酸酯的β-消除反应可以用于制备烯烃,这些烯烃可以用于制备聚合物和医药中间体。
γ-位羟基化反应可以用于合成γ-羟基酮,这些化合物具有抗病毒和抗癌活性。
第二章_烷基化反应

R2NH R3N
R2NH +
跟卤代烃的反应是主要方法之一, 缺点:易得到混合胺
1.伯氨的常用制备方法
(1):氨水和卤代烃的反应
O OH Br 70% NH2 NH3(70mol) O OH
用大大过量的氨水,抑制进一步反应, 该方法在工业中应用广泛
NO2 Cl O2N NH3/AcONH4 170 oC, 6h O2N NO2 NH2
CCl4/AlCl3 10-30 oC,3 h Cl Cl O H2O 80ຫໍສະໝຸດ O OH 95% CH2CH2CN
冠状动脉扩张药派克西林中间体
O OH C6H6/AlCl3 25 C, 4 h
o
冠状动脉扩张药普尼拉明中间体
MeO MeO CN AlCl3 MeO MeO
镇痛药延胡索乙素中间体
1.反应机理 :C+ 离子对芳环的亲电进攻
差向异构
OH CH3 cis CH3I/NaH/THF 50℃, 1 h OCH3 CH3 cis (100%)
OH CH3 trans
CH3I/NaH/THF 50℃, 1 h
OCH3 CH3 trans (100%)
多卤代物的醚化
CHCl3 + 3 CH3ONa CH(OCH3)3
CCl4
+
多卤代烃,甲醛,环氧乙烷
二.格氏试剂的C-烷基化 格氏试剂
OMe
Case:
NHBoc OH
PhCH2Br
NHBoc O
H N O O
二、酚的O-烷基化
1.卤代烃为烃化剂
CONH2 OH EtBr/NaOH 80~100℃, 19.6 x 10exp4 Pa CONH2 OEt
羰基化合物α—碳的不对称烷基化研究进展

作者: 刘永红;刘晓岚
作者机构: 兰州大学化学系3023信箱
出版物刊名: 天水师范学院学报
页码: 36-57页
主题词: 烷基化反应;羰基化合物;不对称;高立体选择性;手性试剂;光学产率;化学产率;有机合成;手性中心;Michael加成反应
摘要: 本文从羰基化合物α—碳的不对称烷基化所采用的方法(采用手性及潜手性试剂、底物,手性熔剂,手性催化剂以及为提高光学产率而采用空间体积大的骨架、引入抗衡离子、引入螫合环稳定构型或构象等等)为出发点,根据此类反应中间体的特点,把1988年至1994年间羰基化合物α—碳的不对称烷基化分为:通过锂、钠、钾、锌、钛等金属的烯醇盐,通过烯醇硫盐,通过烯醇硅醚,以及通过烯胺、亚胺、腙等含氮有机化合物为中间体等四部分进行简要的评述.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
图2.10所示的反应是anti -1,3—不对称诱导的一个实例。
2.3.4 降冰片体系
许多烷基化反应的例子涉及降冰片环系,在此环系中烯醇可以是环 内的或是环外的。由于是刚性的环系,无论是环内的或环外的烯醇 都显现出高度的不对称诱导(图2.11)。
2.4 配位型的环内手性传递
配位型的手性烯醇体系,金属离子在固定原有的手性和烯醇部分之 间的立体化学关系至关重要。
90(R)
2.4.5 手性腙的烷基化
Enders腙试剂法:SAMP/RAMP,可分别由(S)-脯氨酸和(R)-谷氨 酸大量制备。
例,合成切叶蚁警戒信息素31 :
2.4.6 恶唑啉体系的烷基化
容易从2-氨基乙醇衍生物和羧酸制备,原料易得,使用于宽的反应温 度范围,对各种试剂稳定,用作合成羧酸的潜在前体。
Evans型试剂:一种基于金属离子配位作用的,由脯氨醇型手性辅剂 衍生的两种形式的烯醇锂的非对映选择性烷基化,为合成α-取代羧 酸提供了一条有效的路线。对于烯醇体系16,烷基化反应优优先从 Si—面发生,而对于17,优先从Re—面发生。这个反应的特点是从 同一底物(16或17)开始,在烷基化产物酸水解以后,可以得到一对对 映体。
Evans酰亚胺制备烷基酸或相应化合物:
Evans试剂有效合成:
2.4.4 手性烯胺的烷基化 手性环己酮亚胺26 能进行高非对映选择性的烷基化。
PhCH 2 H
N
OMe
1. LDA 2.RX
O R H
Байду номын сангаас
26 RX Me 2SO 4 C 3H7I
CH 2=CHCH 2Br
e.e. 82(R)
95(R)
制备对映选择性纯的氨基酸:50用N-[二(甲硫基)次甲基]甘氨酸甲 酯酰化。
磺内酰胺50是优良的手性辅剂。例,氯化亚铜(I)催化的氯化烷基镁 对α,β—二取代E—烯磺内酰胺57的1,4—加成的不对称诱导。
2.5 季碳手性中心的形成
许多生物活性的天然产物含有季碳原子。Meyers发展了一种不对称 季碳原子的立体选择性引入的新方法。该方法基于γ-酮酸和(S)-颉 氨醇衍生的手性双环内酰胺的交替锂化和烷基化。
手性化合物如羧酸、酯、内酯和醇都可通过恶唑啉化学制备,手性
中心和其它官能团的位置可由制备的方式加以控制。恶唑啉方法学 在有机合成中提供了一种有用的工具。
例,合成欧洲松锯蜂性信息素:
二取代羧酸的制备:
2.4.7 酰基磺内酰胺体系 Oppolzer发展的酰基磺内酰胺50。合成α,α-二取代羧酸衍生物:
2.2 手性传递
手性烯醇:环内烯醇、环外烯醇和配位型环内烯醇
OM
OM
Z Y
X
Z
X
* R
* YR
环内烯醇
OM
Z X
R*
Y
环外烯醇
2.2.1 环内手性传递
原有不对称中心通过环共价键连接到烯醇的两个点,烯醇的几何构 型保持不变并与不对称中心的诱导无直接关联。
2.2.2 环外手性传递
虽然形成的不对称中心通过共价链连接到烯醇上,但手性传递和烯 醇间的立体化学关系并非固定。因为就构象而言,原有的手性部分 并不是通过连接到发生取代作用的三角中心的共价键固定在两个或 多个接触点上的。由于构象可变性的结果,常难以预料这类反应的 立体选择性。
脯氨醇手性辅剂不易回收,与水混溶,引入两个甲基形成叔醇18, 后处理容易回收。通过改变在酰基和烷基中的R`和R``先后反应顺序, 同样可获得羧酸的二个对映体。
C2对称性的吡咯烷21:
2.4.3 酰亚胺体系
酰亚胺化合物22、23( Evans试剂),不对称烷基化或醇醛缩合反应 的有效手性辅剂。
2.4.1β-羟基酸
在烯醇化过程中这些底物可以形成E烯醇或者Z烯醇,对这两种烯醇 体系的任何一种而言,配位作用在确定烯醇对映面的选择中都可能 是决定性的。如图2.12所示,由于Re面进攻Z-或E-烯酵,结果都形 成了主要的组分。
存在羟基Z-烯醇是主要的,不存在羟基E-烯醇是主要的。
实例:
2.4.2 脯氨醇型
OH NH2
(S)-缬氨醇
1.LDA 2.R``X
R
+
O HOOC
R
R
O
O
1.LDA
N
2.R`X
N
R`
O O
γ-酮酸
R O
N
双环内酰胺
R` R`` H2SO4,BuOH
(R=Ph)
O R`` R`
Ph
CO2Bu
O
硝基烯胺能与多种亲核试剂反应,生成加成—消除产物。
包括酮、醛和羧酸衍生物的各种羰基化合物构成了一类具酸性质子 的羰基化合物,其酸性为pKa自25至35(在DMSO中)的范围。羰基化 合物的代表性pKa值列于表2.1。按照羰基化合物的pKa值可以采用不 同的方法产生烯醇。
从羰基化合物产生烯醇,选用的碱要满足两个条件: 1.足够的碱性。 2.碱必须有空间位阻,以便阻碍该碱对羰基中心的亲核进攻。 金属胺化物满足以上条件: 1.二异丙基氨基锂LDA(在有机化学中被认为是最重要的碱) 2. 异丙基环己基氨基锂LICA 3.四甲基哌啶锂LTMP 4.硅烷基胺化物
第二章 羰基化合物的α-烷基化和催化烷基化反应
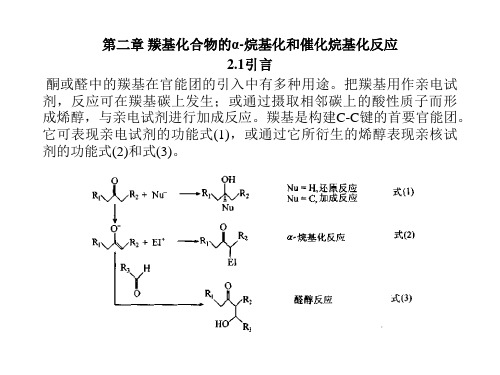
2.1引言
酮或醛中的羰基在官能团的引入中有多种用途。把羰基用作亲电试 剂,反应可在羰基碳上发生;或通过摄取相邻碳上的酸性质子而形 成烯醇,与亲电试剂进行加成反应。羰基是构建C-C键的首要官能团。 它可表现亲电试剂的功能式(1),或通过它所衍生的烯醇表现亲核试 剂的功能式(2)和式(3)。
亲电试剂进攻发生在烯醇的二个非对映面上,即,A进攻和E进攻, 产生酮产物13A和13E。通过假定的椅型过渡态得到的酮13A,可以 认为比酮13E优先形成,后者是由船型过渡态生成的。通过椅型过渡 态形成13A的“坚键烷基化”的能垒相对铰小。
2.3.3 五元环(环内型)
五元环内烯醇的非对映选择性烷基化反应中,1,3—和1,2—不对 称诱导都可能发生。在这种情况下,anti—诱导的烷基化过渡态主要 受空间位阻的控制方式所支配。
2.2.3 配位型的环内手性传递
环内手性传递和环外手性传递相结合而产生的一个概念性思想。本 章重点。
2.3 环内手性传递 2.3.1 六元环(环外型)
10E遭受烷基的空间张力R-X(OM),亲电试剂更有利于竖键进攻10A, 而不是平键进攻较不稳定的10E.
图2.6给出一些实例。
2.3.2 六元环(环内型)