细菌内毒素
细菌内毒素标准品

细菌内毒素标准品
细菌内毒素是一种由细菌产生的有毒物质,它可以引起机体的
免疫反应,甚至导致严重的疾病。
因此,研究和检测细菌内毒素的
标准品具有重要意义。
在医药领域,细菌内毒素标准品被广泛应用
于药物研发、临床诊断和疾病治疗等方面。
首先,细菌内毒素标准品的研究对于药物研发具有重要意义。
许多药物的研发需要进行严格的毒性测试,以确保其安全性和有效性。
细菌内毒素标准品可以作为药物毒性测试的标准物质,帮助科
研人员评估药物的毒性水平,为药物研发提供重要参考。
其次,细菌内毒素标准品在临床诊断中也起着至关重要的作用。
许多疾病的诊断需要通过检测患者体内的细菌内毒素水平来进行判断。
因此,标准的细菌内毒素标准品可以帮助临床医生准确地诊断
疾病,为患者提供及时有效的治疗方案。
此外,细菌内毒素标准品还可以用于疾病治疗的研究中。
一些
疾病的治疗需要针对细菌内毒素进行干预,以阻断其对机体的损害。
因此,研究和生产高质量的细菌内毒素标准品对于开发新的疾病治
疗手段具有重要意义。
综上所述,细菌内毒素标准品在药物研发、临床诊断和疾病治疗中都扮演着重要的角色。
随着医学科研水平的不断提高,对细菌内毒素标准品的需求也将不断增加。
因此,我们有必要加强对细菌内毒素标准品的研究和生产,以满足医药领域对高质量标准品的需求,为保障人民健康作出更大的贡献。
细菌内毒素

细菌内毒素检查法一、细菌内毒素检查法的定义:●本法是利用鲎试剂与细菌内毒素产生凝聚反应的机理,以判断供试品中的细菌内毒素限度是否符合规定的一种方法。
二、背景介绍:●1、细菌内毒素●2、热原●3、鲎1、细菌内毒素●细菌内毒素是一种革兰氏阴性细菌细胞壁的产物,当其死亡或菌体裂解时释放出的一类具有多种生物活性的毒性物质特性:●(1).致热性:内毒素作用人体细胞,使之释放内源性热原,刺激下丘脑体温调节中枢,引起发热反应。
●(2).耐热性:需250度干热30分钟才能彻底灭活。
●(3).分子极性:多糖链亲水,脂肪链疏水,在水中呈不均匀分布。
●(4).鲎反应:能与鲎试剂发生多级酶促反应,形成凝胶。
2、热原2.1热原是指临床上引起哺乳动物发热反应的物质2.2细胞分裂素(IL-1, IL-2, IL-6, IL-8 )内源性热原{产生细胞分裂素的物质内毒素热原外源性热原{非内毒素热原(病毒、细菌、真菌、抗体-抗原复合物、细胞分裂素)2.3热原和内毒素的关系:2.3.1热原是否就是内毒素?在学术上仍有争议,热原不仅是细菌内毒素。
但在药检的范畴,细菌内毒素是主要的热原物质,可以说无内毒素就无热原,控制内毒素就是控制热原。
3、鲎3.1鲎(horseshoe crab)是一类与三叶虫(现在只有化石)一样古老的动物。
鲎的祖先出现在地质历史时期古生代的泥盆纪,当时恐龙尚未崛起,原始鱼类刚刚问世,随着时间的推移,与它同时代的动物或者进化、或者灭绝,而惟独只有鲎从4 亿多年前问世至今仍保留其原始而古老的相貌,所以鲎有“活化石”之称。
又具有很高的药用价值。
3.2鲎试剂鲎的血液中含有铜离子,它的血液是蓝色的。
鲎血液颜色呈蓝色,是因为鲎血浆的主要成分是血蓝蛋白。
这种蓝色血液的提取物——“鲎试剂”。
鲎试剂是由海洋生物鲎的血液提取物制成的“鲎试剂”,能够准确、快速地检测人体是否因细菌感染而致病;鲎试剂在制药行业中,用于检测细菌内毒素。
目前使用的鲎试剂分为美洲鲎试剂和东方鲎鲎试剂两大类。
细菌内毒素

细菌内毒素
细菌内毒素是一种由细菌分泌的有毒物质。
它主要由细菌的细胞壁、胞外膜或细胞内产生,并在细菌感染或死亡时释放出来。
细菌内毒素的化学结构和生物活性因细菌种类而异。
它们可以直接伤害宿主细胞,引发免疫反应或导致中毒。
一些细菌内毒素可以穿透宿主细胞膜,并干扰细胞内代谢和信号传导通路,导致细胞功能异常甚至死亡。
细菌内毒素可以引起许多疾病,包括细菌感染引起的疾病如脓毒症、痢疾以及肺炎等。
它们还可能导致食物中毒、肠胃炎和其他细菌相关的疾病。
针对细菌内毒素的治疗方法包括抗生素治疗、中和细菌内毒素的抗体和免疫疗法,以及相关的疫苗预防措施。
需要注意的是,某些细菌内毒素具有非常强的毒性,可能对人类和动物的健康造成严重威胁。
因此,对细菌内毒素的研究和监测非常重要,以便及时采取控制和预防措施。
《细菌内毒素检查》课件

其他检测方法
热原质鲎试剂盒法
利用鲎试剂与内毒素结合后加热,通 过凝胶形成或浊度变化来检测内毒素 的含量。该方法适用于注射剂、输液 等样品。
高效液相色谱法
利用高效液相色谱技术分离内毒素, 通过紫外吸收或荧光检测器来检测内 毒素的含量。该方法适用于生物制品 、血液制品等样品。
03
细菌内毒素检查的应用
实验后的处理和安全防护
废液的处理
实验后产生的废液应按照实验室规定进行处理, 不得随意倾倒或排放。
实验垃圾的处理
实验后产生的垃圾应按照实验室规定进行分类和 处理。
ABCD
仪器的清洗和维护
实验后应对使用的仪器进行清洗和维护,以确保 其性能和精度。
个人防护
实验人员应穿戴个人防护装备,如实验服、手套 、口罩等,以降低交叉污染和感染的风险。
饮用水监测
01
通过细菌内毒素检查,可以检测饮用水中的细菌内毒素含量,
确保饮用水安全。
污水处理监测
02
在污水处理过程中,对出水进行细菌内毒素检查,可以评估污
水处理效果和出水质量。
环境样品监测
03
对土壤、空气等环境样品进行细菌内毒素检查,可以了解环境
中的细菌污染状况,为环境治理提供依据。
04
细菌内毒素检查的注意事 项
开发新型检测方法和技术
开发多组分检测技术
将多种细菌内毒素相关分子同时纳入检测范围,实现多组分的同 时检测,提高检测效率和准确性。
开发新型免疫学检测方法
利用新型免疫学技术,如单克隆抗体、免疫磁珠等,提高检测的特 异性和灵敏度。
开发新型生物信息学技术
利用生物信息学手段,对细菌内毒素相关基因和蛋白质进行高通量 筛选和鉴定,为新型检测方法的开发提供有力支持。
细菌内毒素检验方法验证方案

细菌内毒素检验方法验证方案一、背景介绍细菌内毒素是一种由细菌产生的有毒分子,它可以引起中毒反应并对人类健康造成危害。
因此,准确检测和验证细菌内毒素的方法非常重要。
本方案旨在验证细菌内毒素检验方法的准确性和可靠性。
二、实验目的本实验旨在验证细菌内毒素检验方法,确保其能够准确地检测细菌内毒素的含量。
三、实验材料和设备1. 细菌内毒素检验试剂盒2. 细菌培养基3. 细菌菌种4. 培养皿5. 恒温培养箱6. 试管7. 离心机8. 显微镜9. 无菌培养室四、实验步骤1. 准备工作a. 清洁实验桌面和实验器材,防止污染。
b. 准备所需要的材料和设备,并确保其干净和无菌。
2. 细菌培养a. 随机取一支细菌菌种采样放入培养基中并均匀涂抹于培养皿上。
b. 将培养皿置于恒温培养箱中,以适当的温度和时间培养细菌。
3. 细菌内毒素检验a. 取一份细菌内毒素检验试剂盒,根据说明书的指导进行试剂的调配和处理。
b. 等待试剂反应完成后,根据试剂盒的说明书进行结果的解读。
4. 结果分析a. 观察试剂盒结果,根据颜色变化和强度判断细菌内毒素的含量。
b. 结果符合预期的样本被认为是阳性,否则为阴性。
五、质量控制1. 测量前需进行必要的质量控制,包括试剂的稳定性和标准品的准确性。
六、风险控制1. 实验过程中需佩戴个人防护装备,如实验手套和口罩,以减少潜在的健康风险。
七、数据处理与结果1. 将实验所得数据进行整理和统计,并进行数据分析和结果的解读。
八、讨论与结论1. 讨论实验结果与预期结果之间的差异,分析可能的原因,并提出改进和优化的建议。
2. 结论验证细菌内毒素检验方法的准确性和可靠性。
九、参考文献参考实验所使用的细菌内毒素检验方法的相关文献。
以上是细菌内毒素检验方法验证方案的内容,该方案通过详细的步骤和操作说明,确保了实验的准确性和可靠性。
通过验证该方法,将可为细菌内毒素检验提供可靠的数据支持,为相关领域的研究和应用提供技术支持。
中国药典 细菌内毒素检验方法

中国药典细菌内毒素检验方法
中国药典细菌内毒素检验方法中,按以下步骤进行实验。
首先,制备试样溶液。
将待测样品溶解在合适的溶剂中,并通过过滤将溶液过滤至少两次以去除杂质。
然后,制备标准溶液。
将内毒素标准品按照指定浓度配制成溶液。
接下来,准备试管。
将标准溶液和试样溶液分别装入两个试管中,并加入适量的无菌生理盐水作为空白对照。
然后,进行混合。
轻轻旋转试管混合样品,确保溶液均匀混合。
接着,培养细菌。
将特定的细菌株接种在培养基中,分别加入试管中的样品溶液,并在适当条件下进行培养。
培养结束后,观察结果。
通过观察培养基的变化,比较试管中的样品溶液和空白对照的差异,判断样品中是否存在细菌内毒素。
最后,记录结果。
根据观察结果,记录样品的检验结果,并进行数据分析。
细菌内毒素检测标准

细菌内毒素检测标准
细菌内毒素检测的标准通常由相关监管机构或行业组织制定。
以下是一些常见的细菌内毒素检测标准:
1. 中国标准:GB/T 18979-2003《食品中内毒素的测定鲎试剂法》是中国关于细菌内毒素检测的国家标准,该标准规定了使用鲎试剂法测定食品中内毒素的试验方法和要求。
2. 美国标准:FDA(美国食品药品监督管理局)制定了关于细菌内毒素检测的联邦法规,如21 CFR 11
3.320(f),规定了食品中内毒素的检测方法和限量。
3. 欧洲标准:欧洲食品安全局(EFSA)对细菌内毒素的检测方法有详细的指南,如针对水产品中的副溶血性弧菌内毒素的检测方法。
4. ISO标准:ISO 11298-1《医学器械细菌内毒素的测定》是国际标准化组织(ISO)制定的关于细菌内毒素检测的国际标准。
这些标准通常涵盖了检测方法、试验条件、结果分析等方面,以确保检测结果的准确性和可靠性。
需要注意的是,不同国家和地区的标准可能存在差异,因此在进行细菌内毒素检测时,应遵循当地相关法规和标准要求。
微生物学药学专业内毒素致病菌

医学ppt
13
Vi抗原
因与毒力有关而命名为Vi抗原。
由聚-N-乙酰-D-半乳糖胺糖醛酸组成。
不稳定,经60℃加热、石碳酸处理或人工传代培养易破坏 或丢失。
新从患者标本中分离出的伤寒杆菌、丙型副伤寒杆菌等有此 抗原。
Vi抗原存在于细菌表面,可阻止O抗原与其相应抗体的反应。
医学ppt
17
肠热症(伤寒和副伤寒)
细菌到达小肠后,侵入肠壁淋巴组织,在其中大量繁殖,并进 血流, 引起第一次菌血症。病人有发热、全身不适、乏力等。
细菌随血流至骨髓、肝、脾、肾、胆囊、皮肤等并在其中繁殖,并再 次进入血流,引起第二次菌血症。此期症状明显,病人持续高热,相 对缓脉,肝脾肿大及全身中毒症状,皮肤出现玫瑰疹。血中的白细胞数量 显著下降。
内毒素致病菌
医学ppt
1
内毒素
内毒素存在于菌体内,是菌体的结构成 份。
细菌在生活状态时不释放出来,只有当 菌体自溶或用人工方法使细菌裂解后才 释放,故称内毒素。
大多数革兰氏阴性都有内毒素,如沙门 氏菌、痢疾杆菌、大肠杆菌、奈瑟氏球 菌等
医学ppt
2
内毒素的作用
内毒素对组织细胞的选择性不强,不同 革兰氏阴性细菌的内毒素,引起的病理 变和临床症状大致相同。 ①发热反应 ②糖代谢紊乱 ③血管舒缩机能紊乱 ④弥漫性血管内凝血
医学ppt
39
所致疾病
志贺氏菌引起的细菌性痢疾是最常见的 消化道传染病,夏秋季多见。
日光直接照射30分钟,56-60℃10分即被杀死, 对酸、高温和化学消毒剂很敏感,1%石炭酸 中15-30分钟即被杀死。
对氯霉素、磺胺类、链霉素敏感,但易产生耐 药性。
细菌内毒素的概念
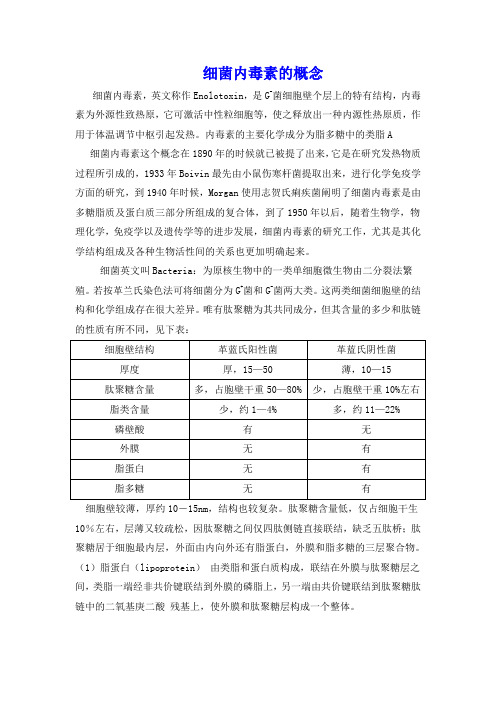
细菌内毒素的概念细菌内毒素,英文称作Enolotoxin,是G-菌细胞壁个层上的特有结构,内毒素为外源性致热原,它可激活中性粒细胞等,使之释放出一种内源性热原质,作用于体温调节中枢引起发热。
内毒素的主要化学成分为脂多糖中的类脂A细菌内毒素这个概念在1890年的时候就已被提了出来,它是在研究发热物质过程所引成的,1933年Boivin最先由小鼠伤寒杆菌提取出来,进行化学免疫学方面的研究,到1940年时候,Morgan使用志贺氏痢疾菌阐明了细菌内毒素是由多糖脂质及蛋白质三部分所组成的复合体,到了1950年以后,随着生物学,物理化学,免疫学以及遗传学等的进步发展,细菌内毒素的研究工作,尤其是其化学结构组成及各种生物活性间的关系也更加明确起来。
细菌英文叫Bacteria:为原核生物中的一类单细胞微生物由二分裂法繁殖。
若按革兰氏染色法可将细菌分为G+菌和G-菌两大类。
这两类细菌细胞壁的结构和化学组成存在很大差异。
唯有肽聚糖为其共同成分,但其含量的多少和肽链的性质有所不同,见下表:细胞壁较薄,厚约10-15nm,结构也较复杂。
肽聚糖含量低,仅占细胞干生10%左右,层薄又较疏松,因肽聚糖之间仅四肽侧链直接联结,缺乏五肽桥;肽聚糖居于细胞最内层,外面由内向外还有脂蛋白,外膜和脂多糖的三层聚合物。
(1)脂蛋白(lipoprotein)由类脂和蛋白质构成,联结在外膜与肽聚糖层之间,类脂一端经非共价键联结到外膜的磷脂上,另一端由共价键联结到肽聚糖肽链中的二氧基庚二酸残基上,使外膜和肽聚糖层构成一个整体。
(2)外膜(outer membrane)是革兰氏阴性菌细胞壁的重要结构,位于肽聚糖的外侧,其结构类似细胞膜,为液态的磷脂双层,其中镶嵌一些特异蛋白质,穿透外膜的内外双层,呈液态镶嵌体。
外膜中间有微小孔道,容许水溶性的小分子通过,以进行细胞内外的物质运输和交换。
除此之外,外膜还能防止胰蛋白酶和溶菌酶等进入,起到保护性屏障作用。
细菌内毒素名词解释

细菌内毒素名词解释细菌内毒素是指细菌产生的一类具有毒性的生物化学物质。
它们主要由蛋白质组成,具有广泛的生物活性和毒性效应。
细菌内毒素主要由细菌细胞外分泌或细胞溶解时释放到周围环境中,进而散播到宿主体内。
细菌内毒素可以分为两类:内毒素A和内毒素B。
内毒素A是脂多糖(lipopolysaccharide,LPS)的组成部分,主要存在于革兰氏阴性细菌的细胞外壳上。
内毒素B则是脂质A(lipid A),它是内毒素A分子中与糖链结合的脂肪酰基和糖胺核酸的组成部分。
内毒素B通常具有强烈的生物活性和毒性效应。
细菌内毒素的毒性主要通过结合宿主细胞受体来实现。
内毒素A结合到宿主细胞表面的Toll样受体4(TLR4)上,进而激活炎症信号通路,释放炎症介质,如肿瘤坏死因子-α(TNF-α)、白细胞介素-1(IL-1)、白细胞介素-6(IL-6)等,引发炎症反应。
而内毒素B则通过与细胞表面的CD14受体结合,通过下游信号分子的激活引起炎症反应。
细菌内毒素可以诱导多种炎症反应,如发热、血管扩张、血小板聚集、血小板凝集等。
在严重感染或败血症的情况下,内毒素的大量释放和持续存在,会导致机体的广泛炎症反应,甚至引发多器官功能障碍综合征(MODS)以及休克,危及生命。
对于细菌内毒素的控制和清除,目前主要包括以下几个方面的策略:抗炎症治疗,通过抑制炎症介质的释放和炎症信号通路的阻断来减轻内毒素引发的炎症反应;抗生素治疗,通过抗生素的使用来控制感染源细菌数量的增殖,减少内毒素的产生;内毒素清除,通过血液净化或清除内毒素的治疗药物来清除体内的细菌内毒素。
综上所述,细菌内毒素是一类具有强烈毒性和生物活性的蛋白质物质,它们的存在和释放会引起机体的炎性反应和病理变化,甚至危及生命。
因此,对于细菌内毒素的研究和控制具有重要的意义,并为治疗严重感染和败血症提供了新的思路和方法。
细菌内毒素常用检测方法

细菌内毒素常用检测方法细菌内毒素常用检测方法细菌内毒素是细菌代谢产生的有毒物质,可以对人体产生不良反应,包括炎症、细胞毒性和免疫反应。
因此,对细菌内毒素的检测非常重要。
本文将按照类别介绍几种常用的细菌内毒素检测方法。
1. 生物学方法生物学方法是通过生物实验来检测细菌内毒素的存在。
这种方法广泛应用于细菌内毒素的检测中。
常用的方法包括小鼠中毒试验、兔子毒性试验和果蝇反应试验。
这些方法基于动物对细菌内毒素的不同反应,可以初步确定细菌内毒素的存在。
2. 免疫学方法免疫学方法是利用抗体来检测细菌内毒素的存在。
这种方法是一种快速、敏感和特异的方法。
常用的免疫学方法包括酶联免疫吸附试验(ELISA)和放射免疫分析法(RIA)。
这些方法基于抗体与细菌内毒素的结合来确定细菌内毒素的存在。
3. 生化方法生化方法是利用化学试剂来检测细菌内毒素的存在。
这种方法是一种快速、高效和特异的方法。
常用的生化方法包括高效液相色谱法(HPLC)和气相色谱法(GC)。
这些方法基于细菌内毒素的化学结构和性质来确定细菌内毒素的存在。
4. 破坏性方法破坏性方法是利用物理或化学方法来破坏细菌内毒素,使其失去活性。
这种方法是一种快速、高效和安全的方法。
常用的破坏性方法包括加热处理、辐射治疗和酸碱处理。
这些方法可以将细菌内毒素破坏成无活性的物质,使其不能对人体产生危害。
5. 分子生物学方法分子生物学方法是一种新型的检测方法,利用PCR、DNA芯片等技术来检测细菌内毒素的存在。
这种方法具有高灵敏度、高特异性和快速的优点。
常用的分子生物学方法包括PCR法和DNA芯片技术。
这些方法可以将细菌内毒素的基因级别分析,确定其存在。
综上所述,细菌内毒素的检测方法有很多种。
我们可以根据实际需要和需求选择合适的检测方法。
同时,我们也需要不断地研究和发展新型的检测方法,以提高检测的灵敏度和准确度,从而更好地保障人体健康。
细菌内毒素检测方法

细菌内毒素检测方法细菌内毒素是一种由细菌产生的毒素,它们可以导致各种疾病和感染。
为了检测细菌内毒素的存在,有几种常用的方法。
1. 酶联免疫吸附试验(ELISA)酶联免疫吸附试验是一种常用的检测细菌内毒素的方法。
它基于抗体与特定细菌内毒素结合的原理。
首先,在试验板上固定特异性的抗体,然后加入样本溶液。
如果样本中含有目标细菌内毒素,它们将与固定的抗体结合。
接下来,通过冲洗去除未结合的物质,并加入与抗体结合的酶标记二抗。
最后,染色物质被加入,形成颜色反应,测量其光密度即可检测细菌内毒素的存在。
2. 质谱分析质谱分析是一种高分辨率的检测细菌内毒素的方法。
它使用质谱仪来分析样品中的化学成分。
首先,样品通过质谱中的电离源产生离子。
然后,质谱仪以不同的方式对离子进行分离和检测,以确定其质量和相对丰度。
通过与已知细菌内毒素的质谱图进行比对,可以确定样品中是否存在目标内毒素。
3. 生物感应器生物感应器是一种基于生物反应的检测细菌内毒素的方法。
它使用已知对细菌内毒素具有特异性反应的生物分子,如细胞、抗体或酶等。
当目标内毒素存在时,它们与生物分子结合,触发特定的生物反应。
通过测量或监测这些反应的信号变化,可以确定细菌内毒素的存在。
4. 培养试验培养试验是一种传统的检测细菌内毒素的方法。
它通过将样品接种到特定培养基上,培育细菌并观察是否产生内毒素。
一般来说,细菌内毒素的产生需要一定的时间,而且对培养条件和培养基的选择也有一定的要求。
因此,这种方法可能需要更长的时间和更复杂的实验条件。
上述方法各有优缺点,选择适合的方法需要根据具体情况和需求进行。
综合考虑灵敏度、准确性、快速性和成本等因素,可以选择最适合的方法来检测细菌内毒素的存在。
细菌内毒素介绍

细菌内毒素介绍细菌内毒素你了解多少?1.热点引导细菌内毒素,英文称作Endotoxin,是G-菌细胞壁个层上的特有结构,内毒素为外源性致热原,它可激活中性粒细胞等,使之释放出一种内源性热原质,作用于体温调节中枢引起发热,细菌内毒素的主要化学成分为脂多糖。
革兰氏阴性菌感染的危害性主要源自其释放的内毒素。
动物传染病多种细菌皆可产生内毒素,如大肠杆菌、沙门氏菌、巴氏杆菌、胸膜肺炎放线杆菌等,它们通过外源性感染和肠道系统内源性转位两种途径进入血液循环导致内毒素血症,是机体致病的重要毒力因子。
很多严重疾病(如损伤、烧伤、心、肾、消化系统、呼吸系统、肿瘤、自动免疫等方面的疾病)都直接或间接与革兰阴性菌感染及其释放的内毒素有关。
内毒素血症、休克和弥漫性血管内凝血等,LPS进入机体后可先后诱导细胞因子和黏附分子等炎性介质产生和释放,引起内皮细胞的损伤和屏障功能的改变,导致全身性炎症反应综合征和脓毒症,严重者可导致低血压、中毒性休克、弥漫性血管内凝血(DIC)、急性呼吸窘迫综合征(ARDS)、多器官功能衰竭(MOF),甚至死亡。
白细胞数目改变,LPS初期可使血液中白细胞数目急剧下降,数小时后骨髓粒细胞又大量释放入血引发白细胞增多症。
该过程会导致动物免疫系统紊乱,增加病原体感染几率。
此外,内毒素还能导致怀孕动物流产、早产或死胎;抑制红细胞生成;具有致敏和抗敏作用等。
2.保健误区生产实践中许多人认为细菌病可用抗菌素治疗,只要通过药敏试验筛选出敏感药物,即可产生理想疗效,其实并非如此。
因为多种因素会导致药敏结果与实际效果不一致,如药物剂量、用法、血清型等。
细菌内毒素在人医上研究较多,但在兽医临床很少有人去考虑抗生素诱导细菌释放内毒素,以及某些革兰氏阴性菌会产生内毒素的问题,以致治疗效果差。
虽然现在防治内毒素损伤的方法和途径多种多样,如LPS抗体、细菌通透性增加蛋白、细胞因子拮抗剂、抗内毒素蛋白等,但尚无一种非常有效的手段。
细菌内毒素检测试验方法

细菌内毒素检测试验方法英文回答:Bacterial Endotoxin Testing Methods.Bacterial endotoxins are pyrogenic substances that can cause fever, chills, and other symptoms in humans. They are produced by gram-negative bacteria and can be found in a variety of products, including medical devices, pharmaceuticals, and food.There are a number of different methods for testing for bacterial endotoxins. The most common method is the Limulus amebocyte lysate (LAL) test. The LAL test is based on the fact that the amebocytes of the horseshoe crab (Limulus polyphemus) are sensitive to endotoxins. When endotoxins are present, the amebocytes release a clotting factor that can be detected by a spectrophotometer.Other methods for testing for bacterial endotoxinsinclude:The rabbit pyrogen test: This test is based on thefact that rabbits are sensitive to endotoxins. When endotoxins are injected into a rabbit, they can cause a fever. The rabbit pyrogen test is less sensitive than the LAL test, but it is more specific.The in vitro cytokine assay: This test measures the release of cytokines by human cells that have been exposed to endotoxins. Cytokines are proteins that are involved in the inflammatory response. The in vitro cytokine assay is more sensitive than the LAL test, but it is less specific.The choice of which method to use for testing for bacterial endotoxins depends on the specific application. The LAL test is the most commonly used method because it is simple, rapid, and inexpensive. The rabbit pyrogen test is more specific than the LAL test, but it is more expensive and time-consuming. The in vitro cytokine assay is the most sensitive method, but it is also the most expensive and time-consuming.中文回答:细菌内毒素检测试验方法。
细菌内毒素计算公式

细菌内毒素计算公式
摘要:
1.细菌内毒素的概念和含义
2.细菌内毒素的来源和生成
3.细菌内毒素的检测方法和计算公式
4.细菌内毒素的危害和应用
5.细菌内毒素的防治措施
正文:
一、细菌内毒素的概念和含义
细菌内毒素(Endotoxin)是一种由细菌产生的外源性致热原,主要存在于G-菌细胞壁的脂多糖成分中。
当细菌死亡解体后,脂多糖成分会被释放,进一步分解为内毒素。
内毒素可以激活中性粒细胞等免疫细胞,导致机体释放出一系列细胞因子,如肿瘤坏死因子、白细胞介素(IL-1)、IL-6、IL-8、前列腺素、凝血素、干扰素、血小板激活因子等。
二、细菌内毒素的来源和生成
细菌内毒素主要来源于G-菌,如大肠杆菌、铜绿假单胞菌等。
这些细菌在生长过程中,其细胞壁中的脂多糖成分会不断生成和释放。
当细菌死亡或受到外界压力时,脂多糖成分会被分解,释放出内毒素。
三、细菌内毒素的检测方法和计算公式
目前,检测细菌内毒素的方法主要有鲎试剂法、凝胶法、比浊法等。
其中,鲎试剂法是最常用的一种方法。
鲎试剂法的计算公式如下:
内毒素含量(EU/ml)=(C×V×1000)/(S×M)
其中,C 为鲎试剂的浓度(IU/ml),V 为供试品的体积(ml),S 为鲎试剂的灵敏度(EU/ml),M 为供试品的质量(g)。
四、细菌内毒素的危害和应用
细菌内毒素在体内过量积累时,会导致发热、低血压、心动过速、休克、多器官功能衰竭甚至死亡。
然而,在适量范围内,细菌内毒素可以激活免疫系统,对机体产生有益作用。
因此,细菌内毒素在医学和科研领域具有广泛的应用。
细菌内毒素检查操作规程

细菌内毒素检查操作规程细菌内毒素检查操作规程一、实验目的细菌内毒素检查是为了检测样本中是否存在细菌产生的内毒素,可以作为细菌感染的诊断依据和治疗效果评估的指标。
二、实验材料和设备1. 细菌培养基和培养物2. 内毒素检测试剂盒3. 显微镜和载玻片4. 离心机5. 温箱和恒温水浴6. 试管、培养皿、移液器等常规实验用品三、实验步骤1. 培养细菌:将待检测的细菌接种到适当的培养基上,放入温箱中以适当的温度和时间进行培养,使细菌生长至对应的生长期。
2. 收集细菌:将培养好的细菌转移到离心管中,使用离心机将细菌离心沉淀。
3. 细菌裂解:将细菌沉淀加入合适的裂解缓冲液中,使用适当的方法将细菌完全裂解。
4. 澄清液分离:使用离心机将细菌裂解液离心,将上清液转移到新的试管中。
5. 内毒素检测:按照内毒素检测试剂盒说明书的要求,将所得上清液加入试剂盒中,进行内毒素的检测。
6. 结果解读:根据试剂盒的指示,观察试剂盒中的颜色变化或读取光密度等指标,判断样品中是否存在细菌内毒素。
7. 结果验证:使用正负对照样品验证结果的准确性和可靠性。
四、实验注意事项1. 实验过程需要遵守无菌操作的要求,避免细菌污染。
2. 实验中使用的试剂和培养基要在有效期内,并按照要求保存。
3. 实验过程中应注意个人防护,避免接触到有害物质。
4. 操作前要充分了解试剂盒的使用方法和读取结果的标准。
5. 结果解读时要考虑到阳性和阴性对照的结果,以免误判。
6. 使用的设备要事先进行验证和校准,以保证实验结果的准确性和可靠性。
五、实验结果记录和分析1. 记录实验所使用的试剂批号和有效期。
2. 记录实验过程中的注意事项和操作细节。
3. 记录实验结果,包括阳性和阴性对照的结果。
4. 对实验结果进行分析和解释,判断样品中是否存在细菌内毒素。
六、实验结果的报告和存档1. 编写实验报告,包括实验目的、材料和方法、结果分析、结论等内容。
2. 报告中要附带实验记录、数据分析和结果验证等材料。
内毒素

科技名词定义中文名称:内毒素英文名称:endotoxin其他名称:细菌内毒素定义1:只在细菌被破坏时才被释放的细菌毒素。
某些革兰阴性菌(如布鲁菌、肠杆菌、奈瑟菌、弧菌等)外膜相关的热稳定毒素,主要成分是脂多糖,在固有免疫及B细胞的多克隆激活中发挥作用。
应用学科:免疫学(一级学科);概论(二级学科);固有免疫(三级学科)定义2:革兰氏阴性菌胞壁成分中的脂多糖,由脂质A、核心寡糖和1条具有重复结构、长度可变的O-抗原糖链三部分组成。
应用学科:生物化学与分子生物学(一级学科);氨基酸、多肽与蛋白质(二级学科)定义3:由革兰氏阴性菌所合成的一种存在于细菌细胞壁外层、只有在细菌死亡和裂解后才释出的有毒物质。
应用学科:水产学(一级学科);水产生物病害及防治(二级学科)内毒素是革兰氏阴性细菌细胞壁中的一种成分,叫做脂多糖。
脂多糖对宿主是有毒性的。
内毒素只有当细菌死亡溶解或用人工方法破坏菌细胞后才释放出来,所以叫做内毒素。
其毒性成分主要为类脂质A。
内毒素位于细胞壁的最外层、覆盖于细胞壁的黏肽上。
各种细菌的内毒素的毒性作用较弱,大致相同,可引起发热、微循环障碍、内毒素休克及播散性血管内凝血等。
内毒素耐热而稳定,抗原性弱。
革兰氏阴性菌的菌体中存在的毒性物质的总称。
是多种革兰氏阴性菌的细胞壁热、微循环障碍、内毒素休克及播散性血管内凝血等。
内毒素耐热而稳定,抗原性弱。
可刺激机体产生抗体,但无中和作用,形成抗毒素,经甲醛处理不能成为类毒素。
内毒素是革兰氏阴性细菌细胞壁中的一种成分,叫做脂多糖。
脂多糖对宿主是有毒性的。
内毒素只有当细菌死亡溶解或用人工方法破坏菌细胞后才释放出来,所以叫做内毒素。
内毒素不是蛋白质,因此非常耐热。
在100℃的高温下加热1小时也不会被破坏,只有在160℃的温度下加热2到4个小时,或用强碱、强酸或强氧化剂加温煮沸30分钟才能破坏它的生物活性。
与外毒素不同之处在于:内毒素不能被稀甲醛溶液脱去毒性成为类毒素;把内毒素注射到机体内虽可产生一定量的特异免疫产物(称为抗体),但这种抗体抵消内毒素毒性的作用微弱。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia. Those portions that are not harmonized are marked with symbols () to specify this fact.这一章节已经和欧洲药典(EP)与日本药典(JP)统一。
没有统一的部分已经用标记出来。
The Bacterial Endotoxins Test (BET) is a test to detect or quantify endotoxins from Gram-negative bacteria using amoebocyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus).细菌内毒素检查法(BET)是通过鲎的阿米巴细胞溶解产物来检测或定量来自革兰氏阴性菌的内毒素。
There are three techniques for this test: the gel-clot technique, which is based on gel formation; the turbidimetric technique, based on the development of turbidity after cleavage of an endogenous substrate; and the chromogenic technique, based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any of the three techniques for the test. In the event of doubt or dispute, the final decision is made based upon the gel-clot technique unless otherwise indicated in the monograph for the product being tested. The test is carried out in a manner that avoids endotoxin contamination.细菌内毒素检查法有3种:凝胶法,基于凝胶的形成;浊度法,BACTERIAL ENDOTOXINS TEST USP32Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopeia and/or the Japanese Pharmacopeia. Those portions that are not harmonized are marked with symbols () to specify this fact.This chapter provides a test to detect or quantify bacterial endotoxins that may be present in or on the sample of the article(s) to which the test is applied. It uses Limulus Amebocyte Lysate (LAL) obtained from the aqueous extracts of circulating amebocytes of horseshoe crab (Limulus polyphemus or Tachypleus tridentatus) which has been prepared and characterized for use as an LAL Reagent.1There are two types of techniques for this test: the gel-clot techniques, which are based on gel formation, and the photometric techniques. The latter include a turbidimetric method, which is based on the development of turbidity after cleavage of an endogenous substrate, and a chromogenic method, which is based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any one of these techniques, unless otherwise indicated in the monograph. In case of dispute, the final decision is based on the gel-clot techniques, unless otherwise indicated in the monograph.In the gel-clot techniques, the reaction endpoint is determined from dilutions of the material under test in direct comparison with parallel dilutions of a reference endotoxin, and quantities of endotoxin are expressed in USP Endotoxin Units (USP-EU). [NOTE—One USP-EU is equal to one IU of endotoxin.]Because LAL Reagents have been formulated to be used also for turbidimetric or colorimetric tests, such tests may be used to comply with the requirements. These tests require the establishment of a standard regression curve; the endotoxin content of the test material is determined by interpolation from the curve. The procedures include incubation for a preselected time of reacting endotoxin and control solutions with LAL Reagent and reading of the spectrophotometric light absorbance at suitable wavelengths. In the endpoint turbidimetric procedure the reading is made immediately at the end of the incubation period. In the endpoint colorimetric procedure the reaction is arrested at the end of the preselected time by the addition of an enzyme reaction-terminating agent prior to the readings. In the turbidimetric and colorimetric kinetic assays the absorbance is measured throughout the reaction period and rate values are determined from those readings.APPARATUS AND GLASSWAREDepyrogenate all glassware and other heat-stable materials in a hot-air oven using a validated process.2Commonly used minimum time and temperature settings are 30 minutes at 250. If employing plastic apparatus, such as microplates and pipet tips for automatic pipetters, use only that which has been shown to be free of detectable endotoxin and not to interfere with the test. [NOTE—In this chapter, the term “tube” includes any other receptacle such as a micro-titer well.]PREPARATION OF THE STANDARD ENDOTOXIN STOCK SOLUTION AND STANDARD SOLUTIONSThe USP Endotoxin RS has a defined potency of 10,000 USP Endotoxin Units (EU) per vial. Constitute the entire contents of 1 vial of the RSE with 5 mL of LAL Reagent Water3, mix intermittently for 30 minutes, using a vortex mixer, and use this concentrate for making appropriate serial dilutions. Preserve the concentrate in a refrigerator for making subsequent dilutions for not more than 14 days. Mix vigorously, using a vortex mixer, for not less than 3 minutes before use. Mix each dilution for not less than 30 seconds before proceeding to make the next dilution. Do not store dilutions, because of loss of activity by adsorption, in the absence of supporting data to the contrary.Preparatory TestingUse an LAL Reagent of confirmed label sensitivity.The validity of test results for bacterial endotoxins requires an adequate demonstration that specimens of the article or of solutions, washings, or extracts thereof to which the test is to be applied do not of themselves inhibit or enhance the reaction or otherwise interfere with the test. Validation is accomplished by performing the inhibition or enhancement test described under each of the three techniques indicated. Appropriate negative controls are included. Validation must be repeated if the LAL Reagent source or the method of manufacture or formulation of the article is changed.Preparation of Sample SolutionsPrepare sample solutions by dissolving or diluting drugs or extracting medical devices using LAL Reagent Water. Some substances or preparations may be more appropriately dissolved, diluted, or extracted in other aqueous solutions. If necessary, adjust the pH of the solution (or dilution thereof) to be examined so that the pH of the mixture of the LAL Reagent and sample falls within the pH range specified by the LAL Reagent manufacturer. This usually applies to a product with a pH in the range of to . The pH may be adjusted using an acid, base, or suitable buffer as recommended by the LAL Reagent manufacturer. Acids and bases may be prepared from concentrates or solids with LAL Reagent Water in containers free of detectable endotoxin. Buffers must be validated to be free of detectable endotoxin and interfering factors.DETERMINATION OF MAXIMUM VALID DILUTION (MVD)The Maximum Valid Dilution is the maximum allowable dilution of a specimen at which the endotoxin limit can be determined. It applies to injections or to solutions for parenteral administration in the form constituted or diluted for administration, or, where applicable, to the amount of drug by weight if the volume of the dosage form for administration could be varied. The general equation to determine MVD is:MVD = (Endotoxin limit × Concentration of sample solution)/()where the concentration of sample solution and are as defined below. Where the endotoxin limit concentration is specified in the individual monograph in terms of volume (in EU per mL), divide the limit by , which is the labeled sensitivity (in EU per mL) of the LAL Reagent, to obtain the MVD factor. Where the endotoxin limit concentration is specified in the individual monograph in terms of weight or Units of active drug (in EU per mg or in EU per Unit), multiply the limit by the concentration (in mg per mL or in Units per mL) of the drug in the solution tested or of the drug constituted according to the label instructions, whichever is applicable, and divide the product of the multiplication by , to obtain the MVD factor. The MVD factor so obtained is the limit dilution factor for the preparation for the test to be valid.ESTABLISHMENT OF ENDOTOXIN LIMITSThe endotoxin limit for parenteral drugs, defined on the basis of dose, is equal to K/M,4where K is the threshold human pyrogenic dose of endotoxin per kg of body weight, and M is equal to the maximum recommended human dose of product per kg of body weight in a single hour period.The endotoxin limit for parenteral drugs is specified in individual monographs in units such as EU/mL, EU/mg, or EU/Unit of biological activity.GEL-CLOT TECHNIQUESThe gel-clot techniques detect or quantify endotoxins based on clotting of the LAL Reagent in the presence of endotoxin. The concentration of endotoxin required to cause the lysate to clot under standard conditions is the labeled sensitivity of the LAL Reagent. To ensure both the precision and validity of the test, tests for confirming the labeled LAL Reagent sensitivity and for interfering factors are described under Preparatory Testing for the Gel-Clot Techniques.Preparatory Testing for the Gel-Clot TechniquesTest for Confirmation of Labeled LAL Reagent Sensitivity—Confirm the labeled sensitivity using at least 1 vial of the LAL Reagent lot. Prepare a series of two-fold dilutions of the USP Endotoxin RS in LAL Reagent Water to give concentrations of 2, , , and , where is as defined above. Perform the test on the four standard concentrations in quadruplicate and include negative controls. The test for confirmation of lysate sensitivity is to be carried out when a new batch of LAL Reagent is used or when there is any change in the experimental conditions that may affect the outcome of the test.Mix a volume of the LAL Reagent with an equal volume (such as aliquots) of one of the standard solutions in each test tube. When single test vials or ampuls containing lyophilized LAL Reagent are used, add solutions directly to the vial or ampul.Incubate the reaction mixture for a constant period according to directions of the LAL Reagent manufacturer (usually at 37 ± 1for 60 ± 2 minutes), avoiding vibration. To test the integrity of the gel, take each tube in turn directly from the incubator and invert it through about 180 in one smooth motion. If a firm gel has formed that remains in place upon inversion, record the result as positive. A result is negative if an intact gel is not formed. The test is not valid unless the lowest concentration of the standard solutions shows a negative result in all replicate tests.The endpoint is the last positive test in the series of decreasing concentrations of endotoxin. Calculate the mean value of the logarithms of the endpoint concentration and then the antilogarithm of the mean value using the following equation:Geometric Mean Endpoint Concentration = antilog (S e / f)where S e is the sum of the log endpoint concentrations of the dilution series used, and f is the number of replicate test tubes. The geometric mean endpoint concentration is the measured sensitivity of the LAL Reagent (in EU/mL). If this is not less than and not more than 2, the labeled sensitivity is confirmed and is used in tests performed with this lysate.Interfering Factors Test for the Gel-Clot Techniques—Prepare solutions A, B, C, and D as shown in Table 1, and perform the inhibition/enhancement test on the sample solutions at a dilution less than the MVD, not containing any detectable endotoxins, following the procedure in the Test for Confirmation of Labeled LAL Reagent Sensitivity above. The geometric mean endpoint concentrations of solutions B and C are determined using the equation in that test.Table 1. Preparation of Solutions for the Inhibition/Enhancement Test for Gel-ClotTechniquesThis test must be repeated when any condition that is likely to influence the test results changes. The test is not valid unless Solutions A and D show no reaction and the result of Solution C confirms the labeled sensitivity.If the sensitivity of the lysate determined in the presence of the sample solution under test of Solution B is not less than and not greater than 2, the sample solution does not contain factors which interfere under the experimental conditions used. Otherwise, the sample solution to be examined interferes with the test.If the sample under test does not comply with the test at a dilution less than the MVD, repeat the test using a greater dilution, not exceeding the MVD. The use of a more sensitive lysate permits a greater dilution of the sample to be examined and this may contribute to the elimination of interference.Interference may be overcome by suitable treatment, such as filtration, neutralization, dialysis, or heating. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assaydescribed below using the preparation to be examined to which USP Endotoxin RS has been added and which has been subjected to the selected treatment.Gel-Clot Limit TestThis test is used when a monograph contains a requirement for endotoxin limits.Procedure—Prepare Solutions A, B, C, and D as shown in Table 2, and perform the test on these solutions following the procedure in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques.Table 2. Preparation of Solutions for the Gel-Clot Limit TestInterpretation—The test is not valid unless both replicates of positive control Solutions B and C are positive and those of negative control Solution D are negative. The preparation under test complies with the test when a negative result is found for both tubes containing Solution A. The preparation under test does not comply with the test when a positive result is found for both tubes containing Solution A.Repeat the test when a positive result is found for 1 tube containing Solution A and a negative result for the other one. The preparation under test complies with the test when a negative result is found for both tubes containing Solution A in the repeat result. If the test is positive for the preparation under test at a dilution less than the MVD, the test may be repeated at a dilution not greater than the MVD.Gel-Clot AssayThis assay quantifies bacterial endotoxins in sample solutions by titration to an endpoint.Procedure—Prepare Solutions A, B, C, and D as shown in Table 3, and test these solutions by following the procedure in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques.Table 3. Preparation of Solutions for the Gel-Clot AssayCalculation and Interpretation—The test is not valid unless the following conditions are met: (1) both replicates of negative control Solution D are negative;(2) both replicates of positive product control Solution B are positive; and (3) the geometric mean endpoint concentration of Solution C is in the range of to 2.To determine the endotoxin concentration of Solution A, calculate the endpoint concentration for each replicate series of dilutions by multiplying each endpoint dilution factor by . The endotoxin concentration in the sample is the geometric mean endpoint concentration of the replicates (see the formula given in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques). If the test is conducted with a diluted sample solution, calculate the concentration of endotoxin in the original sample solution by multiplying by the dilution factor. If none of the dilutions of the sample solution is positive in a valid assay, report the endotoxin concentration as less than (if the diluted sample was tested, less than times the lowest dilution factor of the sample.) If all dilutions are positive, the endotoxin concentration is reported as equal to or greater than the greatest dilution factor multiplied by ., initial dilution factor times 8 times in Table 3).The article meets the requirements of the test if the concentration of endotoxin is less than that specified in the individual monograph.PHOTOMETRIC TECHNIQUESThe turbidimetric method measures increases in turbidity. Depending on the test principle used, this technique is classified as either endpoint-turbidimetric or kinetic-turbidimetric. The endpoint-turbidimetric technique is based on the quantitative relationship between the concentration of endotoxins and the turbidity(absorbance or transmission) of the reaction mixture at the end of an incubation period. The kinetic-turbidimetric technique is a method to measure either the onset time needed to reach a predetermined absorbance of the reaction mixture or the rate of turbidity development.The chromogenic method measures the chromophore released from a suitable chromogenic peptide by the reaction of endotoxins with the LAL Reagent. Depending on the test principle employed, this technique is classified as either endpoint-chromogenic or kinetic-chromogenic. The endpoint-chromogenic technique is based on the quantitative relationship between the concentration of endotoxins and the release of chromophore at the end of an incubation period. The kinetic-chromogenic technique is a method to measure either the onset time needed to reach a predetermined absorbance of the reaction mixture or the rate of color development.All photometric tests are carried out at the incubation temperature recommended by the LAL Reagent manufacturer, which is usually 37 ± 1.Preparatory Testing for the Photometric TechniquesTo assure the precision or validity of the turbidimetric and chromogenic techniques, preparatory tests are conducted to verify that the criteria for the standard curve are valid and that the sample solution does not inhibit or enhance the reaction. Revalidation for the test method is required when conditions that are likely to influence the test result change.Verification of Criteria for the Standard Curve—Using the Standard Endotoxin Solution, prepare at least three endotoxin concentrations to generate the standard curve. Perform the test using at least three replicates of each standard endotoxin concentration according to the manufacturer's instructions for the LAL Reagent (with regard to volume ratios, incubation time, temperature, pH, etc.). If the desired range in the kinetic methods is greater than two logs, additional standards should be included to bracket each log increase within the range of the standard curve. The absolute value of the correlation coefficient, |r|, must be greater than or equal to for the range of endotoxin concentrations indicated by the manufacturer of the LAL Reagent.Interfering Factors Test for the Photometric Techniques—Select an endotoxin concentration at or near the middle of the endotoxin standard curve. Prepare Solutions A, B, C, and D as shown in Table 4. Perform the test on Solutions A, B, C, and D at least in duplicate following the instructions for the LAL Reagent used (with regard to volume of sample and LAL Reagent, volume ratio of sample to LAL Reagent, incubation time, etc.).Table 4. Preparation of Solutions for the Inhibition/Enhancement Test forPhotometric TechniquesCalculate the mean recovery of the added endotoxin by subtracting the mean endotoxin concentration in the solution (if any) from that containing the added endotoxin. In order to be considered free of interfering factors under the conditions of the test, the measured concentration of the endotoxin added to the sample solution must be within 50% to 200% of the known added endotoxin concentration after subtraction of any endotoxin detected in the solution without added endotoxin.When the endotoxin recovery is out of the specified ranges, the interfering factors must be removed as described in the Interfering Factors Test for the Gel-Clot Techniques under Preparatory Testing for the Gel-Clot Techniques.Repeating the Interfering Factors Test for the Gel-Clot Techniques validates the treatment.Procedure for the Photometric TechniquesFollow the procedure described in the Interfering Factors Test for the Photometric Techniques under Preparatory Testing for the Photometric Techniques.Calculation for the Photometric TechniquesCalculate the endotoxin concentration of each of the replicates of test Solution A using the standard curve generated by positive control series C. The test is not valid unless the following conditions are met: (1) the results of control series C comply with the requirements for validation defined under Verification of Criteria for the Standard Curve under Preparatory Testing for the Photometric Techniques;(2) the endotoxin recovery, calculated from the concentration found in Solution B after subtracting the endotoxin concentration found in Solution A is within 50 to 200%; and (3) the result of negative control series D does not exceed the limit of the blank value required in the description of the LAL Reagent used.Interpretation of Results from the Photometric TechniquesIn photometric assays, the preparation under test complies with the test if the mean endotoxin concentration of the replicates of Solution A, after correction for dilution and concentration, is less than the endotoxin limit for the product.1 LAL Reagent reacts with some -glucans in addition to endotoxins. Some preparations that are treated will not react with -glucans and must be used for samples that contain glucans.2 For a validity test of the procedure for inactivating endotoxins, see Dry-Heat Sterilization under Sterilization and Sterility Assurance of Compendial Articles 1211. Use an LAL Reagent having a sensitivity of not less than Endotoxin Unit per mL.3Sterile Water for Injection or other water that shows no reaction with the specific LAL Reagent with which it is to be used, at the limit of sensitivity of such reagent.4K is 5 USP-EU/kg for any route of administration other than intrathecal (for which K is USP-EU/kg body weight). For radiopharmaceutical products not administered intrathecally the endotoxin limit is calculated as 175/V, where V is the maximum recommended dose in mL. For intrathecally administered radiopharmaceuticals, the endotoxin limit is obtained by the formula 14/V. For formulations (usually anticancer products) administered on a per square meter of body surface, the formula is K/M, where K= 5 EU/kg and M is the (maximum dose/m2/hour × m2)/70 Kg.。