依诺肝素钠标准(USP 39)
核准日期2006年12月
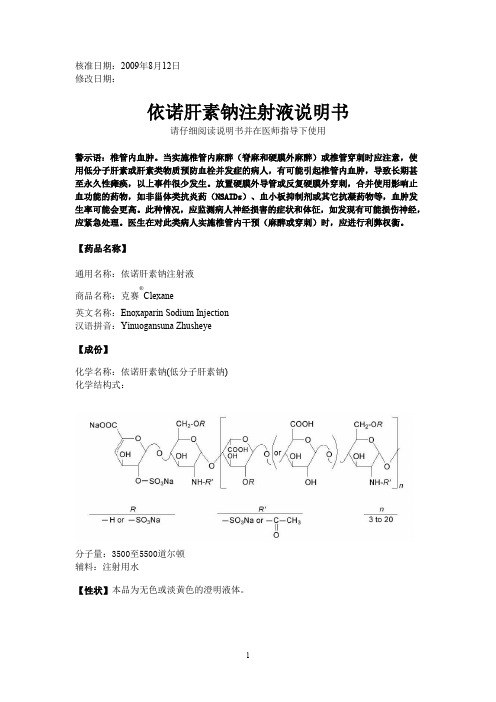
核准日期:2009年8月12日修改日期:依诺肝素钠注射液说明书请仔细阅读说明书并在医师指导下使用警示语:椎管内血肿。
当实施椎管内麻醉(脊麻和硬膜外麻醉)或椎管穿刺时应注意,使用低分子肝素或肝素类物质预防血栓并发症的病人,有可能引起椎管内血肿,导致长期甚至永久性瘫痪,以上事件很少发生。
放置硬膜外导管或反复硬膜外穿刺,合并使用影响止血功能的药物,如非甾体类抗炎药(NSAIDs)、血小板抑制剂或其它抗凝药物等,血肿发生率可能会更高。
此种情况,应监测病人神经损害的症状和体征,如发现有可能损伤神经,应紧急处理。
医生在对此类病人实施椎管内干预(麻醉或穿刺)时,应进行利弊权衡。
【药品名称】通用名称:依诺肝素钠注射液商品名称:克赛®Clexane英文名称:Enoxaparin Sodium Injection汉语拼音:Yinuogansuna Zhusheye【成份】化学名称:依诺肝素钠(低分子肝素钠)化学结构式:分子量:3500至5500道尔顿辅料:注射用水【性状】本品为无色或淡黄色的澄明液体。
【适应症】2000 Axa IU 和4000 Axa IU注射液:•预防静脉血栓栓塞性疾病(预防静脉内血栓形成) ,特别是与骨科或普外手术有关的血栓形成。
6000 Axa IU, 8000 Axa IU 和10000 Axa IU注射液:• 治疗已形成的深静脉栓塞,伴或不伴有肺栓塞,临床症状不严重,不包括需要外科手术或溶栓剂治疗的肺栓塞。
• 治疗不稳定性心绞痛及非Q波心肌梗死,与阿司匹林合用。
• 用于血液透析体外循环中,防止血栓形成。
• 治疗急性ST段抬高型心肌梗死,与溶栓剂联用或同时与经皮冠状动脉介入治疗(PCI)联用。
【规格】(1)0.2ml :2000 AxaIU (2)0.4ml: 4000 AxaIU (3)0.6ml: 6000 AxaIU (4)0.8ml :8000 AxaIU (5)1.0ml :10000 AxaIU【用法用量】预防静脉血栓栓塞性疾病,治疗深静脉栓塞,治疗不稳定性心绞痛及非Q波心肌梗死时应采用深部皮下注射给予依诺肝素;血液透析体外循环时为血管内途径给药;对于ST段抬高型急性心肌梗死,初始的治疗为静脉注射,随后改为皮下注射治疗。
美国药典肝素钠质量标准浅析

版,2 0 1 3 年5 月生效 。现就美 国药典收载 的肝素钠 质量标 准溶 液 。取 多硫 酸软骨素 对照 品,溶于标准溶 液 ,浓度 为 准进行 介绍和分 析说明 ,并 与中 国药典2 0 1 0 年版 二部收载 1% ( w / w),作 为系统适用 性溶液 。取供 试品 ,溶 于0 . 0 2
肝 素 钠 为 糖 胺 聚 糖 类 抗 凝 血 药 , 是 世 界 上 最 有 效 和 取 , 生 产 过 程 均 应 符 合 现 行 版 《 药 品生产质量 管理规范 》
临床用 量最大 的抗凝血药 物之一 ,临床上用于 心脑血管疾 要求 。生产 工艺要经病 毒灭活验证 ,并能去 除有害 的污 染 病 和 血液透 析 治疗 ,还具 有 降血 脂 、抗 中膜 平滑 肌 细 胞 物 ,生产过程 中应确保不被外来物质污染 。 ( S MC)增 生 、促进 血 纤蛋 白溶 解等 作用 。美 国药典 收 2鉴 别 载 了肝素钠 的质量标准 ,2 0 0 8 年 肝素钠事件 后 ,美 国数 次 2 . 1核磁 共振 波谱法
的 肝 素 钠 质 量 标 准 进 行 比较 。 1 定 义 % ( w / v ) 三 甲基 硅 烷 氘 代 丙 酸 钠 重 水 溶 液 , 浓 度 不 低 于 2 O mg / mL,作 为 供 试 品 溶 液 。使 用 脉 冲 ( 傅 里 叶 变 换 )核
肝 素 钠 是 一 种 硫 酸 化 糖 胺 聚 糖 的 钠 盐 , 其 存 在 形 式 磁 共 振 波 谱 仪 进 行 H N MR分 析 ,频 率 不 低 于 5 0 0 MH z , 使 为 分 子 量 各 异 的 不 均 一 性 分 子 的 混 合 物 , 具 有 对 抗 凝 血 级 用 不 低 于 1 6 次扫 描、9 O 。脉冲和 2 0 s 延 迟 获 得 自 由感 应 衰
达肝素钠标准(USP 39)

Ratio: A240/A445, 1.30–1.50for Labeling 〈7〉, Labels and Labeling for Injectable Prod-•B. The retention time of the major peak of the Sample ucts•(CN 1-May-2016).solution corresponds to that of the Standard solution, asADDITIONAL REQUIREMENTS obtained in the Assay.ASSAYChange to read:•P ROCEDURE[N OTE—Use freshly prepared Standard solution and Sam-•P ACKAGING AND S TORAGE:•Preserve as described in Pack-ple solution, protected from light.]aging and Storage Requirements 〈659〉, Injection Packaging, Mobile phase: Acetonitrile and water (3:2)Sterile solids packaging; protect from light.•(CN 1-May-2016).Standard solution: 250µg/mL of USP Dactinomycin RS•L ABELING: Label it to include the statement “Protect from in Mobile phaselight.”Sample solution: 250µg/mL of dactinomycin from•USP R EFERENCE S TANDARDS〈11〉Dactinomycin for Injection diluted with Mobile phase.USP Dactinomycin RSFilter, if necessary, to obtain a clear solution. [N OTE—USP Endotoxin RSPrepare the solution by adding a suitable aliquot of Mo-bile phase to one container of Dactinomycin forInjection.]Chromatographic system(See Chromatography 〈621〉, System Suitability.)Mode: LC Add the following:Detector: UV 254 nmColumn: 3.9-mm × 30-cm; packing L1Flow rate: 2.5mL/minv Dalteparin SodiumInjection size: 10µLSystem suitability [9041-08-1].Sample:Standard solution[N OTE—The retention time for dactinomycin is 6 min.]Suitability requirementsColumn efficiency: NLT 1200 theoretical platesTailing factor: NMT 2Relative standard deviation: NMT 3.0%AnalysisSamples:Standard solution and Sample solutionCalculate the percentage of C62H86N12O16 in the por-tion of Dactinomycin for Injection taken:Result = (r U/r S) × (C S/C U) × 100r U= peak response from the Sample solutionr S= peak response from the Standard solutionC S= concentration of USP Dactinomycin RS in theStandard solution (µg/mL)DEFINITIONC U= nominal concentration of dactinomycin in the Dalteparin Sodium is the sodium salt of a low molecularSample solution (µg/mL)weight heparin obtained by nitrous acid depolymerization Acceptance criteria: 90.0%–120.0%of heparin from porcine intestine or intestinal mucosa.Heparin source material used in the manufacture of SPECIFIC TESTSDalteparin Sodium complies with the compendial require-•P H 〈791〉: 5.5–7.5, in the solution constituted as directedments stated in the Heparin Sodium monograph.in the labelingDalteparin Sodium is produced by a validated manufac-•L OSS ON D RYING〈731〉: Dry a portion in vacuum at aturing and purification procedure under conditions shown pressure not exceeding 5mm of mercury at 60° for 3 h:to minimize the presence of species containing the N–NO it loses NMT 4.0% of its weight.group. The majority of the components have a 2-O-sulfo-α-L-idopyranosuronic acid structure at the non-reducing Change to read:end and a 6-O-sulfo-2,5-anhydro-D-mannitol structure atthe reducing end of their chains. The weight-average mo-•O THER R EQUIREMENTS: It meets the requirements under lecular weight (M w) ranges between 5600 Da and 6400•Injections and Implanted Drug Products 〈1〉•(CN 1-May-2016).Da, with a characteristic value of about 6000 Da. The •B ACTERIAL E NDOTOXINS T EST〈85〉: NMT 100.0 USP Endo-percentage of chains lower than molecular weight 3000 toxin Units/mg of dactinomycin.Da is NMT 13.0%, and the percentage of chains higher •S TERILITY T ESTS〈71〉: Meets the requirements when than molecular weight 8000 Da ranges between 15.0% tested as directed for Test for Sterility of the Product to be and 25.0%. The degree of sulfation is NLT 1.8/disaccha-Examined, Membrane Filtration, each container being con-ride unit. The potency is NLT 110 and NMT 210 Anti-stituted aseptically by injecting Sterile Water for Injection Factor X a International Units (IU)/mg of activity, calculated through the stopper, and the entire contents of all the on the dried basis. The anti-factor IIa activity is NLT 35 containers being collected aseptically with the aid of IU/mg and NMT 100 IU/mg, calculated on the dried ba-200mL of Fluid A before filtering.sis. The ratio of anti-factor Xa activity to anti-factor IIaactivity is between 1.9 and 3.2.Change to read:IDENTIFICATION•A.1H NMR S PECTRUM•C ONSTITUTED S OLUTION: At the time of use, it meets the Standard solution: Dissolve 15mg of USP Dalteparin requirements for •Injections and Implanted Drug Products Sodium RS in 0.7mL of deuterium oxide with deuter-〈1〉, Specific Tests, Completeness and clarity of solutions and ated trimethylsilylpropionic (TSP) acid sodium salt. Thesample is freeze-dried to remove exchangeable P MonographsRedissolve the sample and repeat the freeze-drying step centage of chains lower than the molecular weight twice more before transferring the sample into an NMR3000 Da (M3000) is NMT 13.0%, and the percentage of tube.chains higher than the molecular weight 8000 Da Sample solution: Dissolve 15mg of Dalteparin Sodium(M8000) ranges between 15.0% and 25.0%.in 0.7mL of deuterium oxide (99.9%) with deuterated•C. A NTI-F ACTOR X A TO A NTI-F ACTOR II A R ATIOTSP. The sample is freeze-dried to remove exchangea-(See Anti-Factor Xa and Anti-Factor IIa Assays for Unfrac-ble protons. Redissolve the sample and repeat the tionated and Low Molecular Weight Heparins 〈208〉, Anti-freeze-drying step twice more before transferring the Factor Xa and Anti-Factor IIa Assays for Low Molecular sample into an NMR tube.Weight Heparins.)Instrumental conditions Acceptance criteria: The ratio of the numerical value of (See Nuclear Magnetic Resonance Spectroscopy 〈761〉.)the anti-factor Xa activity, in Anti-Factor X a IU/mg, to Mode: NMR, pulsed (Fourier transform)the numerical value of the anti-factor IIa activity, in Frequency: NLT 500MHz for 1H Anti-Factor II a IU/mg, as determined by the Anti-Factor Temperature: 30°Xa Activity and Anti-Factor IIa Activity assays, is NLT 1.9 System suitability and NMT 3.2, respectively.Samples:Standard solution and Sample solution•D. I DENTIFICATION T ESTS—G ENERAL: Meets the require-Transfer the Standard solution and the Sample solution to ments for Sodium ContentNMR tubes of 5mm in diameter. Using a pulsedASSAY(Fourier transform) NMR spectrometer operating at•A NTI-F ACTOR X A A CTIVITYNLT 500MHz for 1H, acquire a free induction decay(See Anti-Factor Xa and Anti-Factor IIa Assays for Unfrac-(FID) with NLT 32 scans using a 90° pulse, an acquisi-tionated and Low Molecular Weight Heparins 〈208〉, Anti-tion time of NLT 2 s, and at least a 10-s delay. ForFactor Xa and Anti-Factor IIa Assays for Low Molecular each sample, an initial short spectrum is collected (1Weight Heparins, Anti-Factor Xa Activity for Low Molecular scan), and the water resonance is then suppressed byWeight Heparin.)selective irradiation during the relaxation delay. FinalAnalysis: Proceed as directed in the chapter.spectra are recorded over 32 scans. For all samples,Acceptance criteria: The potency is NLT 110 and NMT the TSP methyl signal should be set to 0.00ppm. Re-210 Anti-Factor X a IU/mg on the dried basis.cord the 1H NMR spectrum of the Standard solution.Collect the 1H NMR spectrum with a spectral window OTHER COMPONENTSof at least 10 to −2ppm and without spinning. The•N ITROGEN D ETERMINATION, Method II 〈461〉: 1.5%–2.5% Standard solution shall be run at least daily when the on the dried basisSample solution is being run. All spectra are phased,•S ODIUM C ONTENTand linear baseline correction is applied to all spectra Cesium chloride solution: 1.27mg/mL of cesium chlo-before peak identification.ride in 0.1 M hydrochloric acidSuitability requirements Standard solution A: 0.0025% of sodium chloride in Chemical shift: The TSP methyl signal should be set Cesium chloride solutionto 0.00ppm for all samples.Standard solution B: 0.0050% of sodium chloride in Chemical shifts for system suitability: The ppm val-Cesium chloride solutionues for the methyl group of N-acetyl, the H-2 of N-Standard solution C: 0.0075% of sodium chloride in sulfo glucosamine, the H-2 of glucuronic acid plus Cesium chloride solution3-O-sulfo glucosamine, the H-1 of iduronic acid, and Sample solution: Transfer 50.0mg of Dalteparin So-the H-1 of 3-O sulfo glucosamine of dalteparin in the dium to a 100-mL volumetric flask, and dissolve in and Standard solution are present at 2.05, 3.28, 3.39,dilute with Cesium chloride solution to volume.5.01, and 5.51, respectively. Two additional signals,Analysiscorresponding to the H-1 of the 2-O-sulfo iduronic Samples:Cesium chloride solution, Standard solution A, acid linked to the terminal 2, 5-anhydromannitol and Standard solution B, Standard solution C, and Sample the H-1 of 2-O-sulfo iduronic acid are located at solution5.18–5.22ppm. The ppm values of these signals do Concomitantly determine the absorbances of the Ce-not differ by more than ±0.03ppm, Standard solution.sium chloride solution (blank), the Sample solution, and [N OTE—Depending on specific sample makeup and the Standard solutions at 330.3 nm, using a sodium instrument parameters, including the field strength hollow-cathode lamp and an air–acetylene flame. Us-of the NMR instrument, the two signals associated ing the absorbances of Standard solutions A, B, and C, with the H-1 of 2-O-sulfo iduronic acid at determine the sodium content in the Sample solution5.18–5.22ppm may appear well separated or as a after an appropriate blank correction.main signal with a shoulder.]Acceptance criteria: 10.5%–13.5% on the dried basis AnalysisSample:Sample solution IMPURITIESRecord the 1H NMR spectra of the Sample solution.•L IMIT OF N ITRITESAcceptance criteria: The ppm values for the methyl Mobile phase: Dissolve 13.6g of sodium acetate trihy-group of N-acetyl, the H-2 of N-sulfo glucosamine, the drate in 900mL of water in a 1000-mL volumetric flask.H-2 of glucuronic acid plus 3-O-sulfo glucosamine, the Adjust with orthophosphoric acid to a pH of 4.3, and H-1 of iduronic acid and the H-1 of the 2-O-sulfo dilute with water to 1000mL. Filter through a 0.45-µm iduronic acid linked to the terminal anhydromannitol,membrane.the H-1 of 2-O-sulfo iduronic acid and the H-1 of 3-O Nitrite stock standard solution: Dissolve 0.075g of so-sulfo glucosamine of dalteparin in the Sample solution dium nitrite in a 1000-mL volumetric flask with carbon are present at 2.05, 3.28, 3.39, 5.01, 5.18–5.22, and dioxide-free water (0.05g/L of nitrite).5.51, respectively. The ppm values of these signals do Nitrite standard solution: Dilute 1mL of Nitrite stocknot differ by more than ±0.03ppm.standard solution in a 100-mL volumetric flask with car-•B. M OLECULAR W EIGHT D ISTRIBUTION AND W EIGHT-A VERAGE bon dioxide-free water (500 ng/mL of nitrite).M OLECULAR W EIGHT Calibration standard solutions: Dilute Nitrite standard (See Low Molecular Weight Heparin Molecular Weight De-solution in carbon dioxide-free water to prepare four so-terminations 〈209〉.)lutions with the final nitrite concentrations of 2.5, 5, Acceptance criteria: The weight-average molecular15, and 25 ng/mL.weight (M w) ranges between 5600 Da and 6400 Da,with a characteristic value of about 6000 Da. The per-Sample solution: Weigh 80.0mg of Dalteparin Sodium r SA= response of boron from Standard solution A into a 20-mL volumetric flask, and dissolve in carbon r C= response of boron from the Calibration solution dioxide-free water.r B= response of boron from the BlankChromatographic system Acceptance criteria: NMT 1ppm(See Chromatography 〈621〉, System Suitability.)SPECIFIC TESTSMode: LC•A NTI-F ACTOR II A A CTIVITYDetector: Electrochemical detector containing a work-(See Anti-Factor Xa and Anti-Factor IIa Assays for Unfrac-ing electrode (glassy carbon type) with the potentialtionated and Low Molecular Weight Heparins, 〈208〉, Anti-of +1.00 V against a silver–silver chloride referenceFactor Xa and Anti-Factor IIa Assays for Low Molecular electrodeWeight Heparins, Anti-Factor IIa Activity for Low Molecular Column: 3-mm × 15-cm; 5-µm packing L92Weight Heparin.)Column temperature: 30±5°Acceptance criteria: NLT 35 and NMT 100 Anti-Factor Column regeneration: 1M sodium chloride (NaCl) atII a IU/mg on the dried basis0.5mL/min for about 1 h. After regeneration, wash•M OLAR R ATIO OF S ULFATE TO C ARBOXYLATE the column with water and re-equilibrate with MobileMobile phase: Carbon dioxide-free water phase.Sample solution: 50mg of Dalteparin Sodium in 10mL Flow rate: 0.5mL/minof carbon dioxide-free waterInjection volume: 25µLChromatographic systemRun time: 10 min(See Chromatography 〈621〉, System Suitability.) System suitabilityMode: LCSamples:Calibration standard solutions and SampleDetector: IonsolutionColumn: Two columns: one 1.5-cm × 2.5-cm column, Suitability requirementspacked with an anion-exchange resin L64packing, and Column efficiency: NLT 4000 theoretical plates forone 1.5-cm × 7.5-cm column, packed with a cation-the nitrite peak for all Calibration solutions and Sampleexchange resin L65packing.1 The outlet of the anion-solution runsexchange column is connected to the inlet of the cat-Tailing factor: Between 0.8 and 1.2 for all Calibrationion-exchange column.solutions and Sample solution runsFlow rate: 1mL/minRelative standard deviation: Inject Calibration stan-Analysisdard solutions with 25 ng/mL concentration at leastSample:Sample solutionsix times. Calculate the relative standard deviation %[N OTE—Regenerate the anion-exchange column and the (%RSD) of the nitrite peak areas of the last six injec-cation-exchange column with 1N sodium hydroxide tions. The %RSD is NMT 2%.and 1N hydrochloric acid, respectively, between two Analysisinjections.]Samples:Calibration standard solutions and SampleWith the valve in the inject position, inject the Sample solutionsolution into the anion-exchange column, and collect Plot the areas of the nitrite peaks from the chromato-the eluate from the cation-exchange column in a grams of the Calibration standard solutions against re-beaker at the outlet until the ion detector reading re-spective concentrations of nitrite. Draw a best-fit re-turns to the baseline value. Quantitatively transfer the gression line through the points. The correlationeluate to a titration vessel containing a magnetic stir-coefficient is NLT 0.995. Calculate the concentrationring bar, and dilute with carbon dioxide-free water to of nitrite from the areas of the nitrite peak in theabout 60mL. Position the titration vessel on a mag-chromatogram of the Sample solution.netic stirrer, and immerse the electrodes. Note the ini-Acceptance criteria: NMT 5ppmtial conductivity reading, and titrate with approxi-•B ORONmately 0.1 N sodium hydroxide added in 100-µL [N OTE—Use only plastic labware, avoid glass.]portions. [N OTE—Prepare the sodium hydroxide solu-Blank: 1% (v/v) solution of nitric acid in watertion in carbon dioxide-free water.] Record the buret Calibration solution: Prepare a 11.4-µg/mL solution ofreading and the conductivity meter reading after each USP Boric Acid RS in the Blank.addition of the sodium hydroxide solution.Standard solution A: Dissolve 0.2500g of USP LowPlot the conductivity measurements on the y-axis Molecular Weight Heparin for Boron Analysis RS inagainst the volumes of sodium hydroxide added on about 2mL of water, add 100µL of nitric acid, andthe x-axis. The graph will have three linear sections—dilute with the Blank to 10.00mL.an initial downward slope, a middle slight rise, and a Standard solution B: Dissolve 0.2500g of USP Lowfinal rise. For each of these sections, draw the best-fit Molecular Weight Heparin for Boron Analysis RS instraight lines using linear regression analysis. At the about 2mL of Blank, add 10µL of a 5.7-mg/mL solu-points where the first and second straight lines inter-tion of USP Boric Acid RS, and dilute with the Blank tosect and where the second and third lines intersect,10.00mL. This solution contains 1µg/mL of boron.draw perpendiculars to the x-axis to determine the Sample solution: Dissolve 0.2500g of Dalteparin So-volumes of sodium hydroxide taken up by the sample dium in about 2mL of water, add 100µL of nitric acid,at those points. The point where the first and second and dilute with the Blank to 10.00mL.lines intersect corresponds to the volume of sodium Analysishydroxide taken up by the sulfate groups (V S). The Samples:Blank, Calibration solution, Standard solutionpoint where the second and third lines intersect corre-A, Standard solution B, and Sample solutionsponds to the volume of sodium hydroxide consumed Boron is determined by measurement of the emissionby the sulfate and the carboxylate groups together from inductively coupled plasma (ICP) at 249.733 nm(V T).or a suitable wavelength. Use an appropriate appara-Calculate the molar ratio of sulfate to carboxylate: tus with settings that have been optimized as directedby the manufacturer.Result = V S/(V T−V S) Calculate the content of boron in Dalteparin Sodiumusing the following correction factor:1The procedure is based on analyses performed with two columns: one 1.5-cm × 2.5-cm packed with anion-exchange resin Dowex 1X8 (200–400 mesh)and the other 1.5-cm × 7.5-cm packed with cation-exchange resin DowexF = (r SB – r SA) × 2/(r C – r B)50WX2 (100–200 mesh).r SB= response of boron from Standard solution BAcceptance criteria: The molar ratio of sulfate to car-Percentageboxylate is NLT 1.8.Concentra-(%,•P H 〈791〉: 5.5–8.0 for a 1.0% solution in water tion for comparison•L OSS ON D RYING 〈731〉Standard (µg RS perwith test Sample: 1gsolutionDilution mL)specimen)Analysis: Dry the Sample under vacuum at 70° for 6 h.A (1 in 2)500 1.0Acceptance criteria: NMT 10%B (1 in 4)2500.5•B ACTERIAL E NDOTOXINS T EST 〈85〉: It contains NMT 0.01C (1 in 10)1000.2USP Endotoxin Unit/IU of anti-factor Xa activity.D(1 in 20)500.1ADDITIONAL REQUIREMENTS•P ACKAGING AND S TORAGE : Preserve in tight, light-resistant Test solution—Dissolve an accurately weighed quantity of containers, and store below 40°, preferably at room Danazol in Solvent to obtain a solution containing 50mg temperature.per mL.•L ABELING : Label to state the number of Anti-factor X a In-Procedure—Apply separately 5µL of the Test solution and ternational Units of activity per mg.5µL of each Standard solution to a suitable thin-layer chro-•USP R EFERENCE S TANDARDS 〈11〉matographic plate (see Chromatography 〈621〉) coated with USP Boric Acid RSa 0.25-mm layer of chromatographic silica gel mixture. Posi-USP Dalteparin Sodium RS tion the plate in a chromatographic chamber and develop USP Endotoxin RSthe chromatograms in a solvent system consisting of a mix-USP Low Molecular Weight Heparin for Bioassays RSture of cyclohexane and ethyl acetate (7:3) until the solvent USP Low Molecular Weight Heparin for Boron Analysis RS front has moved about three-fourths of the length of the USP Low Molecular Weight Heparin Molecular Weight plate. Remove the plate from the developing chamber, mark Calibrant RSthe solvent front, and allow the solvent to evaporate in v USP39warm, circulating air. Examine the plate under short-wave-length UV light. Expose the plate to iodine vapors for 5min-utes. Compare the intensities of any secondary spots ob-served in the chromatogram of the Test solution with those of the principal spots in the chromatograms of the Standard solutions: the sum of the intensities of secondary spots ob-Danazoltained from the Test solution corresponds to not more than 1.0% of related compounds, with no single impurity corre-sponding to more than 0.5%.Assay—Dissolve about 100mg of Danazol, accuratelyweighed and previously dried, in about 50mL of alcohol ina 100-mL volumetric flask, swirl until dissolved, dilute with alcohol to volume, and mix. Transfer 2.0mL of this solution C 22H 27NO 2337.46to a 100-mL volumetric flask, dilute with alcohol to volume,Pregna-2,4-dien-20-yno[2,3-d ]isoxazol-17-ol, (17α)-.and mix. Similarly, dissolve an accurately weighed quantity 17α-Pregna-2,4-dien-20-yno[2,3-d ]isoxazol-17-ol of USP Danazol RS in alcohol to obtain a Standard solution [17230-88-5].having a known concentration of about 20µg per mL. Con-comitantly determine the absorbances of both solutions in » Danazol contains not less than 97.0percent 1-cm cells at the wavelength of maximum absorbance at and not more than 102.0percent of C 22H 27NO 2,about 285 nm, using alcohol as the blank. Calculate the quantity, in mg, of C 22H 27NO 2 in the portion of Danazol calculated on the dried basis.taken by the formula:Packaging and storage—Preserve in tight, light-resistant containers.5C (A U /A S )USP Reference standards 〈11〉—in which C is the concentration, in µg per mL, of USPUSP Danazol RS Danazol RS in the Standard solution; and A U and A S are the Identification—absorbances of the solution of Danazol and the Standard A: Infrared Absorption 〈197K 〉.solution, respectively.B: Ultraviolet Absorption 〈197U 〉—Solution: prepared as directed in the Assay .Specific rotation 〈781S 〉: between +21° and +27°.Test solution: 10mg per mL, in chloroform.Danazol CapsulesLoss on drying 〈731〉—Dry it at a pressure not exceeding 5mm of mercury at 60° to constant weight: it loses not » Danazol Capsules contain not less thanmore than 2.0% of its weight.90.0percent and not more than 110.0percent of Chromatographic purity—the labeled amount of C 22H 27NO 2.Solvent—Prepare a mixture of chloroform and methanol (9:1).Packaging and storage—Preserve in well-closed contain-Standard solutions—Dissolve an accurately weighed quan-ers.tity of USP Danazol RS in Solvent to obtain a solution having USP Reference standards 〈11〉—a known concentration of 1mg per mL. Dilute quantita-USP Danazol RStively with Solvent to obtain Standard solutions having the Identification—Shake the contents of a sufficient number following compositions:of Capsules, equivalent to about 50mg of Danazol, with 50mL of chloroform, and filter. Evaporate the filtrate on a steam bath with the aid of a stream of nitrogen to dryness:the IR absorption spectrum of a potassium bromide disper-sion of the residue, previously dried, exhibits maxima at the。
低分子肝素药物的质量标准综述

低分子肝素药物的质量标准综述作者:张利赟来源:《科技传播》2010年第16期摘要本文通过对国内外低分子肝素质量标准、生产工艺、结构特点、药理作用等比较和总结,为我国低分子肝素产业的进一步发展提供参考。
关键词低分子肝素;质量标准;生产工艺;结构;药理中图分类号R9 文献标识码A 文章编号 1674-6708(2010)25-0091-02肝素是临床常用的抗凝血药,但在临床使用中暴露出很多缺点,如:剂量个体差异大,自发性出血倾向,停药后血栓易复发等,还有可引起血小板减少症,长期应用可产生暂时性脱发、骨质疏松和自发性骨折等不良反应[1]。
随着对肝素的深入研究,特别是对以往认为无活性的低分子组分对Xa因子和凝血酶(thrombin)的作用差异的认识,为开发LMWH的提供了理论依据,20世纪70年代末,LMWH进入临床。
低分子量肝素(low-molecular-weight heparin,LMWH)是未分级肝素(unfractionated heparin,UFH)中具有较低分子量的组分或片段,具有与UFH相同的母体结构。
UFH是一种高度硫酸化的糖胺聚糖,分子量范围为3 000~30 000。
LMWH是近20年发展起来的新一代肝素类抗血栓药物,是采用适当方法将肝素分级或降解得到的具有较低分子量的肝素组分或片段3 000~8 000。
在临床使用中,与肝素比较,LMWH抗血栓作用强,抗凝血作用弱于肝素,且具有皮下注射吸收良好、生物利用度高、体内半衰期长、出血倾向小,按体重给药,抗凝效果可以预测等优点[2],目前已逐步取代肝素应用于临床。
国外已有超低分子肝素研究[3],其平均分子量为2 000~3 000,抗FXa/FIIa>30,已经进入III期临床研究。
1 国内外几种LMWH的质量标准综述目前,国外LMWH商品约有十多种,我国进口的有6种,国内仿制的四类新药生产企业约有十几家。
我国有低分子肝素钠部颁标准。
依诺肝素钠相对分子量的EP与USP分析方法对比研究

依诺肝素钠相对分子量的EP与USP分析方法对比研究伯小霞;崔慧斐【期刊名称】《中国生化药物杂志》【年(卷),期】2012(33)6【摘要】目的以国产依诺肝素钠和其原研药为样品,比较欧洲药典(EP)和美国药典(USP)测定依诺肝素钠相对分子质量(Mr)方法的差异,为国内该品种的药典标准制定提供参考.方法采用这两种方法分别检测了10批国产依诺肝素钠原料药和2批原研药注射液LOVENOX的Mr情况.结果 USP方法测得平均Mr均较EP方法结果偏高在200以内,Mr2 000以下组分比例EP方法偏高,而Mr2000 ~8 000组分比例USP方法偏高,但差距均在4%以内.国产依诺肝素钠Mr与原研药的基本一致.结论 EP和USP方法分析依诺肝素钠Mr结果较为接近.%Purpose To analyze the difference of the relative molecular weight (Mr) analysis method of Enoxaparin Sodium in european Pharmacopoeia ( EP) and the United States Pharmacopoeia ( USP ) through the analysis of domestic samples and the branded, and to provide some opinions for Enoxaparin Sodium domestic standard. Methods Ten batches of Enoxaparin Sodium samples and 2 batches brand Injection Lovenox Enoxaparin Sodium were analyzed in EP and USP method. Results The average Mr by EP method was higher than that by USP method with difference of less than 200. The percentage of less than 2 000 by EP method was higher,and that of 2 000 - 8 000 by USP method was higher and both differences within 4% . The Mr of domestic samples was comparable to the branded. Conclusion The resultsof Mr of Enoxaparin Sodium by EP method were closed to those by USP method.【总页数】4页(P818-821)【作者】伯小霞;崔慧斐【作者单位】山东大学药学院,山东济南250012;东营天东生化工业有限公司,山东东营257067;山东大学药学院,山东济南250012;(山东大学)国家糖工程技术研究中心,山东济南250012【正文语种】中文【中图分类】R927.1【相关文献】1.不同阿胶酶解液相对分子量分布及补血升白作用对比研究 [J], 李敏;代龙;庞萌萌;田晨颖;刘庆丰;于帅;王少平;刘国飞;刘玉军;高鹏2.对比研究宽分布对照品测定依诺肝素钠分子量 [J], 徐勤娟;崔洪萌;3.不同分子量聚合物驱相对渗透率曲线对比研究 [J], 李斌会; 杨清彦; 贾忠伟; 徐联玉; 王晓芳4.依诺肝素钠注射液分子量与分子量分布比较研究 [J], 由鹏飞;薛维丽;杭宝健;李春焕;王玉团;陈晓;咸瑞卿5.依诺肝素钠注射液分子量与分子量分布比较研究 [J], 由鹏飞;薛维丽;杭宝健;李春焕;王玉团;陈晓;咸瑞卿因版权原因,仅展示原文概要,查看原文内容请购买。
伊诺肝素钠

HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use Lovenox safely and effectively. See full prescribing information for Lovenox.Lovenox® (enoxaparin sodium injection) for subcutaneous and intravenous useInitial U.S. Approval: 1993WARNING : SPINAL/EPIDURAL HEMATOMASee full prescribing information for complete boxed warning . • Enoxaparin use in patients undergoing spinal/epidural anesthesia or spinal puncture increases the risk of spinal or epidural hematoma, which may cause long-term or permanent paralysis (5.5) • Risk is increased by:o Indwelling epidural catheters for analgesia (5.5) o Drugs affecting hemostasis [e.g., nonsteroidal anti-inflammatory drugs, platelet inhibitors, anticoagulants] (5.5, 7)o Traumatic or repeated spinal or epidural puncture (5.5)-----------------------RECENT MAJOR CHANGES ------------------Indications and Usage (1.4), 5/2007 Dosage and Administration (2) 5/2007ST-segment Elevation Myocardial Infarction Warnings and Precautions (5.2) 5/2007 Percutaneous coronary revascularization procedures------------------INDICATIONS AND USAGE -------------------Lovenox is a low molecular weight heparin [LMWH] indicated for:• Prophylaxis of deep vein thrombosis (DVT) in abdominalsurgery, hip replacement surgery, knee replacement surgery, or medical patients with severely restricted mobility during acute illness (1.1)• Inpatient treatment of acute DVT with or without pulmonaryembolism (1.2)• Outpatient treatment of acute DVT without pulmonaryembolism. (1.2)• Prophylaxis of ischemic complications of unstable angina andnon-Q-wave myocardial infarction [MI] (1.3)• Treatment of acute ST-segment elevation myocardialinfarction [STEMI] managed medically or with subsequent percutaneous coronary intervention [PCI] (1.4)-------------------DOSAGE AND ADMINISTRATION--------------Indication Standard Regimen (2.1, 2.3) Severe RenalImpairment (2.2)DVT prophylaxis in abdominal surgery 40 mg SC once daily 30 mg SC once daily DVT prophylaxis in knee replacement surgery30 mg SC every 12 hours 30 mg SC once daily DVT prophylaxis in hip replacement surgery 30 mg SC every 12 hours or 40 mg SConce daily30 mg SC once daily DVT prophylaxis in medical patients 40 mg SC once daily 30 mg SC once daily Inpatient treatment of acute DVT with or without pulmonary embolism 1 mg/kg SC every 12 hours or 1.5 mg/kg SC once daily (with warfarin)1 mg/kg SC once daily Outpatient treatment acute DVT without of hours (with warfarin) g SC oncedaily pulmonary embolism 1 mg/kg SC every 12 1 mg/k Unstable angina and g SC once non-Q-wave MI 1 mg/kg SC every 12hours (with aspirin) 1 mg/k daily Acute STEMI in patients <75 years of CI, see 1)] hours with aspirin) by g SC once daily age[For dosing insubsequent P Dosage andAdministration (2.30 mg single IV bolus plus a 1 mg/kg SC dose followed by 1 mg/kg SC every 12 30-mg single IV bolus plus a 1 mg/kgSC dose followed 1 mg/k Acute STEMI in patients ≥75 years of 12 hours (no bolus) daily (no bolus) age0.75 mg/kg SC every 1 mg/kg SC once Do not use as intramuscular injection.For subcutaneous use, do not mix with other injections or fusions. --------------------DOSAGE FORMS AND STRENGTHS-------- • 60 mg/0.6 mL, 80 mg/0.8AINDICATIONS ----------------- i-platelet) oval5.3) arin or other LMWHs (5.6)notMultiple-dose formulations contain benzyl alcohol (5.8) --- adverse reactions (>1%) were bleeding, anemia, rombocytopenia, elevation of serum aminotransferase, diarrhea, ADVERSE REACTIONS, contact nofi-aventis at 1-800-633-1610 or FDA at 1-800-FDA-1088 or /medwatch. in--100mg/mL concentration (3.1):• Prefilled syringes: 30 mg/0.3 mL, 40 mg/0.4 mLGraduated prefilled syringes:mL,100 mg/1 mL• Multiple-dose vial: 300 mg/3 mL 150mg/mL concentration (3.2):• Graduated prefilled syringes: 120 mg/0.8 mL, 150 mg/1 mL------------------------------CONTR• Active major bleeding (4.1)Thrombocytopenia with a positive in vitro test for ant • antibody in the presence of enoxaparin sodium (4.2 Hypersensitivity to enoxaparin sodium (4.3)•• Hypersensitivity to heparin or pork products (4.4)-----------------------WARNINGS AND PRECAUTIONS ----------• Use caution in conditions with increased risk of hemorrhage(5.1)• Obtain hemostasis at the puncture site before sheath rem after percutaneous coronary revascularisation (5.2)• Use caution with concomitant medical conditions (• Use caution in case of history of heparin-inducedthrombocytopenia (5.4)• Monitor thrombocytopenia of any degree closely (5.5) •Do not exchange with hep • Pregnant women with mechanical prosthetic heart valves adequately studied (5.7)•• Periodic blood counts recommended (5.9)-----------------------------ADVERSE REACTIONS ----------------Most common th and nauseaTo report SUSPECTED sa w-----------------------------DRUG INTERACTIONS ----- hich may enhance hemorrhage risk prior to itiation of Lovenox or conduct close clinical and laboratory --------------Discontinue agents w in monitoring (5.9, 7).----------------USE IN SPECIFIC POP ------ULATIONS ------------ patients with• Hepatic Impairment (8.8)• Low-weight patients: Observe for signs of bleeding (8.9)• Severe renal impairment: Adjust dose for creatinine clearance <30 mL/min (2.2)See 17 for PATIENT COUNSELING INFORMATIONRevised: [m/year]WARNIN 1 INDIC ction)2 NGTHSon 4 5 ures ty with Other Heparinswith Mechanical Prosthetic Heart7 CTIONS8 USE IN SPECIFIC POPULATIONS8.1 Pregnancyical Prosthetic Heart Valves ent 11 12 LOGY13 OLOGYlity 14 Acute ardialections or subsections omitted from the full prescribing ation are not listedFULL PRESCRIBING INFORMATION: CONTENTS* G - SPINAL / EPIDURAL HEMATOMAS ATIONS AND USAGEProphylaxis of deep vein thrombosis in patients1.1 undergoing surgery and in medical patients with severely restricted mobility during acute illness Treatment of acute deep vein thrombosis1.2 1.3 Prophylaxis of ischemic complications of unstableangina and non-Q-wave myocardial infar 1.4 Treatment of acute ST-segment Elevation Myocardial Infarction (STEMI DOSAGE AND ADMINISTRATION2.1 Adult dosage 2.2 Renal impairment2.3 Geriatric patients with acute STEMI 2.4 Administration3 3.1 100-mg/mL concentrati DOSAGE FORMS AND STRE 3.2 150-mg/mL concentration CONTRAINDICATIONSWARNINGS AND PRECAUTIONS5.1 Increased risk of Hemorrhage5.2 Percutaneous coronary revascularization proced 5.3 Use of Lovenox with Concomitant Medical Conditions 5.4 History of heparin-induced thrombocytopenia 5.5 ThrombocytopeniaInterchan 5.6 geabili 5.7 Pregnant Women Valves5.8 Benzyl alcohol 5.9 Laboratory tests6 6.1 Clinical StudiesADVERSE REACTIONS6.2 Post-marketing experience DRUG INTERA 8.3 Nursing Mothers 8.4 Pediatric Use 8.5 Geriatric Use8.6 Patients with Mechan 8.7 Renal impairment 8.8 Hepatic impairm 8.9 Low-weight Patients 10 OVERDOSAGE DESCRIPTIONCLINICAL PHARMACO 12.1 Mechanism of Action 12.2 Pharmacodynamics 12.3 PharmacokineticsNONCLINICAL TOXIC 13.1 Carcinogenesis, Mutagenesis, Impairment of Ferti 13.2 Animal ToxicologyICAL TRIALS EX CLIN PERIENCE14.1 Prophylaxis of deep vein thrombosis followingabdominal surgery14.2 Prophylaxis of deep vein thrombosis following Hip orKnee Replacement surgeryProphylaxis of deep vein thr 14.3 ombosis in MedicalPatients with Severely Restricted Mobility during Illness14.4 Treatment of acute deep vein thrombosis with orwithout pulmonary embolism14.5 Prophylaxis of ischemic complications in unstableangina and non-Q-wave myocardial infarction14.6 Treatment of acute ST-segment Elevation Myoc Infarction (STEMI)16 HOW SUPPLIED/STORAGE AND HANDLING 17 PATIENT COUSELING INFORMATION*S inform12FULL PRESCRIBING INFORMATIONWARNING: SPINAL / EPIDURAL HEMATOMAS3When neuraxial anesthesia (epidural/spinal anesthesia) or spinal puncture is employed, 4patients anticoagulated or scheduled to be anticoagulated with low molecular weight 5heparins or heparinoids for prevention of thromboembolic complications are at risk of 6developing an epidural or spinal hematoma which can result in long-term or permanent 7paralysis.89The risk of these events is increased by the use of indwelling epidural catheters for 10administration of analgesia or by the concomitant use of drugs affecting hemostasis such as 11non steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, or other 12anticoagulants. The risk also appears to be increased by traumatic or repeated epidural or 13spinal puncture.1415Monitor patients for signs and symptoms of neurological impairment. If neurologic 16compromise is noted, urgent treatment is necessary.1718Consider the potential benefit versus risk before neuraxial intervention in patients 19anticoagulated or to be anticoagulated for thromboprophylaxis [see Warnings and 20Precautions (5.1) and Drug Interactions (7)].2122232425262728293031323334353637383940414243 1 INDICATIONS AND USAGE1.1Prophylaxis of deep vein thrombosisLovenox is indicated for the prophylaxis of deep vein thrombosis, which may lead to pulmonary embolism:•in patients undergoing abdominal surgery who are at risk for thromboembolic complications [see Clinical Trials Experience 14.1].•in patients undergoing hip replacement surgery, during and following hospitalization.•in patients undergoing knee replacement surgery.•in medical patients who are at risk for thromboembolic complications due to severely restricted mobility during acute illness.1.2Treatment of Acute Deep Vein ThrombosisLovenox is indicated for:•the inpatient treatment of acute deep vein thrombosis with or without pulmonary embolism, when administered in conjunction with warfarin sodium;•the outpatient treatment of acute deep vein thrombosis without pulmonary embolism when administered in conjunction with warfarin sodium.1.3Prophylaxis of Ischemic Complications of Unstable Angina and Non-Q-wave Myocardial InfarctionLovenox is indicated for the prophylaxis of ischemic complications of unstable angina and non-Q-wave myocardial infarction, when concurrently administered with aspirin.44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 1.4 Treatment of acute ST- segment Elevation Myocardial Infarction (STEMI)Lovenox has been shown to reduce the rate of the combined endpoint of recurrent myocardial infarction or death in patients with acute STEMI receiving thrombolysis and being managed medically or with Percutaneous Coronary Intervention (PCI).2 DOSAGE AND ADMINISTRATIONAll patients should be evaluated for a bleeding disorder before administration of Lovenox, unless the medication is needed urgently. Since coagulation parameters are unsuitable for monitoring Lovenox activity, routine monitoring of coagulation parameters is not required [see Warnings and Precautions (5.9)].For subcutaneous use, Lovenox should not be mixed with other injections or infusions. For intravenous use (i.e., for treatment of acute STEMI), Lovenox can be mixed with normal saline solution (0.9%) or 5% dextrose in water.Lovenox is not intended for intramuscular administration.2.1 Adult DosageAbdominal Surgery: In patients undergoing abdominal surgery who are at risk for thromboembolic complications, the recommended dose of Lovenox is 40 mg once a day administered by SC injection with the initial dose given 2 hours prior to surgery. The usual duration of administration is 7 to 10 days; up to 12 days administration has been administered in clinical trials. 65 66 67 68 69 70Hip or Knee Replacement Surgery: In patients undergoing hip or knee replacement surgery, the recommended dose of Lovenox is 30 mg every 12 hours administered by SC injection. Provided that hemostasis has been established, the initial dose should be given 12 to 24 hours after surgery. For hip replacement surgery, a dose of 40 mg once a day SC, given initially 12 (±3) hours prior to surgery, may be considered. Following the initial phase of thromboprophylaxis in hip replacement surgery patients, it is recommended that continued prophylaxis with Lovenox 40 mg once a day is administered by SC injection for 3 weeks. The usual duration of administration is 7 to 10 days; up to 14 days administration has been administered in clinical trials. 71 72 73 74 75 76 77 78 79 80Medical Patients During Acute Illness: In medical patients at risk for thromboembolic complications due to severely restricted mobility during acute illness, the recommended dose of Lovenox is 40 mg once a day administered by SC injection. The usual duration of administration is 6 to 11 days; up to 14 days of Lovenox has been administered in the controlled clinical trial. 81 82 83 84 85 86Treatment of Deep Vein Thrombosis With or Without Pulmonary Embolism: In outpatient treatment , patients with acute deep vein thrombosis without pulmonary embolism who can be 87 88treated at home, the recommended dose of Lovenox is 1 mg/kg every 12 hours administered SC. In inpatient (hospital) treatment , patients with acute deep vein thrombosis with pulmonary embolism or patients with acute deep vein thrombosis without pulmonary embolism (who are not candidates for outpatient treatment), the recommended dose of Lovenox is 1 mg/kg every 12 hours administered SC or 1.5 mg/kg once a day administered SC at the same time every day. In both outpatient and inpatient (hospital) treatments, warfarin sodium therapy should be initiated when appropriate (usually within 72 hours of Lovenox). Lovenox should be continued for a minimum of 5 days and until a therapeutic oral anticoagulant effect has been achieved (International Normalization Ratio 2.0 to 3.0). The average duration of administration is 7 days; up to 17 days of Lovenox administration has been administered in controlled clinical trials. 89 90 91 92 93 94 95 96 97 98 99 100Unstable Angina and Non-Q-Wave Myocardial Infarction: In patients with unstable angina or non-Q-wave myocardial infarction, the recommended dose of Lovenox is 1 mg/kg administered SC every 12 hours in conjunction with oral aspirin therapy (100 to 325 mg once daily). Treatment with Lovenox should be prescribed for a minimum of 2 days and continued until clinical stabilization. The usual duration of treatment is 2 to 8 days; up to 12.5 days of Lovenox has been administered in clinical trials. [See Warnings and Precautions (5.2) and Clinical Trials Experience (14.5)]. 101 102 103 104 105 106 107 108Treatment of acute ST-segment Elevation Myocardial Infarction: In patients with acute ST-segment Elevation Myocardial Infarction, the recommended dose of Lovenox is a single IV bolus of 30 mg plus a 1 mg/kg SC dose followed by 1 mg/kg administered SC every 12 hours (maximum 100 mg for the first two doses only, followed by 1 mg/kg dosing for the remaining doses). Dosage adjustments are recommended in patients ≥75 years of age [see Dosage and Administration (2.3)]. 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132When administered in conjunction with a thrombolytic (fibrin-specific or non-fibrin specific), Lovenox should be given between 15 minutes before and 30 minutes after the start of fibrinolytic therapy. All patients should receive acetylsalicylic acid (ASA) as soon as they are identified as having STEMI and maintained with 75 to 325 mg once daily unless contraindicated. In the pivotal clinical study, the Lovenox treatment duration was 8 days or until hospital discharge, whichever came first. An optimal duration of treatment is not known, but it is likely to be longer than 8 days.For patients managed with Percutaneous Coronary Intervention (PCI): If the last Lovenox SC administration was given less than 8 hours before balloon inflation, no additional dosing is needed. If the last Lovenox SC administration was given more than 8 hours before balloon inflation, an IV bolus of 0.3 mg/kg of Lovenox should be administered [see Warnings and Precautions (5.2)].2.2 Renal ImpairmentAlthough no dose adjustment is recommended in patients with moderate (creatinine clearance 30-50 mL/min) and mild (creatinine clearance 50-80 mL/min) renal impairment, all such patients should be observed carefully for signs and symptoms of bleeding.133 134 135 136 137 The recommended prophylaxis and treatment dosage regimens for patients with severe renal impairment (creatinine clearance <30 mL/min) are described in Table 1 [see Use in SpecificPopulations (8.6) and Clinical Pharmacology (12.3)].Table 1Dosage Regimens for Patients with Severe Renal Impairment(creatinine clearance <30mL/minute)Indication DosageRegimen Prophylaxis in abdominal surgery 30 mg administered SC once dailyProphylaxis in hip or knee replacement surgery 30 mg administered SC once dailyProphylaxis in medical patients during acute illness 30 mg administered SC once dailyInpatient treatment of acute deep vein thrombosis withor without pulmonary embolism, when administered inconjunction with warfarin sodium1 mg/kg administered SC once dailyOutpatient treatment of acute deep vein thrombosiswithout pulmonary embolism, when administered inconjunction with warfarin sodium1 mg/kg administered SC once dailyProphylaxis of ischemic complications of unstableangina and non-Q-wave myocardial infarction, whenconcurrently administered with aspirin1 mg/kg administered SC once dailyTreatment of acute ST-segment Elevation MyocardialInfarction in patients <75 years of age30 mg single IV bolus plus a 1 mg/kgSC dose followed by 1 mg/kgadministered SC once daily.Treatment of acute ST-segment Elevation MyocardialInfarction in geriatric patients ≥75 years of age1 mg/kg administered SC once daily(no initial bolus)138 139140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 2.3 Geriatric patients with acute ST-Elevation Myocardial InfarctionFor treatment of acute ST-segment Elevation Myocardial Infarction in geriatric patients ≥75 years of age, do not use an initial IV bolus. Initiate dosing with 0.75 mg/kg SC every 12 hours (maximum 75 mg for the first two doses only, followed by 0.75 mg/kg dosing for the remaining doses)[see Use in Specific Populations (8.5) and Clinical Pharmacology (12.3)].No dose adjustment is necessary for other indications in geriatric patients unless kidney function is impaired [see Dosage and Administration (2.2)].2.4 AdministrationLovenox is a clear, colorless to pale yellow sterile solution, and as with other parenteral drug products, should be inspected visually for particulate matter and discoloration prior to administration.The use of a tuberculin syringe or equivalent is recommended when using Lovenox multiple-dose vials to assure withdrawal of the appropriate volume of drug.156157 158 159 160 161 162 163 Lovenox must not be administered by intramuscular injection. Lovenox is intended for use under the guidance of a physician.For subcutaneous administration, patients may self-inject only if their physicians determine that it is appropriate and with medical follow-up, as necessary. Proper training in subcutaneous injection technique (with or without the assistance of an injection device) should be provided. Subcutaneous Injection Technique: Patients should be lying down and Lovenox administered by deep SC injection. To avoid the loss of drug when using the 30 and 40 mg prefilled syringes, do not expel the air bubble from the syringe before the injection. Administration should be alternated between the left and right anterolateral and left and right posterolateral abdominal wall. The whole length of the needle should be introduced into a skin fold held between the thumb and forefinger; the skin fold should be held throughout the injection. To minimize bruising, do not rub the injection site after completion of the injection.164 165 166 167 168 169 170171 172 173 174 175 176 177 178 Lovenox prefilled syringes and graduated prefilled syringes are available with a system that shields the needle after injection.1. Remove the needle shield by pulling it straight off the syringe (see Figure A). If adjusting the dose is required, the dose adjustment must be done prior to injecting the prescribed dose to the patient.Figure A179 180181 182 183 184 2. Inject using standard technique, pushing the plunger to the bottom of the syringe (see FigureB).Figure B185 186187 188 3. Remove the syringe from the injection site keeping your finger on the plunger rod (see FigureC).189Figure C190 191 192 193 194 195 196198199 200 201 2024. Orient the needle away from you and others, and activate the safety system by firmly pushing the plunger rod. The protective sleeve will automatically cover the needle and an audible “click” will be heard to confirm shield activation (see Figure D).Figure D1975. Immediately dispose of the syringe in the nearest sharps container (see Figure E).Figure E203 204 205 206 207 208 209 210 211 212 213NOTE:• The safety system can only be activated once the syringe has been emptied.• Activation of the safety system must be done only after removing the needle from the patient’s skin.• Do not replace the needle shield after injection. • The safety system should not be sterilized.Activation of the safety system may cause minimal splatter of fluid. For optimal safety activate the system while orienting it downwards away from yourself and others.Intravenous (Bolus) Injection Technique: For intravenous injection, the multiple-dose vial should be used. Lovenox should be administered through an intravenous line. Lovenox should not be mixed or co-administered with other medications. To avoid the possible mixture of Lovenox with other drugs, the intravenous access chosen should be flushed with a sufficient amount of saline or dextrose solution prior to and following the intravenous bolus administration of Lovenox to clear the port of drug. Lovenox may be safely administered with normal saline solution (0.9%) or 5% dextrose in water.214 215 216 217 218 219 220 221222223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 3 DOSAGE FORMS AND STRENGTHSLovenox is available in two concentrations:3.1 100 mg per mL-Prefilled Syringes 30 mg / 0.3 mL, 40 mg / 0.4 mL-Graduated Prefilled Syringes 60 mg / 0.6 mL, 80 mg / 0.8 mL, 100 mg / 1 mL-Multiple-Dose Vials 300 mg / 3 mL3.2 150 mg per mL-Graduated Prefilled Syringes 120 mg / 0.8 mL, 150 mg / 1 mL4 CONTRAINDICATIONS•Active major bleeding.•Thrombocytopenia associated with a positive in vitro test for anti-platelet antibody in the presence of enoxaparin sodium.•Known hypersensitivity to enoxaparin sodium (e.g., pruritus, urticaria, anaphylactoid reactions) [see Adverse Reactions (6.2)].•Known hypersensitivity to heparin or pork products.•Known hypersensitivity to benzyl alcohol (which is in only the multi-dose formulation of Lovenox).5 WARNINGS AND PRECAUTIONS5.1 Increased Risk of HemorrhageCases of epidural or spinal hematomas have been reported with the associated use of Lovenox and spinal/epidural anesthesia or spinal puncture resulting in long-term or permanent paralysis. The risk of these events is higher with the use of post-operative indwelling epidural catheters or by the concomitant use of additional drugs affecting hemostasis such as NSAIDs[see boxed Warning, Adverse Reactions (6.2) and Drug Interactions (7)].Lovenox should be used with extreme caution in conditions with increased risk of hemorrhage, such as bacterial endocarditis, congenital or acquired bleeding disorders, active ulcerative and angiodysplastic gastrointestinal disease, hemorrhagic stroke, or shortly after brain, spinal, or ophthalmological surgery, or in patients treated concomitantly with platelet inhibitors.Major hemorrhages including retroperitoneal and intracranial bleeding have been reported. Some of these cases have been fatal.Bleeding can occur at any site during therapy with Lovenox. An unexplained fall in hematocrit or blood pressure should lead to a search for a bleeding site.5.2Percutaneous coronary revascularization proceduresTo minimize the risk of bleeding following the vascular instrumentation during the treatment of unstable angina, non-Q-wave myocardial infarction and acute ST-segment elevation myocardial267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 infarction, adhere precisely to the intervals recommended between Lovenox doses. It is important to achieve hemostasis at the puncture site after PCI. In case a closure device is used, the sheath can be removed immediately. If a manual compression method is used, sheath should be removed 6 hours after the last IV/SC Lovenox. If the treatment with enoxaparin sodium is to be continued, the next scheduled dose should be given no sooner than 6 to 8 hours after sheath removal. The site of the procedure should be observed for signs of bleeding or hematoma formation [see Dosage and Administration (2.1)].5.3 Use of Lovenox with Concomitant Medical ConditionsLovenox should be used with care in patients with a bleeding diathesis, uncontrolled arterial hypertension or a history of recent gastrointestinal ulceration, diabetic retinopathy, and hemorrhage.5.4 History of Heparin-induced ThrombocytopeniaLovenox should be used with extreme caution in patients with a history of heparin-induced thrombocytopenia.5.5ThrombocytopeniaThrombocytopenia can occur with the administration of Lovenox.Moderate thrombocytopenia (platelet counts between 100,000/mm3 and 50,000/mm3) occurred at a rate of1.3% in patients given Lovenox, 1.2% in patients given heparin, and 0.7% in patients given placebo in clinical trials.Platelet counts less than 50,000/mm3 occurred at a rate of 0.1% in patients given Lovenox, in 0.2% of patients given heparin, and 0.4% of patients given placebo in the same trials. Thrombocytopenia of any degree should be monitored closely. If the platelet count falls below 100,000/mm3, Lovenox should be discontinued. Cases of heparin-induced thrombocytopenia with thrombosis have also been observed in clinical practice. Some of these cases were complicated by organ infarction, limb ischemia, or death [see Warnings and Precautions (5.4)].5.6 Interchangeability with Other HeparinsLovenox cannot be used interchangeably (unit for unit) with heparin or other low molecular weight heparins as they differ in manufacturing process, molecular weight distribution, anti-Xa and anti-IIa activities, units, and dosage. Each of these medicines has its own instructions for use.5.7 Pregnant Women with Mechanical Prosthetic Heart ValvesThe use of Lovenox for thromboprophylaxis in pregnant women with mechanical prosthetic heart valves has not been adequately studied. In a clinical study of pregnant women with mechanical prosthetic heart valves given enoxaparin (1 mg/kg twice daily) to reduce the risk of thromboembolism, 2 of 8 women developed clots resulting in blockage of the valve and leading to maternal and fetal death. Although a causal relationship has not been established these deaths may have been due to therapeutic failure or inadequate anticoagulation. No patients in the。
依诺肝素钠药学研究资料基本要求

发布日期20101110栏目化药药物评价>>综合评价标题依诺肝素钠药学研究资料基本要求作者余立、赵慧玲、陆益红、张震、黄晓龙部门审评四部正文内容依诺肝素钠(Enoxaparin Sodium)为低分子肝素类(low molecular weight heparin,简称LMWH)品种之一,EP、BP、USP等均已收载,我国也已有依诺肝素钠注射液进口,执行进口药注册标准(JX20000267,规格20mg/0.2ml、40mg/0.4、60mg/0.6ml、80mg/0.8ml、100mg/1.0ml)。
因依诺肝素钠生产的起始原料肝素钠为生物提取的大分子多组分生化药,所以药学研究申报资料除应符合《化学药品注册分类及申报资料要求》[1]外,还应参考《多组分生化药注射剂技术审评基本要求》[2],结合该品种的特点重点注意以下方面。
一.制备工艺欧洲药典(EP)7.0版[3]对依诺肝素钠制备方法简要描述为:依诺肝素钠是一种低分子肝素钠盐,它通过对猪肠粘膜肝素的苄基酯衍生物进行碱解聚而获得。
专利US005389618A[4]即采用这种碱解聚的β-消除法,其基本步骤为:以猪粘膜肝素为起始原料,经肝素季铵盐制备、肝素苄酯的制备、对肝素苄酯进行碱解聚、以酸中和、醇沉淀,精制、脱色,脱水干燥,得到依诺肝素钠成品。
由于依诺肝素钠的结构与组成比较复杂,在结构确证与质量标准控制上存在一定的不确定性,所以起始原料和生产过程的研究与控制就尤为重要。
除按一般的化学药要求提供工艺研究资料外,还需要重点提供以下信息:1.起始原料的来源和质量起始原料应来源于猪肠粘膜,并在相关内控标准中予以明确。
1)使用已获批准上市的肝素钠作为起始原料应提供肝素钠原料药的批准证明文件、药品质量标准、检验报告、原料药生产企业的营业执照、《药品生产许可证》、《药品生产质量管理规范》认证证书、销售发票、供货协议等复印件,以及有关病毒灭活方法及其选择依据、病毒灭活验证资料、申报单位对肝素钠的供应商进行审计的资料。
依诺肝素

商品名:克赛/Clexane英文名:Enoxaparin通用名:依诺肝素【理化特性】成分及含量:每个注射器:20mg40mg60mg80mg100mg依诺肝素钠:20mg40mg60mg80mg100mg加注射用水至0.2ml0.4ml0.6ml0.8ml 1.0ml剂型:无菌无致热源可注射液,已预装入注射器。
药理治疗分类抗栓剂/低分子肝素 (B:血液,造血器官)【药理作用】药效动力学特性:本品为具有高活性抗Xa(100 I.U./mg)作用和低活性抗IIa 或抗凝血酶(28 I.U./mg)作用的低分子肝素. 在不同适应证所需的剂量下, 本品并不延长出血时. 在预防剂量时, 本品对APTT没有明显改变. 既不影响血小板聚集也不影响纤维蛋白原与血小板的结合.药代动力学特性:药代动力学参数源于对血浆中抗Xa因子活性的研究.生物利用度:皮下注射本品可迅速并完全被吸收, 本品的生物利用度接近95%.分布:皮下注射本品3小时后达最大血浆活性. 抗Xa活性存在于血管内.生物转化:本品主要在肝脏代谢.消除:使用40 mg 本品时其抗Xa 活性的半衰期约为4.4小时. 使用60mg 或 80mg 本品时约为4小时.排泄:本品经尿排出. 在老年患者中消除半衰期略延长.赋形剂成分:注射用水【适应证】-20 mg和40 mg注射液:.预防静脉栓塞性疾病 (防止静脉内血栓形成) 尤其是与某些手术有关的栓塞.用于血液透析体外循环中,防止血栓形成-60 mg, 80 mg和100 mg注射液:.治疗深静脉血栓形成.治疗急性不稳定性心绞痛及非Q波心肌梗死,与阿司匹林同用【用法及用量】1 mg依诺肝素产生相当于100 I.U抗Xa活性为预防及治疗目的而使用依诺肝素时应采用深部皮下注射给药,用于血液透析体外循环时为血管内途径给药禁止肌肉内注射皮下用药须知:预装药液注射器可供直接使用。
在注射之前勿将注射器内的气泡排出。
应于患者平躺后进行注射。
依诺肝素钠新版美国药典标准

Revision Bulletin1Official December 1, 2008Add the following:the dried basis. The ratio of anti-factor X a activity to anti-factor II a activity is between 3.3 and 5.3.Packaging and storage—Preserve in tight containers, and store be-low 40°, preferably at room temperature.sEnoxaparin SodiumUSP Reference standards 〈11〉—USP Benzyl Alcohol RS . USP En-dotoxin RS . USP Enoxaparin Sodium RS . USP Enoxaparin Sodium Solution for Bioassays RS . USP Enoxaparin Sodium MolecularWeight Calibrant A RS. USP Enoxaparin Sodium Molecular Weight Calibrant B RS.Identification—A: Ultraviolet Absorption 〈197U 〉—Solution:500µg per mL.Medium:0.01N hydrochloric acid. The spectra exhibit maxima at 231±2 nm.B:13C NMR spectrum (see Nuclear Magnetic Resonance 〈761〉)—Standard solution—Dissolve 200mg of USP Enoxaparin Sodium RS in a mixture of 0.2mL of deuterium oxide and 0.8mL of water.Add 0.05mL of deuterated methanol to serve as an internal reference.Test solution—Dissolve 200mg of Enoxaparin Sodium in a mix-ture of 0.2mL of deuterium oxide and 0.8mL of water. Add 0.05mL of deuterated methanol.[9041-08-1].Procedure—Transfer the Standard solution and the Test solution to NMR tubes of 5-mm diameter. Using a pulsed (Fourier trans-Change to read:form) NMR spectrometer operating at not less than 75MHz for 13C,record the 13C NMR spectra of the Standard solution and the Test » Enoxaparin Sodium is the sodium salt of a de-solution at 40°. The spectra are similar.polymerized heparin. It is obtained by alkaline de-C:The ratio of the numerical value of the anti-factor X a activ-polymerization of heparin benzyl ester. The starting ity, in Anti-Factor X a IU per mg, to the numerical value of the anti-factor II a activity, in Anti-Factor II a IU per mg, as determined by material, heparin, is obtained exclusively from porcine the Assay (anti-factor X a activity) and the Anti-factor II a activity,intestinal mucosa. •Heparin source material used in the respectively, is not less than 3.3 and not more than 5.3.manufacture of Enoxaparin Sodium complies with the D:Molecular weight distribution and weight-average molecu-compendial requirements stated in the Heparin Sodium lar weight—monograph.•(RB 01-Dec-2008) Enoxaparin Sodium consists Mobile phase—Prepare a 0.5M lithium nitrate solution. Pass of a complex set of oligosaccharides that have not yet through a membrane filter having a porosity of 0.45 µm or less, and degas with helium.been completely characterized. The majority of the Standard solution—Dissolve about 10mg of USP Enoxaparin components have a 4-enopyranose uronate structure at Sodium RS, accurately weighed, in 1mL of Mobile phase .the non-reducing end of their chain. About 20 percent Test solution—Dissolve about 10mg of Enoxaparin Sodium, ac-of the materials contain a 1,6-anhydro derivative on the curately weighed, in 1mL of Mobile phase.reducing end of the chain, the range being between 15Chromatographic system (see Chromatography 〈621〉)—The and 25 percent. The weight-average molecular weight high performance size exclusion chromatograph is equipped with a of Enoxaparin Sodium is 4,500 Da, the range being be-differential refractive index detector, a 6- × 40-mm guard column tween 3,800 and 5,000 Da; about 16 percent have a and two 7.8- × 300-mm analytical columns in series, both analytical and guard columns prepacked with packing L59, and used at room molecular weight of less than 2,000 Da, the range being temperature. The flow rate is about 0.6mL per minute maintained between 12.0 and 20.0 percent; about 74 percent have a constant to ±1.0%.molecular weight between 2,000 and 8,000 Da, the Procedure—Reconstitute one vial each of USP Enoxaparin So-range being between 68.0 and 82.0 percent. Not more dium Molecular Weight Calibrant A RS and USP Enoxaparin So-than 18.0 percent have a molecular weight higher than dium Molecular Weight Calibrant B RS in 1mL of Mobile phase.Separately inject 20 µL of USP Enoxaparin Sodium Molecular 8,000 Da. When prepared as a solution, the solution is Weight Calibrant A RS and USP Enoxaparin Sodium Molecular analyzed for clarity and degree of color using a vali-Weight Calibrant B RS, record the chromatograms, and measure the dated method. The degree of sulfation is not less than retention times. Inject in duplicate 20 µL of each of the Standard 1.8 per disaccharide unit. It has a potency of not less solution and the Test solution, and record the chromatograms for a than 90 and not more than 125 Anti-Factor X a Interna-length of time to ensure complete elution, including salt and solvent peaks. Calculate the total area under each of the Standard solution tional Units (IU) per mg, and not less than 20.0 and not and Test solution chromatograms, excluding salt and solvent peaks more than 35.0 Anti-Factor II a IU per mg, calculated onat the end.Calibration curve—Plot the retention times on the x -axis against the peak molecular weights on the y -axis for the peaks in the chro-matograms of USP Enoxaparin Sodium Molecular Weight Calibrant A RS and USP Enoxaparin Sodium Molecular WeightCopyright 2008 The United States Pharmacopeial Convention All Rights Reserved.2 Revision BulletinOfficial December 1, 2008Calibrant B RS, and fit the data to a third-order polynomial using a Mobile phase:carbon dioxide-free water.suitable gel permeation chromatography (GPC) software.Test solution—Dissolve an accurately weighed quantity of about Calculations—Compute the data, using the same GPC software 50mg of Enoxaparin Sodium in 10mL of carbon dioxide-free and determine the weight-average molecular weight, M W , for each water.of the duplicate chromatograms of the Standard solution and the Chromatographic system—The liquid chromatographic system Test solution, and take the average for each solution. Correct the consists of two peristaltic pumps, a six-port injection valve, an ion mean value of M W to the nearest 50. The Chromatographic system detector, and two columns—one 1.5- × 2.5-cm column packed with is suitable if M W of USP Enoxaparin Sodium RS is within 150 Da an anion-exchange resin L64 packing and one 1.5- × 7.5-cm column of the labeled M W value. The M W for the Test solution is between packed with a cation-exchange resin L65 packing. The outlet of the 3,800 and 5,000 Da. Using the same software, determine for each of anion-exchange column is connected to the inlet of the cation-ex-the duplicate Test solution chromatograms the percentage of Enox-change column. The flow rate is about 1mL per minute.aparin Sodium chains with molecular weights lower than 2000 Da,Procedure—[NOTE —Regenerate the anion-exchange column M 2000, the percentage of Enoxaparin Sodium chains with molecular and the cation-exchange column with 1N sodium hydroxide and weights in the range 2000 to 8000 Da, M 2000–8000, and the percentage 1N hydrochloric acid, respectively, between two injections.] Inject of Enoxaparin Sodium chains with molecular weights greater than the Test solution into the anion-exchange column, and collect the 8000 Da, M 8000. Average the duplicate values and express to the eluate from the cation-exchange column in a beaker at the outlet nearest 0.5%. M 2000 is between 12.0% and 20.0%, M 2000–8000 is be-until the ion detector reading returns to the baseline value. Quantita-tween 68.0% and 82.0%, and M 8000 is not more than 18.0%.tively transfer the eluate to a titration vessel containing a magnetic E:It responds to the test for Sodium 〈191〉.stirring bar, and dilute with carbon dioxide-free water to about Specific absorbance (see Spectrophotometry and Light-Scattering 60mL. Position the titration vessel on a magnetic stirrer and im-〈851〉)—merse the electrodes. Note the initial conductivity reading and ti-trate with approximately 0.1N sodium hydroxide added in 100-µL Test solution—Dissolve 50.0mg of Enoxaparin Sodium in portions. [NOTE —Prepare the sodium hydroxide solution in carbon 100mL of 0.01N hydrochloric acid.dioxide-free water.] Record the burette reading and the conductivity Procedure—Obtain the UV spectra of the Standard solution and meter reading after each addition of the sodium hydroxide solution.the Test solution between 200nm and 300nm against 0.01N hy-Calculations—Plot the conductivity measurements on the y -axis drochloric acid blank. Calculate the specific absorbance at the against the volumes of sodium hydroxide added on the x -axis. The wavelength of maximum absorbance at 231±2 nm, with reference graph will have three linear sections—an initial downwards slope, a to the dried substance, using the following formula:middle slight rise, and a final rise. For each of these sections draw the best-fit straight lines using linear regression analysis. At the A × 100×1000 / [M × l × (100 – E )]points where the first and second straight lines intersect and where in which A is the absorbance at the wavelength of maximum ab-the second and third lines intersect, draw perpendiculars to the x-sorbance; M is the weight, in mg, of Enoxaparin Sodium in the Test axis to determine the volumes of sodium hydroxide taken up by the solution; l is the pathlength (typically l = 1cm); and E is the loss on sample at those points. The point where the first and second lines drying, in percent. The specific absorbance is between 14.0 and intersect corresponds to the volume of sodium hydroxide taken up 20.0, calculated on the dried basis.by the sulfate groups (V S ). The point where the second and third Bacterial endotoxins 〈85〉—It contains not more than 0.01 USP lines intersect corresponds to the volume of sodium hydroxide con-Endotoxin Unit per IU of anti-factor X a activity.sumed by the sulfate and the carboxylate groups together (V T ). Cal-pH 〈791〉:between 6.2 and 7.7 of a 10.0% solution in water.culate the molar ratio of sulfate to carboxylate by the formula:Loss on drying 〈731〉—Dry 1g in a vacuum at 70° for 6 hours: it V S /(V T – V S )loses not more than 10.0% of its weight.The molar ratio of sulfate to carboxylate is not less than 1.8.Nitrogen content, Method II 〈461〉:between 1.8% and 2.5%, cal-culated on the dried basis.Benzyl alcohol content—Heavy metals, Method I 〈231〉—Prepare a 5% solution in water:Mobile phase—Prepare a filtered and degassed mixture of water,the limit is not more than 0.0030%.acetonitrile, and methanol (80:15:5 v/v).Sodium content (see Spectrophotometry and Light-Scattering Standard solution—Dissolve 100mg of USP Benzyl Alcohol RS 〈851〉)—in 200mL of water. Transfer 5mL of this solution to a 25-mL volu-metric flask, and dilute with water to volume.Cesium chloride solution—Prepare a solution of cesium chloride in 0.1N hydrochloric acid containing 1.27mg per mL.Test solution—Weigh 0.5g of Enoxaparin Sodium into a 10-mL volumetric flask, and dissolve in 5.0mL of 1N sodium hydroxide.Standard solutions—Dissolve an accurately weighed quantity of Allow to stand at room temperature for about 1 hour. Add 1.0mL sodium chloride in Cesium chloride solution to obtain a solution of glacial acetic acid, dilute with water to volume, and mix.having a known concentration of about 0.2% sodium. Dilute accu-rately measured volumes of this solution with Cesium chloride so-Chromatographic system (see Chromatography 〈621〉)—The liq-lution having known concentrations of 0.0025%, 0.0050%, and uid chromatograph is equipped with a 256-nm detector and a 4.6-0.0075% of sodium.mm × 15-cm stainless steel column that contains L7 packing. The flow rate is about 1.0mL per minute maintained constant to ±10%.Test solution—Transfer an accurately weighed quantity of about 50.0mg of Enoxaparin Sodium to a 100-mL volumetric flask, and Procedure—Separately inject equal volumes (about 20 µL) of the dissolve in and dilute with Cesium chloride solution to volume.Standard solution and the Test solution, record the chromatograms,and measure the peak responses.Procedure—Concomitantly determine the absorbances of the Ce-sium chloride solution (blank), Test solution, and Standard solu-Calculation—Calculate the percentage of benzyl alcohol in tions at 330.3nm using a sodium hollow-cathode lamp and an Enoxaparin Sodium taken by the formula,air–acetylene flame. Using the absorbances of Standard solutions,determine the sodium content in the Test solution after appropriate (A T × C S )/(A S × C T )blank correction. The sodium content, calculated on the dried basis,in which A T is the benzyl alcohol peak area in the Test solution; C S is between 11.3% and 13.5%.is the concentration, in mg per mL, of benzyl alcohol; A S is the area Molar ratio of sulfate to carboxylate (see Chromatography of the benzyl alcohol peak in the Standard solution; and C T is the〈621〉)—Copyright 2008 The United States Pharmacopeial Convention All Rights Reserved.Revision Bulletin3 Official December 1, 2008concentration, in mg per mL, of Enoxaparin Sodium. The percent- Human antithrombin III solution—Reconstitute a vial of an-age of benzyl alcohol is not more than 0.1%.tithrombin III (see Reagent Specifications in the section Reagents,Indicators, and Solutions) in water to obtain a solution containing 5 Anti-factor II a activity—Antithrombin III Units per mL. Dilute this solution with pH 7.4 Acetic acid solution, pH 7.4 Polyethylene glycol 6000 buffer, pHPolyethylene glycol 6000 buffer to obtain a solution having a con-7.4 Buffer, pH 8.4 Buffer, and Human antithrombin III solution—centration of 1.0 Antithrombin III Unit per mL.Proceed as directed under Assay (anti-factor X a activity), except thatFactor X a solution—Reconstitute an accurately weighed quantity the concentration of the Human antithrombin III solution is 0.5 An-of bovine factor X a (see Reagent Specifications in the section Re-tithrombin III Unit per mL.agents, Indicators, and Solutions) in pH 7.4 Polyethylene glycol Thrombin human solution—Reconstitute thrombin human (see6000 buffer to obtain a solution that gives an increase in absorbance Reagent Specifications in the section Reagents, Indicators and So-value at 405nm of not more than 0.20 absorbance units per minute lutions) in water, and dilute in pH 7.4 Polyethylene glycol 6000when assayed as described below but using as an appropriate vol-buffer to obtain a solution having a concentration of 5 Thrombinume (V, in µL) of pH 7.4 Buffer instead of VµL of the enoxaparin Units per mL.solution.Chromogenic substrate solution—Prepare a solution of a suitableChromogenic substrate solution—Prepare a solution of a suitable chromogenic substrate for an amidolytic test (see Reagent Specifi-chromogenic substrate for amidolytic test (see Reagent Specifica-cations in the section Reagents, Indicators, and Solutions) fortions in the section Reagents, Indicators, and Solutions) for factor thrombin in water to obtain a concentration of about 3 mM. Imme-X a in water to obtain a concentration of about 3 mM. Dilute with diately before use, dilute with pH 8.4 Buffer to 0.5 mM.pH 8.4 Buffer to obtain a solution having a concentration of 0.5 Standard solutions—Dilute USP Enoxaparin Sodium Solution for mM.Bioassays RS with pH 7.4 Buffer to obtain four dilutions havingStandard preparations—Dilute USP Enoxaparin Sodium Solu-concentrations in the range between 0.015 and 0.075 IU of anti-tion for Bioassays RS with pH 7.4 Buffer to obtain four dilutions in factor II a activity per mL.the concentration range between 0.025 and 0.2 USP Anti-Factor X a Test solutions—Proceed as directed under Standard solutions to IU per mL.obtain concentrations of Enoxaparin Sodium similar to those ob-Assay preparations—Proceed as directed for Standard prepara-tained for the Standard solutions.tions to obtain concentrations of Enoxaparin Sodium similar to Procedure—Proceed as directed under Assay (anti-factor X a ac-those obtained for the Standard preparations.tivity), except to use Thrombin human solution instead of Factor X aProcedure—Label 18 suitable tubes: B1 and B2 for blanks; T1, solution and to use the Human antithrombin III solution as de-T2, T3, and T4 each in duplicate for the dilutions of the Assay prep-scribed above.arations; and S1, S2, S3, and S4 each in duplicate for the dilutions Calculations—For each series, calculate the regression of the ab-of the Standard preparations. [NOTE—Treat the tubes in the order sorbance against log concentrations of the Test solutions and of the B1, S1, S2, S3, S4, T1, T2, T3, T4, T1, T2, T3, T4, S1, S2, S3, S4, Standard solutions, and calculate the potency of the enoxaparin so-B2.] To each tube add the same volume, V, (20 to 50 µL) of Human dium in IU of anti-factor II a activity per mg using statistical meth-antithrombin III solution and an equal volume, V, of either the ods for parallel-line assays. The four independent dilution estimates blank, pH 7.4 Buffer, or an appropriate dilution of the Assay prepa-are then combined to obtain the final weighted mean. Then calcu-rations or the Standard preparations. Mix, but do not allow bubbles late the confidence limits. Express the anti-factor II a activity of to form. Incubate at 37° for 1.0 minute. Add to each tube volumeEnoxaparin Sodium per mg, calculated on the dried basis. It has a2V (40 to100 µL) of Factor Xa solution, and incubate for 1.0 min-potency of not less than 20.0 and not more than 35.0 anti-Factor II a ute. Add 5V (100 to 250 µL) volume of Chromogenic substrate so-IU per mg.lution. Stop the reaction after 4.0 minutes with 5V (100 to 250 µL) Assay (anti-factor X a activity)—volume of Acetic acid solution. Measure the absorbance of each so-lution at 405nm using a suitable spectrophotometer (see Spectro-Acetic acid solution—Transfer 42mL of glacial acetic acid to aphotometry and Light-Scattering 〈851〉) against blank B1. The read-100-mL volumetric flask, dilute with water to volume, and mix.ing of blank B2 relative to the blank B1 is not more than ±0.05 pH 7.4 Polyethylene glycol 6000 buffer—Dissolve 6.08g ofabsorbance units.tris(hydroxymethyl)aminomethane and 8.77g of sodium chloride inCalculations—For each series, calculate the regression of the ab-500mL of water. Add 1.0g of polyethylene glycol 6000, adjustsorbance against log concentrations of the Assay preparations and with hydrochloric acid to a pH of 7.4, and dilute with water toof the Standard preparations, and calculate the potency of the 1000mL.enoxaparin sodium in IU of anti-factor X a activity per mL using pH 7.4 Buffer—Dissolve 6.08g of tris(hydroxymethyl)ami-statistical methods for parallel-line assays. The four independent nomethane and 8.77g of sodium chloride in 500mL of water. Ad-log relative potency estimates are then combined to obtain the final just with hydrochloric acid to a pH of 7.4, and dilute with water togeometric mean. Its confidence limits are calculated. Express the 1000mL.anti-factor X a activity of Enoxaparin Sodium per mg, calculated on pH 8.4 Buffer—Dissolve 3.03g of tris(hydroxymethyl)ami-the dried basis. The potency is not less than 90 and not more thannomethane, 5.12g of sodium chloride and 1.40g of edetate sodium125 Anti-Factor Xa IU per mg.s2S(USP31)in 250mL of water. Adjust with hydrochloric acid to a pH of 8.4,and dilute with water to 500mL.Copyright 2008 The United States Pharmacopeial Convention All Rights Reserved.。
【优秀文档推荐下载】依诺肝素钠

“凡大医者,必当安神定志,无欲无求,先发大慈恻隐之心,誓愿普救含灵之苦”-----孙思邈以下为本文具体内容:依诺肝素钠一概述依诺肝素钠(Enoxaparinsodium)为低分子量肝素制剂,其分子量为4000~6000D,有明显的抗凝血因子Xa作用,而抗凝血酶(Ⅱa)作用弱,有很强的抗血栓作用,对血小板聚集无明显影响,出血副作用较常规肝素钠轻。
皮下注射达最大效应的时间为3~5小时,持续时间为24小时,生物利用度为92%,这取决于抗凝因子Xa的活性。
二适应证1.预防深部静脉血栓形成和肺栓塞。
2.治疗已形成的急性深部静脉血栓。
3.在血液透析或血液滤过时,防治体外循环系统中发生血栓或血液凝固。
4.用于狼疮抗体阳性所致的习惯性流产。
三临床应用皮下注射:术前给予20~40mg,术后20~40mg/24小时,疗程10~14日或至血栓形成风险消失,剂量大小视风险的高低而定。
用于已形成的静脉血栓。
皮下注射:每次1mg/kg,每12小时1次。
用于体外循环抗凝。
1mg/kg,若超过4小时,每小时追加上述剂量的1/4。
用于急性冠脉综合征。
发病24小时内开始,皮下注射:每次1mg/kg,每12小时1次。
四不良反应有报道,鞘内硬膜外麻醉和术后置留硬膜外导管的同时,注射本药可导致脊柱内出血,从而引起不同程度的神经损伤(包括长期或永久性的麻痹)。
其余参见肝素钠。
五注意事项发生出血或有严重出血倾向者禁用;要定期进行血小板计数检查;用药过量后可用鱼精蛋白拮抗。
不主张与阿司匹林、非甾体类消炎药或其他抗血小板药物联合应用。
口服抗凝剂、糖皮质激素和右旋糖酐的患者应慎用本品。
本品不宜肌注。
肝素注射液说

体重 Kg 45 50 55 60 65 70 75 80 85 90 95 100
在稀释完成后静脉给药注射体积 需要剂量(30IU/kg) 稀释到300IU/ml时需注射体积
IU
ml
1350
4.5
1500
5
1650
5.5
1800
6
1950
6.5
2100
7
2250
7.5
2400
8
2550
8.5
2700
【用法用量】
预防静脉血栓栓塞性疾病,治疗深静脉栓塞,治疗不稳定性心绞痛及非Q波心肌梗死时应 采用深部皮下注射给予依诺肝素;血液透析体外循环时为血管内途径给药;对于ST段抬高 型急性心肌梗死,初始的治疗为静脉注射,随后改为皮下注射治疗。 本品为成人用药 禁止肌内注射 每毫升注射液含10000 AxaIU,相当于100mg依诺肝素。每毫克(0.01ml)依诺肝素约等于100 AxaIU。
,应延长治疗至静脉血栓栓塞危险消除且患者不需卧床为止。在矫形外科手术中,连续3周
,每日一次给药4000 AxaIU是有益的。
• 在内科治疗患者中,预防静脉血栓栓塞性疾病
依诺肝素推荐剂量为每日一次皮下给药4000
AxaIU
(0.4
ml)。依诺肝素治疗最短应为6天,直至患者不需卧床为止,最长为14天。
3
2
用注射器从输液袋中取出30ml溶液弃除。注入全部6000AxaIU依诺肝素钠预填充注射液 到剩余20ml溶液的输液袋中。轻轻混合输液袋中药物。用注射器吸取所需的稀释液用于 静脉注射。稀释完全后,根据如下公式计算[稀释体积ml=患者体重kg*0.1]或用下表得 出所需注射液的体积。推荐在使用前制备稀释液。
依诺肝素钠说明书翻译
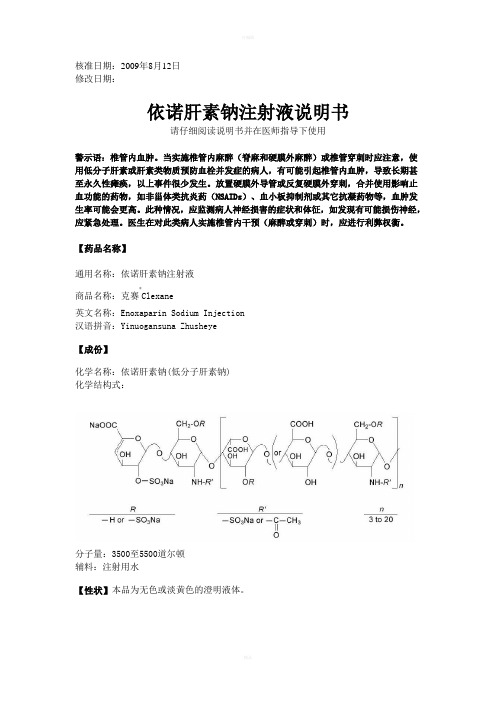
核准日期:2009年8月12日修改日期:依诺肝素钠注射液说明书请仔细阅读说明书并在医师指导下使用警示语:椎管内血肿。
当实施椎管内麻醉(脊麻和硬膜外麻醉)或椎管穿刺时应注意,使用低分子肝素或肝素类物质预防血栓并发症的病人,有可能引起椎管内血肿,导致长期甚至永久性瘫痪,以上事件很少发生。
放置硬膜外导管或反复硬膜外穿刺,合并使用影响止血功能的药物,如非甾体类抗炎药(NSAIDs)、血小板抑制剂或其它抗凝药物等,血肿发生率可能会更高。
此种情况,应监测病人神经损害的症状和体征,如发现有可能损伤神经,应紧急处理。
医生在对此类病人实施椎管内干预(麻醉或穿刺)时,应进行利弊权衡。
【药品名称】通用名称:依诺肝素钠注射液商品名称:克赛®Clexane英文名称:Enoxaparin Sodium Injection 汉语拼音:Yinuogansuna Zhusheye【成份】化学名称:依诺肝素钠(低分子肝素钠) 化学结构式:分子量:3500至5500道尔顿辅料:注射用水【性状】本品为无色或淡黄色的澄明液体。
【适应症】2000 Axa IU 和4000 Axa IU注射液:•预防静脉血栓栓塞性疾病 (预防静脉内血栓形成) ,特别是与骨科或普外手术有关的血栓形成。
6000 Axa IU, 8000 Axa IU 和10000 Axa IU注射液:• 治疗已形成的深静脉栓塞,伴或不伴有肺栓塞,临床症状不严重,不包括需要外科手术或溶栓剂治疗的肺栓塞。
• 治疗不稳定性心绞痛及非Q波心肌梗死,与阿司匹林合用。
• 用于血液透析体外循环中,防止血栓形成。
• 治疗急性ST段抬高型心肌梗死,与溶栓剂联用或同时与经皮冠状动脉介入治疗(PCI)联用。
【规格】(1)0.2ml :2000 AxaIU (2)0.4ml: 4000 AxaIU (3)0.6ml: 6000 AxaIU (4)0.8ml :8000 AxaIU (5)1.0ml :10000 AxaIU【用法用量】预防静脉血栓栓塞性疾病,治疗深静脉栓塞,治疗不稳定性心绞痛及非Q波心肌梗死时应采用深部皮下注射给予依诺肝素;血液透析体外循环时为血管内途径给药;对于ST段抬高型急性心肌梗死,初始的治疗为静脉注射,随后改为皮下注射治疗。
新版的肝素钠欧洲药典标准

NOTE ON THE MONOGRAPHThis monograph has been thoroughly revised further to the contamination events in 2008to update and strengthen the quality standards for unfractionated heparin.The style and presentation have also been updated in line with the latest version of the Style guide.Definition:the minimum potency limit has been raised after an enquiry among European manufacturers in line with the quality of currently marketed heparin batches; only1grade of heparin has been kept as the present2-tiered specification no longer reflects the situation in Europe.Production:the tests for nuclear magnetic resonance spectrometry(NMR)and capillary electrophoresis previously introduced in the1st-step revision applicable from1August 2008have been deleted,as detailed tests are now provided under Identification and Tests;statements have been added to emphasise the need for a reliable quality assurance system throughout production and,based on current practice among European manufacturers,for confirming the identity of the source species as well as the absence of any material issued from other species likely to contaminate the drug substance.This monograph is also revised to harmonise the information related to the source species for substances of human and animal origin and its presentation in monographs.The statement relative to the origin of the substance is moved under Definition accordingly and a paragraph is added regarding the health of the animals used for the preparation of heparin sodium.Identification:the tests for specific optical rotation(shown not to be discriminative enough)and zone electrophoresis have been replaced by the highly specific1H-NMR and strong anion-exchange liquid chromatography(SAX-HPLC)tests;1H-NMR has been selected for its ability not only to allow identification of heparin,but also to alert users to possible contaminations,whatever the nature of the contaminant;identification of the counterion is now based on the test for sodium by atomic absorption spectrometry described under Tests.Nucleotidic impurities:the limit has been tightened,based on current batch data. Protein:the Lowry test method has been introduced to replace the non-specific absorbance test;it is foreseen that this method be replaced by method2of general chapter2.5.33,in line with Ph.Eur.general policy,and users are therefore kindly requested to carry out the Ph.Eur.method and to submit their results during the public enquiry.Related substances:a SAX-HPLC-based test has been introduced,allowing the differentiation of natural contaminants linked to the production process(such as dermatan sulfate and chondroitin sulfate)from chemically synthesised contaminants;a limit for the sum of dermatan sulfate and chondroitin sulfate,which co-elute in this method,is proposed,further to consideration of current batch data.Nitrogen:a lower limit has been added,based on current batch data.Heavy metals:method C has been replaced by method F,in line with the general policy for heavy metals tests.Sulfated ash:in view of the highly specific tests introduced into the monograph,this test has become redundant and has therefore been deleted.XXXX:0333HEPARIN SODIUMHeparinum natricumDEFINITIONPreparation containing the sodium salt of a sulfated glycosaminoglycan present in mammalian tissues.It is prepared either from the lungs of oxen or from the intestinal mucosae of pigs,oxen or sheep.On complete hydrolysis,it liberates D-glucosamine,D-glucuronic acid,L-iduronic acid,acetic acid and sulfuric acid.It has the characteristic property of delaying the clotting of freshly shed blood.The potency of heparin sodium intended for parenteral administration is not less than150IU/mg,calculated with reference to the dried substance.The potency of heparin sodium not intended for parenteral administration is not less than120IU/mg,calculated with reference to the dried substance.Potency:minimum180IU/mg(dried substance).PRODUCTIONIt is prepared either from the lungs of oxen or from the intestinal mucosae of pigs,oxen or sheep.The animals from which heparin sodium is derived must fulfil the requirements for the health of animals suitable for human consumption.All stages of production and sourcing are subjected to a suitable quality assurance system.The identity of the source species and the absence of material from the other species is verified by appropriate testing during production.It is produced by methods of manufacturing designed to minimise or eliminate substances lowering blood pressure and to ensure freedom from contamination by over-sulfated glycosaminoglycans.It complies with the following additional requirements.Nuclear magnetic resonance spectrometry(2.2.33).The1H NMR spectrum obtained with a frequency of at least300MHz complies with the specifications approved by the competent authority.Capillary electrophoresis(2.2.47).The electropherogram obtained complies with the specifications approved by the competent authority.CHARACTERSAppearance:white or almost white,hygroscopic powder.Solubility:freely soluble in water.IDENTIFICATIONA.It delays the clotting of recalcified citrated sheep plasma(see Assay).B.Dissolve0.40g in water R and dilute to10.0ml with the same solvent.The specific optical rotation(2.2.7)is not less than+35.C.Examine by zone electrophoresis(2.2.31)using agarose for electrophoresis R asthe supporting medium.To equilibrate the agarose and as electrolyte solution use a mixture of50ml of glacial acetic acid R and800ml of water R adjusted to pH3by addition of lithium hydroxide R and diluted to1000.0ml with water R.Test solution.Dissolve25mg of the substance to be examined in water R and dilute to10ml with the same solvent.Reference solution.Dilute heparin sodium BRP with an equal volume of water R. Apply separately to the strip2µl to3µl of each solution.Pass a current of1mAto2mA per centimetre of strip width at a potential difference of300V for about10min.Stain the strips using a1g/l solution of toluidine blue R and remove the excess by washing.The ratio of the mobility of the principal band or bands in the electropherogram obtained with the test solution to the mobility of the band in the electropherogram obtained with the reference solution is0.9to1.1.B.Nuclear magnetic resonance spectrometry(2.2.33).Preparation:dissolve40mg of the substance to be examined in0.7ml of a solution containing20µg/ml of deuterated sodium trimethylsilylpropionate R and12µg/ml of sodium edetate R in deuterium oxide R.Comparison:dissolve40mg of heparin sodium for NMR identification CRS in0.7ml of a solution containing20µg/ml of deuterated sodium trimethylsilylpropionate R and12µg/ml of sodium edetate R in deuterium oxide R.If stored,the sodium edetate and deuterated sodium trimethylsilylpropionate solutions must be kept in high-density,natural polyethylene bottles. Apparatus:spectrometer operating at minimum300MHz.Acquisition of1H-NMR spectra:—number of transients:minimum16;it is adjusted until the signal-to-noise ratio is at least2000:1for the heparin methyl signal;—temperature:about25°C;test sample and reference spectra have to be obtained at the same temperature;—acquisition time:2s;—repetition time(acquisition time plus delay):minimum4s;—spectral width:10-12ppm,centred at around4.5ppm;—pulse width:to give a flip angle between30°and90°.Processing:—trimethylsilylpropionate reference signal set at0.00ppm;—exponential line-broadening window function(LB):0.3Hz;—phasing(manually if needed);—baseline correction;—zero filling:equal to data points used in acquisition.Results:—the large heparin sodium signals must be present:2.04ppm,3.27ppm(doublet),4.34ppm,5.22ppm and5.42ppm,all within±0.03ppm;—the1H-NMR spectrum obtained has to be superimposed on the1H-NMR spectrum obtained with heparin sodium for NMR identification CRS;the2spectra arenormalised so as to have a similar intensity;dermatan sulfate with a methyl signal at2.08±0.03ppm may be observed;no unidentified signals larger than4percent compared to the height of the heparin signal at5.42ppm are present in theranges0.10-2.00ppm,2.12-3.20ppm and5.70-8.00ppm;signals from the solvent or process-related substances may be present and have to be identified to be accepted;variations in the intensity of some signal regions of the spectrum of heparin mayoccur:the intensity-variable regions are between3.35ppm and4.55ppm,where the signal pattern is approximately kept but intensity varies.Note:a signal occurring at2.10ppm may be observed and is not to be confused with that of dermatan sulfate.C.Liquid chromatography(2.2.29)as described in the test for related substances with the following modifications.Injection:test solution(a)and reference solution(a).Retention time:dermatan sulfate and chondroitin sulfate=about22min; heparin=about26min;over-sulfated chondroitin sulfate=about33min.System suitability:reference solution(a):—peak-to-valley ratio:minimum1.5,where H p=height above the baseline of the peak due to dermatan sulfate+chondroitin sulfate and H v=height above the baseline of the lowest point of the curve separating this peak from the peak due to heparin. The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.2.heparin3.over-sulfated chondroitin sulfate1.dermatan sulfate+chondroitinsulfateFigure0333.-1.–Chromatogram for identification test C for heparin sodium:referencesolution(a)Results:the principal peak in the chromatogram obtained with test solution(a)is similar in retention time and shape to the principal peak in the chromatogram obtained with reference solution(a).D.The residue obtained in the test for sulfated ash(see Tests)gives reaction(a)of sodium(2.3.1).It complies with the test for sodium(see Tests).TESTSAppearance of solution.The solution is clear(2.2.1)and not more intensely coloured than intensity5of the range of reference solutions of the most appropriate colour(2.2.2, Method II).Dissolve a quantity equivalent to50000IU in water R and dilute to10ml with the same solvent.pH(2.2.3):5.5to8.0.Dissolve0.1g in carbon dioxide-free water R and dilute to10ml with the same solvent. Protein and Nucleotidic impurities.Dissolve40mg in10ml of water R.The absorbance (2.2.25)measured at260nm is not greater than0.150.20and that measured at280nm is not greater than0.15.Protein:maximum0.5per cent(dried substance).Solution A.Mix2volumes of a10g/l solution of sodium hydroxide R and2volumes of a 50g/l solution of sodium carbonate R and dilute to5volumes with water R. Solution B.Mix2volumes of a12.5g/l solution of copper sulfate R and2volumes of a 29.8g/l solution of sodium tartrate R and dilute to5volumes with water R.Solution C.Mix1volume of solution B and50volumes of solution A.Solution D.Dilute a phosphomolybdotungstic reagent(1)2-to4-fold in water R.Suitable dilutions produce solutions of pH10.25±0.25after addition of solutions C and D to the test and reference solutions.Test solution.Dissolve the substance to be examined in water R to obtain a concentration of5mg/ml.Reference solutions.Dissolve bovine albumin R in water R to obtain a concentrationof100mg/ml.Prepare dilutions of the solution in water R to obtain not fewer than5reference solutions having protein concentrations evenly spaced over a suitable range situated between5µg/ml and100µg/ml.Blank:water R.Procedure.To1ml of each reference solution,of the test solution and of the blank,add 5ml of solution C.Allow to stand for10min.Add0.5ml of solution D,mix and allowto stand at room temperature for30min.Determine the absorbances(2.2.25)of the solutions at750nm,using the solution prepared from the blank as compensation liquid. e a2nd-order polynomial regression curve of absorption against concentration.Related substances.Liquid chromatography(2.2.29).Test solution(a).Dissolve an accurately weighed quantity of about50mg of the substance to be examined in5.0ml of water R.Mix using a vortex mixer until dissolution is complete.(1)Folin-Ciocalteu’s phenol reagent from Merck(reference1.09001.0500)is suitable.Test solution(b).Dissolve an accurately weighed quantity of about0.1g of the substance to be examined in1.0ml of water R.Mix using a vortex mixer until dissolution is complete.Mix500µl of the solution and250µl of1M hydrochloric acid,then add50µl of a250mg/ml solution of sodium nitrite R.Mix gently and allow to stand at room temperature for40min before adding200µl of1M sodium hydroxide to stop the reaction. Reference solution(a).Dissolve the contents of a vial of heparin for system suitability CRS in water R to obtain concentrations of10mg/ml of heparin sodium, 0.2mg/ml of dermatan sulfate and0.05mg/ml of over-sulfated chondroitin sulfate.Mix using a vortex mixer to homogenise.Reference solution(b).Mix400µl of a125mg/ml solution of heparin sodium CRS with100µl of water R and mix using a vortex mixer.Add250µl of1M hydrochloric acid,then add50µl of a250mg/ml solution of sodium nitrite R.Mix gently and allow to stand at room temperature for40min before adding200µl of1M sodium hydroxide to stop the reaction.Reference solution(c).Dissolve the contents of a vial of heparin for system suitability CRS in water R to obtain concentrations of50mg/ml of heparin sodium,1mg/ml of dermatan sulfate and0.25mg/ml of over-sulfated chondroitin sulfate.To 500µl of the solution add250µl of1M hydrochloric acid,then add50µl of a250mg/ml solution of sodium nitrite R.Mix gently and allow to stand at room temperature for40min before adding200µl of1M sodium hydroxide to stop the reaction.Reference solutions are stable at room temperature for24h.Precolumn:—size:l=0.05m,Ø=2mm;—stationary phase:anion exchange resin R(13µm)(2).Column:—size:l=0.05m,Ø=2mm;—stationary phase:anion exchange resin R(9µm)(3);—temperature:40°C.Mobile phase:—mobile phase A:dissolve0.80g of sodium dihydrogen phosphate R in2litres of water R and adjust to pH3.0with phosphoric acid R;—mobile phase B:dissolve0.80g of sodium dihydrogen phosphate R in2litres of water R,add280g of sodium perchlorate R(4)and adjust to pH3.0with phosphoric acid R;filter and degas;Time (min)Mobile phase A(per cent V/V)Mobile phase B(per cent V/V)0-10752510-3575→025→10035-400100 Flow rate:0.22ml/min.Detection:spectrophotometer at202nm.(2)AG11-HC from Dionex(reference052963)is suitable.(3)AS11-HC from Dionex(reference052961)is suitable.(4)Normapur from VWR/Prolabo(reference27988.232)is suitable.Equilibration:15min.Injection:20µl of test solution(b)and reference solutions(b)and(c).System suitability:—the chromatogram obtained with reference solution(b)shows no peak at the retention time of heparin;—resolution:minimum3.0between the peaks due to dermatan sulfate+chondroitin sulfate and over-sulfated chondroitin sulfate in the chromatogram obtained with reference solution(c).The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.1.dermatan sulfate+chondroitin sulfate2.over-sulfated chondroitin sulfate Figure0333.-2.–Chromatogram for the test for related substances of heparin sodium:reference solution(c)Results:—sum of dermatan sulfate and chondroitin sulfate:not more than the area of the corresponding peak in the chromatogram obtained with reference solution(c)(2.0per cent);—any other impurity:absence.Nitrogen(2.5.9):not more than1.5per cent to2.5per cent(dried substance),determined on0.100g.Sodium:9.5per cent to12.5per cent(dried substance).Atomic absorption spectrometry(2.2.23,Method I).Test solution.Dissolve50mg of the substance to be examined in a1.27mg/ml solution of caesium chloride R in0.1M hydrochloric acid and dilute to100.0ml with the same solvent.Reference solutions.Prepare reference solutions containing25ppm,50ppm and75ppm of Na,using sodium standard solution(200ppm Na)R diluted with a1.27mg/ml solution of caesium chloride R in0.1M hydrochloric acid.Source:sodium hollow-cathode lamp.Wavelength:330.3nm.Atomisation device:flame of suitable composition(for example11litres of air and2litres of acetylene per minute).Heavy metals(2.4.8):maximum30ppm.0.5g complies with test CF.Prepare the reference solution using1.5ml of lead standard solution(10ppm Pb)R.Loss on drying(2.2.32):maximum8.0per cent,determined on1.000g by drying at60°C over diphosphorus pentoxide R for3h at a pressure not exceeding670Pa.Sulfated ash(2.4.14):30per cent to43per cent,determined on0.20g and calculated with reference to the dried substance.Bacterial endotoxins(2.6.14):less than0.01IU per International Unit of heparin,if intended for use in the manufacture of parenteral preparations without a further appropriate procedure for the removal of bacterial endotoxins.ASSAYCarry out the assay of heparin(2.7.5).The estimated potency is not less than90per cent and not more than111per cent of the stated potency.The confidence limits of the estimated potency(P=0.95)are not less than80per cent and not more than125per cent of the stated potency.STORAGEIn an airtight container.If the substance is sterile,store in a sterile,airtight,tamper-proof container.LABELLINGThe label states:—the number of International Units per milligram;—the animal species of origin;—where applicable,that the substance is suitable for use in the manufacture of parenteral preparations.ReagentsDeuterated sodium trimethylsilylpropionate.C8H92H4NaO2Si.(M r172.3).XXXXXXX. [24493-21-8].Sodium3-(trimethylsilyl)(2,2,3,3-2H4)propionate.TSP-d4.Degree of deuteration:minimum98per cent.White or almost white powder.。
依诺肝素质量标准

依诺肝素质量标准1. 产品描述与定义依诺肝素是一种低分子量肝素,主要应用于肺血栓栓塞和静脉血栓栓塞等疾病的预防和治疗。
依诺肝素以注射剂的形式供应。
2. 性状和外观依诺肝素应呈无色或几乎无色、透明的溶液,不应有可见的异物。
3. 标准要求3.1 粒度依诺肝素注射剂的粒径分布应符合以下要求:- 95%的粒径应小于等于10微米;- 99%的粒径应小于等于15微米。
3.2 透明度依诺肝素注射剂的透明度应符合以下要求:- 使用2英寸玻璃比色皿检测,不得有可见沉淀或浑浊,光透过度应达到98%以上。
3.3 酸碱度(pH)依诺肝素注射剂的酸碱度应符合以下要求:- pH值应在5.0至7.0之间。
3.4 游离钠含量依诺肝素注射剂的游离钠含量应符合以下要求:- 游离钠含量不得超过每毫升15微克。
3.5 杂质依诺肝素注射剂中杂质含量应符合以下要求:- 重金属杂质(如铅、砷、铬等)含量不得超过每毫升5微克;- 其他有害杂质含量不得超过每毫升25微克。
3.6 活性成分含量依诺肝素注射剂的活性成分含量应符合以下要求:- 每毫升依诺肝素注射剂应含有不少于90国际单位的活性成分。
4. 性能验证与测试方法4.1 粒径测试方法:采用光散射法测定依诺肝素颗粒的粒径大小。
4.2 透明度测试方法:使用2英寸玻璃比色皿,对依诺肝素溶液的透明度进行测定。
4.3 酸碱度测试方法:采用电位法测定依诺肝素注射剂的pH值。
4.4 游离钠含量测试方法:采用离子选择电极法测定依诺肝素注射剂中游离钠的含量。
4.5 杂质测试方法:采用相关的色谱法或其他适用的分析方法对依诺肝素注射剂中的重金属和其他有害杂质进行测定。
4.6 活性成分含量测试方法:采用适用的生化法或其他准确的分析方法测定依诺肝素注射剂中的活性成分含量。
5. 包装与储存依诺肝素注射剂应采用符合相关药品包装标准的密封包装,并在阴凉干燥的条件下储存,远离光线和热源。
注:以上依诺肝素质量标准仅供参考,具体标准应根据实际情况和相关法规进行制定。
【VIP专享】依诺肝素钠说明书翻译

依诺肝素钠注射液说明书
请仔细阅读说明书并在医师指导下使用 警示语:椎管内血肿。当实施椎管内麻醉(脊麻和硬膜外麻醉)或椎管穿刺时应注意,使 用低分子肝素或肝素类物质预防血栓并发症的病人,有可能引起椎管内血肿,导致长期甚 至永久性瘫痪,以上事件很少发生。放置硬膜外导管或反复硬膜外穿刺,合并使用影响止 血功能的药物,如非甾体类抗炎药(NSAIDs)、血小板抑制剂或其它抗凝药物等,血肿发 生率可能会更高。此种情况,应监测病人神经损害的症状和体征,如发现有可能损伤神经, 应紧急处理。医生在对此类病人实施椎管内干预(麻醉或穿刺)时,应进行利弊权衡。 【药品名称】 通用名称:依诺肝素钠注射液
ห้องสมุดไป่ตู้
2000 Axa IU 和 4000 Axa IU 注射液:
• 预防静脉血栓栓塞性疾病 (预防静脉内血栓形成) ,特别是与骨科或普外手术有关的
血栓形成。
6000 Axa IU, 8000 Axa IU 和 10000 Axa IU 注射液:
• 治疗已形成的深静脉栓塞,伴或不伴有肺栓塞,临床症状不严重,不包括需要外科
手术或溶栓剂治疗的肺栓塞。
• 治疗不稳定性心绞痛及非 Q 波心肌梗死,与阿司匹林合用。
• 用于血液透析体外循环中,防止血栓形成。
•
治疗急性 ST 段抬高型心肌梗死,与溶栓剂联用或同时与经皮冠状动脉介入治疗
(PCI)联用。
【规格】 (1)0.2ml :2000 AxaIU (2)0.4ml: 4000 AxaIU (3)0.6ml: 6000 AxaIU (4)0.8ml :8000 AxaIU (5)1.0ml :10000 AxaIU
®
商品名称:克赛 Clexane 英文名称:Enoxaparin Sodium Injection 汉语拼音:Yinuogansuna Zhusheye 【成份】 化学名称:依诺肝素钠(低分子肝素钠) 化学结构式:
依诺肝素钠注射液说明书

核准日期:2009年8月12日修改日期:依诺肝素钠注射液说明书请仔细阅读说明书并在医师指导下使用警示语:椎管内血肿。
当实施椎管内麻醉(脊麻和硬膜外麻醉)或椎管穿刺时应注意,使用低分子肝素或肝素类物质预防血栓并发症的病人,有可能引起椎管内血肿,导致长期甚至永久性瘫痪,以上事件很少发生。
放置硬膜外导管或反复硬膜外穿刺,合并使用影响止血功能的药物,如非甾体类抗炎药(NSAIDs)、血小板抑制剂或其它抗凝药物等,血肿发生率可能会更高。
此种情况,应监测病人神经损害的症状和体征,如发现有可能损伤神经,应紧急处理。
医生在对此类病人实施椎管内干预(麻醉或穿刺)时,应进行利弊权衡。
【药品名称】通用名称:依诺肝素钠注射液商品名称:克赛®Clexane英文名称:Enoxaparin Sodium Injection汉语拼音:Yinuogansuna Zhusheye【成份】化学名称:依诺肝素钠(低分子肝素钠)化学结构式:分子量:3500至5500道尔顿辅料:注射用水【性状】本品为无色或淡黄色的澄明液体。
【适应症】2000 Axa IU 和4000 Axa IU注射液:• 预防静脉血栓栓塞性疾病(预防静脉内血栓形成) ,特别是与骨科或普外手术有关的血栓形成。
6000 Axa IU, 8000 Axa IU 和10000 Axa IU注射液:•治疗已形成的深静脉栓塞,伴或不伴有肺栓塞,临床症状不严重,不包括需要外科手术或溶栓剂治疗的肺栓塞。
• 治疗不稳定性心绞痛及非Q波心肌梗死,与阿司匹林合用。
• 用于血液透析体外循环中,防止血栓形成。
•治疗急性ST段抬高型心肌梗死,与溶栓剂联用或同时与经皮冠状动脉介入治疗(PCI)联用。
【规格】(1)0.2ml :2000 AxaIU (2)0.4ml: 4000 AxaIU (3)0.6ml: 6000 AxaIU(4)0.8ml :8000 AxaIU (5)1.0ml :10000 AxaIU【用法用量】预防静脉血栓栓塞性疾病,治疗深静脉栓塞,治疗不稳定性心绞痛及非Q波心肌梗死时应采用深部皮下注射给予依诺肝素;血液透析体外循环时为血管内途径给药;对于ST段抬高型急性心肌梗死,初始的治疗为静脉注射,随后改为皮下注射治疗。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
•L IMIT OF F LUORIDE ADDITIONAL REQUIREMENTS [N OTE—Use plasticware throughout this test.]•P ACKAGING AND S TORAGE: Preserve in tight, light-resistant Buffer: Dissolve 110g of sodium chloride and 1g of containers, and avoid exposure to excessive heat.sodium citrate in 700mL of water in a 2000-mL volu-•USP R EFERENCE S TANDARDS〈11〉metric flask. Cautiously add 150g of sodium hydroxide,USP Enflurane RSand dissolve with shaking. Cool to room temperature,and, while stirring, cautiously add 450mL of glacialacetic acid to the cooled solution. Cool, add 600mL ofisopropyl alcohol, and dilute with water to volume; thepH of this solution is 5.0–5.5.Enoxaparin SodiumStandard stock solution: 1mg/mL of fluoride ion, pre-pared as follows. Transfer 221mg of sodium fluoride, previously dried at 150° for 4 h, to a 100-mL volumet-ric flask. Add 20mL of water, and mix to dissolve. Add 1.0mL of sodium hydroxide solution (1 in 2500), and dilute with water to volume. [N OTE—Store in a tightly closed plastic container.]Standard solution A: 100mL of a solution containing 1µg/mL of fluoride ion in Buffer, from Standard stock solutionStandard solution B: 100mL of a solution containing 3µg/mL of fluoride ion in Buffer, from Standard stock solutionStandard solution C: 100mL of a solution containing 5µg/mL of fluoride ion in Buffer, from Standard stock solutionStandard solution D: 100mL of a solution containing 10µg/mL of fluoride ion in Buffer, fromStandard stocksolution[9041-08-1].Sample stock solution: Enflurane and water (1:1).Shake the Sample stock solution for 5 min, allow theDEFINITIONliquids to separate completely, and use the water layer.Enoxaparin Sodium is the sodium salt of a depolymerized Sample solution: Sample stock solution and Bufferheparin. It is obtained by alkaline depolymerization of (1:1). Use volumetric glassware.heparin benzyl ester. The starting material, heparin, is ob-Electrode system: Use a pH meter capable of a mini-tained exclusively from porcine intestinal mucosa. Heparin mum reproducibility of ±0.2 mV, equipped with a glass-source material used in the manufacture of Enoxaparin sleeved, calomel-fluoride, specific-ion electrode systemSodium complies with the compendial requirements (see pH 〈791〉).stated in the Heparin Sodium monograph. Enoxaparin So-Analysisdium consists of a complex set of oligosaccharides that Samples: Standard solutions and Sample solutionhave not yet been completely characterized. The majority Transfer the solution to a 150-mL beaker, add aof the components have a 4-enopyranose uronate struc-polytef-coated stirring bar, and immerse the elec-ture at the nonreducing end of their chain. About 20% of trodes in the solution. Stir with a magnetic stirrerthe materials contain a 1,6-anhydro derivative on the re-having an insulated top until equilibrium is obtainedducing end of the chain, the range being between 15% (1–2 min), and record the potential, in mv. [N OTE—and 25%. The weight-average molecular weight of Enox-Rinse and dry the electrodes between measurements,aparin Sodium is 4500 Da, the range being between taking care to avoid damaging the crystal of the spe-3800 and 5000 Da; about 16% have a molecular weight cific-ion electrode.]of less than 2000 Da, the range being between 12.0% Plot the logarithm of the fluoride-ion concentrations,and 20.0%; about 74% have a molecular weight between in µg/mL, of the Standard solutions versus the poten-2000 and 8000 Da, the range being between 68.0% and tial, in mV. From the measured potential of the Sam-82.0%. NMT 18.0% have a molecular weight higher thanple solution and the standard curve, determine the8000 Da. When prepared as a solution, the solution is concentration, in µg/mL, of fluoride ions in the Sam-analyzed for clarity and degree of color using a validated ple solution.method. The degree of sulfation is NLT 1.8per disaccha-Acceptance criteria: NMT 10µg/mLride unit. It has a potency of NLT 90 and NMT 125 Anti-•L IMIT OF N ONVOLATILE R ESIDUEFactor Xa International Units (IU)/mg, and NLT 20.0 and Analysis: Allow 10.0mL to evaporate at room tempera-NMT 35.0 Anti-Factor IIa IU/mg, calculated on the dried ture in a tared evaporating dish, dry the residue at 50°basis. The ratio of Anti-Factor Xa activity to Anti-Factor IIa for 2 h, and weigh.activity is between 3.3 and 5.3.Acceptance criteria: NMT 2mg of residue remains.IDENTIFICATIONSPECIFIC TESTS•A. U LTRAVIOLET A BSORPTION〈197U〉•S PECIFIC G RAVITY〈841〉: 1.516–1.519Medium: 0.01 N hydrochloric acid•R EFRACTIVE I NDEX〈831〉: 1.3020–1.3038 at 20°Sample solution: 500µg/mL•W ATER D ETERMINATION, Method I〈921〉: NMT 0.14%Acceptance criteria: The spectra exhibit maxima at •A CIDITY OR A LKALINITY231±2 nm.Sample: Shake 20mL of Enflurane with 20mL of car-bon dioxide-free water for 3 min, and allow the layersto separate. Draw off the water layer, add bromocresol Change to read:purple TS as the indicator, and titrate with 0.010 Nsodium hydroxide or 0.010 N hydrochloric acid.•B.13C NMR S PECTRUMAcceptance criteria: NMT 0.10mL of 0.010 N sodium(See •Nuclear Magnetic Resonance Spectroscopy 〈761〉•(CN hydroxide or NMT 0.60mL of 0.010 N hydrochloric1-May-2016).)acid is required for neutralization.Standard solution: Dissolve 200mg of USP EnoxaparinSodium RS in a mixture of 0.2mL of deuterium oxideand 0.8mL of water. Add 0.05mL of deuterated meth-Acceptance criteria:M2000 is between 12.0% and anol to serve as an internal reference.20.0%, M2000–8000 is between 68.0% and 82.0%, and Sample solution: Dissolve 200mg of Enoxaparin So-M8000 is NMT 18.0%.dium in a mixture of 0.2mL of deuterium oxide and•E. I DENTIFICATION T ESTS—G ENERAL, Sodium〈191〉: Meets0.8mL of water. Add 0.05mL of deuterated methanol.the requirementsAnalysis: Transfer the Standard solution and the SampleASSAYsolution to NMR tubes of 5-mm diameter. Using apulsed (Fourier transform) NMR spectrometer operatingat NLT 75MHz for 13C, record the 13C NMR spectra of Change to read:the Standard solution and the Sample solution at 40°.Acceptance criteria: The spectra are similar.•ANTI-F ACTOR X a A CTIVITY•C. The ratio of the numerical value of the Anti-Factor Xa Acetic acid solution: Glacial acetic acid and water activity, in Anti-Factor Xa IU/mg, to the numerical value(42:58)of the Anti-Factor IIa activity, in Anti-Factor IIa IU/mg, as pH 7.4polyethylene glycol 6000 buffer: Dissolve determined by the Assay (Anti-Factor Xa Activity) and Im- 6.08g of tris(hydroxymethyl)aminomethane and 8.77g purities (Anti-Factor IIa Activity), respectively, is NLT 3.3of sodium chloride in 500mL of water. Add 1.0g of and NMT 5.3.polyethylene glycol 6000, adjust with hydrochloric acid •D. M OLECULAR W EIGHT D ISTRIBUTION AND W EIGHT-A VERAGE to a pH of 7.4, and dilute with water to 1000mL.M OLECULAR W EIGHT pH 7.4 buffer: Dissolve 6.08g of tris(hydroxymethyl)-Mobile phase: Prepare a 0.5 M lithium nitrate solution.aminomethane and 8.77g of sodium chloride in Pass through a membrane filter of 0.45-µm or smaller500mL of water. Adjust with hydrochloric acid to a pH pore size, and degas with helium.of 7.4, and dilute with water to 1000mL.Standard solution: 10mg/mL of USP Enoxaparin So-pH 8.4 buffer: Dissolve 3.03g of tris(hydroxymethyl)-dium RS in Mobile phase aminomethane, 5.12g of sodium chloride, and 1.40g Sample solution: 10mg/mL of Enoxaparin Sodium in of edetate sodium in 250mL of water. Adjust with hy-Mobile phase drochloric acid to a pH of 8.4, and dilute with water to Chromatographic system500mL.(See Chromatography 〈621〉, System Suitability.)Human antithrombin III solution: Reconstitute a vial Mode: Size exclusion LC of antithrombin III (see Reagents, Indicators, and Solu-Detector: Differential refractive index tions—Reagent Specifications) in water to obtain a solu-Column tion containing 5 Antithrombin III Units/mL. Dilute this Guard: 6-mm × 40-mm; packing L59solution with pH 7.4polyethylene glycol 6000 buffer to Analytical: Two 7.8-mm × 300-mm columns in series;obtain a solution having a concentration of 1.0 Anti-packing L59thrombin III Unit/mL.Temperature: Room temperature Factor Xa solution: Reconstitute a weighed quantity of Flow rate: 0.6mL/min maintained constant to ± 1.0%bovine factor Xa (see Reagents, Indicators, and Solu-Analysis: Reconstitute 1 vial each of USP Enoxaparin tions—Reagent Specifications) in pH 7.4polyethylene gly-Sodium Molecular Weight Calibrant A RS and USP col 6000 buffer to obtain a solution that gives an in-Enoxaparin Sodium Molecular Weight Calibrant B RS in crease in absorbance value at 405 nm of NMT 0.20 1mL of Mobile phase. Separately inject 20µL of USP absorbance units/min when assayed as described below Enoxaparin Sodium Molecular Weight Calibrant A RS but using as an appropriate volume, V, the volume in and USP Enoxaparin Sodium Molecular WeightµL of pH 7.4 buffer instead of VµL of the enoxaparin Calibrant B RS, record the chromatograms, and meas-solution.ure the retention times. Inject in duplicate 20µL each Chromogenic substrate solution: Prepare a solution of of the Standard solution and the Sample solution, and a suitable chromogenic substrate for amidolytic test record the chromatograms for a length of time to en-(see Reagents, Indicators, and Solutions—Reagent Specifi-sure complete elution, including salt and solvent peaks.cations) for factor Xa in water to obtain a concentration Calculate the total peak areas under each of the Stan-of about 3 mM. Dilute with pH 8.4 buffer to obtain a dard solution and Sample solution chromatograms, ex-solution having a concentration of 0.5 mM.cluding salt and solvent peaks at the end.Standard solutions: Reconstitute the entire contents of Calibration curve: Plot the retention times on the x-an ampul of USP Enoxaparin Sodium for Bioassays RS axis against the peak molecular weights on the y-axis with water, and dilute with pH 7.4 buffer to obtain four for the peaks from USP Enoxaparin Sodium Molecular dilutions in the concentration range between 0.025 and Weight Calibrant A RS and USP Enoxaparin Sodium Mo-0.2 Anti-Factor Xa IU/mL.lecular Weight Calibrant B RS, and fit the data to a Sample solutions: Proceed as directed for Standard so-third-order polynomial, using suitable gel permeation lutions to obtain concentrations of Enoxaparin Sodium chromatography (GPC) software.similar to those obtained for the Standard solutions.Calculations: Compute the data, using the same GPC Analysissoftware; determine the weight-average molecular Samples:Standard solutions, Sample solutions, Human weight, M w, for each of the duplicate chromatograms of antithrombin III solution, pH 7.4 buffer, Factor Xa solu-the Standard solution and the Sample solution; and take tion, Chromogenic substrate solution, and Acetic acid the average for each solution. Correct the mean value solutionof M w to the nearest 50. The Chromatographic system is Label 18 suitable tubes: B1 and B2 for blanks; T1, T2, suitable if M w for USP Enoxaparin Sodium RS is within T3, and T4 each in duplicate for the dilutions of the 150 Da of the labeled M w value. The M w for the Sample Sample solutions; and S1, S2, S3, and S4 each in dupli-solution is between 3800 and 5000 Da. Using the same cate for the dilutions of the Standard solutions. [NOTE—software, determine for each of the duplicate Sample Treat the tubes in the order B1, S1, S2, S3, S4, T1, T2,solution chromatograms the percentage of Enoxaparin T3, T4, T1, T2, T3, T4, S1, S2, S3, S4, B2.] To each Sodium chains with molecular weights lower than 2000tube add the same volume, V (20–50µL), of Human Da, M2000, the percentage of Enoxaparin Sodium chains antithrombin III solution and an equal volume, V, of with molecular weights in the range 2000–8000 Da,either the blank (pH 7.4 buffer) or an appropriate dilu-M2000–8000, and the percentage of Enoxaparin Sodium tion of the Sample solutions or the Standard solutions.chains with molecular weights greater than 8000 Da,Mix, but do not allow bubbles to form. Incubate at M8000. Average the duplicate values, and express to the37° for 1.0 min. Add to each tube 2V (40–100µL) of nearest 0.5%.Factor Xa solution, and incubate for 1.0 min. Add a 5V Analysis(100–250µL) volume of Chromogenic substrate solu-Samples:Standard solution A, Standard solution B, tion. Stop the reaction after 4.0 min with a 5V Standard solution C, Cesium chloride solution, and Sam-(100–250µL) volume of Acetic acid solution. Measure ple solutionthe absorbance of each solution at 405 nm, using a Concomitantly determine the absorbances of the Ce-suitable spectrophotometer (see •Ultraviolet-Visible sium chloride solution (blank), Sample solution, and Spectroscopy 〈857〉•(CN 1-May-2016)) against blank B1. The Standard solutions at 330.3 nm, using a sodiumreading of blank B2 relative to blank B1 is NMT ± 0.05hollow-cathode lamp and an air–acetylene flame. Us-absorbance unit.ing the absorbances of Standard solutions A–C, deter-Calculations: For each series, calculate the regression mine the sodium content in the Sample solution after of the absorbance against log concentrations of the an appropriate blank correction.Sample solutions and of the Standard solutions, and cal-Acceptance criteria: 11.3%–13.5% on the dried basis culate the potency of the Enoxaparin Sodium in IU ofIMPURITIESAnti-Factor Xa activity/mL, using statistical methods forparallel-line assays. The four independent log relativepotency estimates are then combined to obtain the Delete the following:final geometric mean. Its confidence limits are calcu-lated. Express the Anti-Factor Xa activity of Enoxaparin••H EAVY M ETALS, Method I〈231〉: NMT 30µg/g, using a Sodium/mg. 2.7% solution in water•(Official 1-Jan-2018) Acceptance criteria: The potency is NLT 90 and NMT125 Anti-Factor Xa IU/mg on the dried basis.SPECIFIC TESTS•P H 〈791〉: 6.2–7.7 for a 10.0% solution in water OTHER COMPONENTS•LOSS ON D RYING〈731〉: Dry 1g in a vacuum at 70° for 6•B ENZYL A LCOHOL C ONTENT h: it loses NMT 10.0% of its weight.Mobile phase: Acetonitrile, methanol, and water(3:1:16)Standard solution: 0.1mg/mL of USP Benzyl Alcohol Change to read:RS in waterSample solution: Weigh 0.5g of Enoxaparin Sodium•S PECIFIC A BSORBANCEinto a 10-mL volumetric flask, and dissolve in 5.0mL of(See •Ultraviolet-Visible Spectroscopy 〈857〉•(CN 1-May-2016).) 1N sodium hydroxide. Allow to stand at room temper-Sample solution: 0.5mg/mL of Enoxaparin Sodium in ature for about 1 h. Add 1.0mL of glacial acetic acid,0.01 N hydrochloric aciddilute with water to volume, and mix.Analysis: Obtain the UV spectra of the Standard solution Chromatographic system and the Sample solution between 200 nm and 300 nm (See Chromatography 〈621〉, System Suitability.)against a 0.01 N hydrochloric acid blank.Mode: LC Calculate the specific absorbance at the wavelength of Detector: UV 256 nm maximum absorbance at 231±2 nm, with reference Column: 4.6-mm × 15-cm stainless steel; packing L7to the dried substance:Flow rate: 1.0mL/min, maintained constant to ±10%Injection volume: 20µL Result = A× 100×1000/[M×l× (100 −E)] AnalysisA= absorbance at the wavelength of maximum Samples:Standard solution and Sample solutionabsorbanceCalculate the percentage of benzyl alcohol in the por-M= weight of Enoxaparin Sodium in the Sample tion of Enoxaparin Sodium taken:solution (mg)Result = (r U/r S) × (C S/C U) × 100l= path length (typically 1cm)E= loss on drying (%)r U= peak area of benzyl alcohol from the Sample Acceptance criteria: 14.0–20.0 on the dried basis solution•B ACTERIAL E NDOTOXINS T EST〈85〉: It contains NMT 0.01 r S= peak area of benzyl alcohol from the Standard USP Endotoxin Unit/IU of Anti-Factor Xa activity.solution•A NTI-F ACTOR II a A CTIVITYC S= concentration of benzyl alcohol in the Acetic acid solution, pH 7.4polyethylene glycol 6000Standard solution (mg/mL)buffer, pH 7.4 buffer, pH 8.4 buffer, and HumanC U= concentration of Enoxaparin Sodium in the antithrombin III solution: Proceed as directed in theSample solution (mg/mL)Assay for Anti-Factor Xa Activity, except that the concen-Acceptance criteria: NMT 0.1%tration of the Human antithrombin III solution is 0.5•N ITROGEN D ETERMINATION, Method II〈461〉: 1.8%–2.5%Antithrombin III Unit/mL.on the dried basis Thrombin human solution: Reconstitute thrombinhuman (see Reagents, Indicators and Solutions—ReagentSpecifications) in water, and dilute in pH 7.4polyethylene Change to read:glycol 6000 buffer to obtain a solution having a concen-tration of 5 Thrombin Units/mL.•S ODIUM C ONTENT Chromogenic substrate solution: Prepare a solution of (See •Atomic Absorption Spectroscopy 〈852〉•(CN 1-May-2016).) a suitable chromogenic substrate for an amidolytic test Cesium chloride solution: 1.27mg/mL of cesium chlo-(see Reagents, Indicators, and Solutions—Reagent Specifi-ride in 0.1 N hydrochloric acid cations) for thrombin in water to obtain a concentration Standard solution A: 0.0025% of sodium chloride in of about 3 mM. Immediately before use, dilute with pH Cesium chloride solution8.4 buffer to 0.5 mM.Standard solution B: 0.0050% of sodium chloride in Standard solutions: Reconstitute the entire contents of Cesium chloride solution an ampul of USP Enoxaparin Sodium for Bioassays RS Standard solution C: 0.0075% of sodium chloride in with water, and dilute with pH 7.4 buffer to obtain four Cesium chloride solution dilutions having concentrations in the range between Sample solution: Transfer 50.0mg of Enoxaparin So-0.015 and 0.075 IU of Anti-Factor IIa activity/mL.dium to a 100-mL volumetric flask, and dissolve in anddilute with Cesium chloride solution to volume.Sample solutions: Proceed as directed under Standard ADDITIONAL REQUIREMENTSsolutions to obtain concentrations of Enoxaparin Sodium•P ACKAGING AND S TORAGE: Preserve in tight containers, similar to those obtained for the Standard solutions.and store below 40°, preferably at room temperature.Analysis: Proceed as directed in the Assay for Anti-Factor•USP R EFERENCE S TANDARDS〈11〉Xa Activity, except to use Thrombin human solution in-USP Benzyl Alcohol RSstead of Factor Xa solution and to use Human antithrom-USP Endotoxin RSbin III solution as described P Enoxaparin Sodium RSCalculations: For each series, calculate the regression of USP Enoxaparin Sodium Molecular Weight Calibrant A RS the absorbance against log concentrations of the Sam-USP Enoxaparin Sodium Molecular Weight Calibrant B RS ple solutions and of the Standard solutions, and calculate USP Enoxaparin Sodium for Bioassays RSthe potency of the Enoxaparin Sodium in IU of Anti-Factor IIa activity/mg, using statistical methods for par-allel-line assays. The four independent dilution estimatesare then combined to obtain the final weighted mean.Then calculate the confidence limits. Express the Anti-Enoxaparin Sodium InjectionFactor IIa activity of Enoxaparin Sodium/mg.Acceptance criteria: It has a potency of NLT 20.0 and DEFINITIONNMT 35.0 Anti-Factor IIa IU/mg on the dried basis.Enoxaparin Sodium Injection is a sterile solution of Enox-•M OLAR R ATIO OF S ULFATE TO C ARBOXYLATE aparin Sodium in Water for Injection. Its appearance is Mobile phase: Carbon dioxide-free water analyzed for clarity and degree of color, using a validated Sample solution: 5mg/mL of Enoxaparin Sodium in method. Its potency value is NLT 90% and NMT 110% of carbon dioxide-free water the potency stated on the label in terms of International Chromatographic system Anti-Factor Xa Units (IU). It may contain, in multiple-dose (See Chromatography 〈621〉, System Suitability.)containers, a suitable antimicrobial preservative, such as Mode: LC benzyl alcohol.Detector: IonColumn: Two columns; one 1.5-cm × 2.5-cm column,IDENTIFICATIONpacked with an anion-exchange resin L64packing; and•A.one 1.5-cm × 7.5-cm column, packed with a cation-Analysis: Transfer the total contents of a single-dose exchange resin L65packing. The outlet of the anion-container or 0.4mL from a multiple-dose container to a exchange column is connected to the inlet of the cat-glass test tube, add 2mL of water and 1mL of 2% ion-exchange column.(w/v) protamine sulfate solution, and mix.Flow rate: 1mL/min Acceptance criteria: A creamy white precipitate is Analysis formed.Sample:Sample solution•B. U LTRAVIOLET A BSORPTION〈197U〉[N OTE—Regenerate the anion-exchange column and the Medium: 0.01 N hydrochloric acidcation-exchange column with 1N sodium hydroxide Standard solution: 500µg/mLand 1N hydrochloric acid, respectively, between two Sample solution: Transfer the total content of a single-injections.]dose container or 0.4mL from a multiple-dose con-Inject the Sample solution into the anion-exchange col-tainer to a 100-mL volumetric flask. Dilute with Medium umn, and collect the eluate from the cation-exchange to volume.column in a beaker at the outlet until the ion detector Acceptance criteria: The spectra exhibit maxima at reading returns to the baseline value. Quantitatively231±2 nm.transfer the eluate to a titration vessel containing a•C. I DENTIFICATION T ESTS—G ENERAL, Sodium〈191〉: Meets magnetic stirring bar, and dilute with carbon dioxide-the requirementsfree water to about 60mL. Position the titration vesselASSAYon a magnetic stirrer, and immerse the electrodes.Note the initial conductivity reading, and titrate withapproximately 0.1 N sodium hydroxide added in Change to read:100-µL portions. [N OTE—Prepare the sodium hydroxidesolution in carbon dioxide-free water.] Record the bu-•A NTI-F ACTOR X a A CTIVITYret reading and the conductivity meter reading after Acetic acid solution: Glacial acetic acid and water each addition of the sodium hydroxide solution.(42:58)Calculations: Plot the conductivity measurements on pH 7.4polyethylene glycol 6000 buffer: Dissolve the y-axis against the volumes of sodium hydroxide 6.08g of tris(hydroxymethyl)aminomethane and 8.77g added on the x-axis. The graph will have three linear of sodium chloride in 500mL of water. Add 1.0g of sections—an initial downward slope, a middle slight polyethylene glycol 6000, adjust with hydrochloric acid rise, and a final rise. For each of these sections draw the to a pH of 7.4, and dilute with water to 1000mL.best-fit straight lines, using linear regression analysis. At pH 7.4 buffer: Dissolve 6.08g of tris(hydroxymethyl)-the points where the first and second straight lines in-aminomethane and 8.77g of sodium chloride in tersect and where the second and third lines intersect,500mL of water. Adjust with hydrochloric acid to a pH draw perpendiculars to the x-axis to determine the of 7.4, and dilute with water to 1000mL.volumes of sodium hydroxide taken up by the sample pH 8.4 buffer: Dissolve 3.03g of tris(hydroxymethyl)-at those points. The point where the first and second aminomethane, 5.12g of sodium chloride, and 1.40g lines intersect corresponds to the volume of sodium hy-of edetate sodium in 250mL of water. Adjust with hy-droxide taken up by the sulfate groups (V S). The point drochloric acid to a pH of 8.4, and dilute with water to where the second and third lines intersect corresponds500mL.to the volume of sodium hydroxide consumed by the Human antithrombin III solution: Reconstitute a vial sulfate and the carboxylate groups together (V T).of antithrombin III (see Reagents, Indicators, and Solu-Calculate the molar ratio of sulfate to carboxylate:tions—Reagent Specifications) in water to obtain a solu-tion containing 5 Antithrombin III Units/mL. Dilute this Result = V S/(V T−V S)solution with pH 7.4polyethylene glycol 6000 buffer toobtain a solution having a concentration of 1.0 Anti-Acceptance criteria: The molar ratio of sulfate to car-thrombin III Unit/mL.boxylate is NLT 1.8.。