二代测序数据分析简介
二代测序数据分析简介

• The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used
Quality
Encoding
• Sanger format can encode a Phred quality score from 0 to 93 using ASCII 33 to 126 • Illumina's newest version (1.8) of their pipeline CASAVA will directly produce fastq in Sanger format • Solexa/Illumina 1.0 format can encode a Solexa/Illumina quality score from -5 to 62 using ASCII 59 to 126 • Starting with Illumina 1.3 and before Illumina 1.8, the format encoded a Phred quality score from 0 to 62 using ASCII 64 to 126 • Starting in Illumina 1.5 and before Illumina 1.8, the Phred scores 0 to 2 have a slightly different meaning
二代测序数据分析简介
童春发 2013.12.23
主要内容
• • • • 重测序的原理及流程 数据结构与质量评估 SRA数据库及数据获取 Bowtie2、BWA和SAMtools软件使用
重测序的原理及流程
二代测序技术简介

二代测序技术简介一、什么是二代测序技术?二代测序技术,也被称为高通量测序技术,是一种快速、高效的DNA 或RNA序列测定方法。
相比传统的Sanger测序技术,二代测序技术具有较高的测序效率和容量,能够同时测序数百万到数十亿个碱基对,大大提高了测序的速度和数据产量。
常用的二代测序技术包括Illumina 测序技术、Ion Torrent PGM 测序技术等。
二、Illumina二代测序技术的原理与过程1. 原理Illumina二代测序技术基于桥式扩增和碱基扩增的原理。
DNA样本经过打断、连接和PCR扩增等处理后,将单链DNA固定于特定表面上,并在每个DNA分子之间形成成千上万个桥式扩增复合物。
在模板DNA的存在下,通过逐个反复封闭、复制和荧光标记的方式,进行碱基的逐渐扩增,并利用荧光信号记录测序结果。
2. 过程(1)样本制备:包括DNA或RNA的提取、打断、连接和PCR扩增等步骤,以获得特定长度的DNA片段。
(2)文库构建:将DNA片段连接到Illumina测序芯片上的适配器上,并进行PCR扩增,形成DNA桥式扩增复合物。
(3)测序芯片加载:将DNA桥式扩增复合物置于测序芯片上,使得每个DNA分子都与芯片上的特定区域相结合。
(4)桥式扩增:通过逐个反复封闭、复制和荧光标记的方式进行碱基的逐步扩增,形成簇团。
(5)图像获取:利用高分辨率成像系统拍摄簇团的荧光信号。
(6)数据分析:将图像数据转化为碱基序列,通过比对和组装等算法,得到原始测序数据。
三、Illumina二代测序技术的优势和应用领域1. 优势(1)高通量:能够在较短时间内产生大规模的测序数据。
(2)高准确性:其错误率低于其他二代测序技术,能够提供高质量的测序结果。
(3)可扩展性:适用于不同规模的测序项目,从几个目标区域到整个基因组的测序,具有较高的灵活性。
(4)低成本:相对于传统的Sanger测序技术,具有更低的测序成本。
2. 应用领域(1)基因组学研究:能够对物种的基因组进行全面测序和变异分析,有助于揭示基因组结构和功能。
第二代基因测序技术的原理和应用

第二代基因测序技术的原理和应用引言随着科技的不断发展,人类对基因的研究和探究也越来越深入。
在过去,我们只能使用第一代基因测序技术来了解人体基因的构成和作用,但是随着第二代基因测序技术的出现,为基因领域的研究和应用打开了更加广阔的空间。
在本文中,我们将深入探讨第二代基因测序技术的原理和应用。
第二代基因测序技术的原理第二代基因测序技术是一种基于光学或化学原理的高通量测序技术。
与第一代基因测序技术使用的是Sanger测序方法不同,第二代基因测序技术可以通过平行处理多个DNA分子的测序来提高测序效率和吞吐量,并且在测序速度和准确性方面也有了极大的提升。
第二代基因测序技术的基本原理是将DNA分子切成短片段后,使用测序仪器在一张玻片上进行并行测序。
测序过程中,每个DNA片段都会被放置在玻片的一个位置上,然后通过连续的循环反应进行测序,最后获得DNA序列信息。
这种并行测序的方法不仅大大减少了测序所需的时间和成本,同时还可以提高测序的准确性和稳定性,为后续基因分析和研究提供了更加丰富和有力的原始数据和支持。
第二代基因测序技术的应用第二代基因测序技术的广泛应用使得人类对基因的研究和应用有了更大的发展空间。
下面我们将详细介绍第二代基因测序技术的几个主要应用领域。
1. 基因组测序第二代基因测序技术可以用于全基因组测序和基因组重测序,对于人体基因的筛查、疾病基因定位以及复杂性疾病的研究等都有着重要的应用价值。
例如,通过基因组测序技术,我们可以了解个体基因的构成、基因综合表达、突变信息等数据信息,为基因治疗和疾病预防等提供更为准确和精细的依据和重要的研究基础。
2. 表观基因组测序表观遗传学是一门研究基因组DNA外部化学修饰和红茶结构发生变化的学科,是研究个体遗传信息与环境互动的核心内容。
第二代基因测序技术在表观遗传学领域的应用主要涉及到DNA甲基化的分析和ChIP测序,这些技术可以帮助我们了解个体表观遗传学调控,深入研究个体生长、发育和疾病等方面的关键因素和机制。
quality score 二代测序

quality score 二代测序
Quality Score(质量评分)是二代测序(Next Generation Sequencing,NGS)中用于评估测序数据质量的重要指标。
在NGS中,测序仪会生成大量的测序读段(reads),每个读段包含一定长度的DNA序列信息。
然而,这些读段中可能存在一些错误,这些错误可能是由于测序过程中引入的随机误差、序列特异性或背景噪声等因素导致的。
为了评估这些错误,研究人员引入了质量评分系统。
质量评分是对每个测序碱基(base)的可靠性进行评估的数值,通常使用Phred质评分(Phred quality score)来表示。
Phred质评分是一种对测序数据质量进行量化评估的算法,它基于测序碱基的错误概率来计算质量评分。
在NGS中,测序数据的质量评分对于后续的生物信息学分析非常重要。
高质量的测序数据可以提供更准确、更可靠的基因组、转录组和其他生物信息学分析结果。
因此,在进行NGS数据分析之前,通常需要对测序数据进行质量评估和质量控制,以确保后续分析的准确性和可靠性。
在NGS中,质量评分通常以ASCII码的形式表示,范围从0到93。
其中,0表示最差的质量评分,93表示最好的质量评分。
一般来说,质量评分越高,表示该碱基的测序结果越可靠。
在进行数据分析时,通常会根据不同的阈值对测序数据进行过滤和处理,以确保后续分析的准确性和可靠性。
总之,quality score是二代测序中用于评估测序数据质量的重要指标,对于后续的生物信息学分析非常重要。
二代测序简介

Image Acquisition and Processing
Fluorescence Chemiluminescence
a
Sequential Sequenc e Extensio n Reaction
Polymerase
Ligase 12
2nd Generation Performance
1995 - ABI’s first automated DNA sequencer
2006 - 2nd generation DNA sequencer on market
2007 and beyond – Singleamolecule sequencing
4
Sanger’s
M eLatbheloeddprimer (1)
POLONATOR 连接酶 20G 17 × ~30×2 7天 1周 99% ~40% off
a
13
Roche/454 Life Sciences’ Genome Sequencer
a
14
Roche/454 Workflow
DNA Library Prep
DNA Fragment
End repaired
Enzyme beads
Sulfurylase Luciferase
Packing beads help toa
Pyrosequencing
4 nucleotides sequentially flow in
Incorporation of a nucleotide releases a pyrophosphate (PPi)
GS 454
技术原理 聚合酶/焦磷酸酶
单运数据产量 0.4~0.6Gb
第二代测序数据分析原理

第二代测序数据分析原理第二代测序技术是近年来迅速发展起来的高通量测序技术,能够产生大量的DNA序列数据。
与第一代测序技术相比,第二代测序技术具有更高的产量、更快的速度和更低的成本,成为当前基因组学研究和医学诊断的重要工具之一第二代测序数据分析原理是指对产生的高通量测序数据进行处理和解读的过程。
该过程涉及到数据的质控、序列比对、变异检测和功能注释等多个步骤,以获取对生物学问题回答所需的信息。
下面将详细介绍第二代测序数据分析的原理。
1.数据质控数据质控是第二代测序数据分析的第一步,其目的是剔除低质量的序列,保证后续分析得到的结果的准确性。
主要的质控步骤包括去除低质量碱基、去除接头序列和过滤冗余数据。
这些步骤可以通过使用不同的软件工具来实现,如Trimmomatic、FastQC等。
2.序列比对序列比对是将测序数据与参考基因组进行比对的过程。
参考基因组可以是已知的基因组序列,也可以是人工合成的探针序列。
序列比对主要采用两种方法:短序列比对和长序列比对。
短序列比对常用的算法有Bowtie、BWA等,长序列比对常用的算法有BLAST、GSNAP等。
3.变异检测变异检测是根据测序数据中的变异信息来鉴定样本中存在的单核苷酸多态性(SNP)、插入缺失(indel)等变异类型。
变异检测的过程主要包括变异鉴定、变异筛选和变异注释。
变异鉴定的方法包括泛素缺失、泛素纯化和下一代序列法。
变异筛选使用一系列的过滤条件来减少假阳性的产生,如频率过滤、质量过滤和功能过滤等。
变异注释是将检测到的变异与已有的数据库进行比对,以获取变异的生物学功能信息,如GEMINI、ANNOVAR等。
4.功能注释功能注释是将检测到的变异与基因、通路等功能元件进行关联,从而了解变异对生物学功能的影响。
功能注释的方法包括基因本体论(GO)、通路分析、蛋白质相互作用网络分析等。
这些方法可以帮助研究者理解变异的生物学意义以及变异在特定疾病中的作用机制。
综上所述,第二代测序数据分析原理包括数据质控、序列比对、变异检测和功能注释等多个步骤。
《二代测序简介》PPT课件

陈竺,日本血吸虫基因组
.
10
Next-Gen Platforms
GA – Illumina/Solexa
SBS with reversible fluorescent terminators
GS FLX – Roche/454 Life Sciences
SBS through pyrosequencing
• 宏基因组学(Metagenomics) • 泛基因组学(Pangenomics)
.
3
3
Key Genomics Technologies
1975 - Southern DNA hybridization technique
1977 - Sanger’s chain-termination and Maxam、Gilbert’s
.
6
Limitation of 1st Gen Sequencer
Throughput
Time-consuming separation of chainterminated fragments
Hard to produce massively parallel system based electrophoretic separation
Template DNA immobilized on primer coated capture beads thru hybridization (1 fragment on each bead)
Thermocyle to amplify (forward primer is biotinylated)
– Asymetric Adaptors ligated (one biotinylated)
第二代测序技术在基因组研究中的应用

第二代测序技术在基因组研究中的应用随着科技的不断发展,人类对基因组的研究也越来越深入。
而第二代测序技术的出现,则为基因组研究带来了极大的便利。
本文将探讨第二代测序技术在基因组研究中的应用。
一、第二代测序技术简介第二代测序技术是相对于第一代测序技术而言的。
第一代测序技术通常采用Sanger测序法,其分析速度较慢,成本较高。
而第二代测序技术则采用高通量测序技术,能够在较短的时间内,完成大规模、高精度、高通量的基因测序。
目前,主流的第二代测序技术包括Illumina、Ion Torrent、PacBio和Oxford Nanopore等。
二、RNA测序RNA测序(RNAseq)是利用第二代测序技术对RNA分子进行测序的一种技术。
由于RNA分子的表达量和类型反映了细胞内基因表达的情况,因此RNA测序技术被广泛应用于基因表达谱分析、编码和非编码RNA注释和功能鉴定、转录因子结合位点分析以及基因剪接等研究领域。
此外,在肿瘤的诊断和治疗中,RNA测序技术也被广泛应用,例如通过分析癌细胞和正常细胞的转录组差异,来实现对肿瘤的早期诊断和个性化治疗。
三、基因组学第二代测序技术的高通量特性和较低的成本,使得全基因组测序(WGS)技术逐渐成为基因组学领域中最先进、最具前景的技术之一。
WGS技术可以实现对生命体的整个基因组进行全面、系统的分析,这种全面性的分析能够帮助科学家们更好地理解基因组功能,预测遗传病的影响和评估获得保健信息的可行性。
此外,WGS技术还可以用于对一些罕见疾病或人群的基因组研究,以了解人类基因组的变异情况和进化历史。
四、基因蛋白质互作基因蛋白质互作是一种非常关键的生命过程,它可以通过调节蛋白质间的相互作用,来达到调节基因表达和生命活动的效果。
目前,研究者们通过第二代测序技术,发现了许多与基因蛋白质互作相关的基因和非编码RNA序列,同时也发现了一些新的蛋白质互作路径。
这些发现为我们更加深入地了解细胞内的生命过程和炎症反应提供了有益的信息。
上机-第二代测序中的数据分析-基因组

参考基因组索引建立过程
bwa index 指令更多的用法及 options
5. 拼接组装
● 生成 sai 文件
– $ cd ~/proj1/ – $ bwa aln ref/ref1.fa reads/example1.L.fq > aln_example1.L.sai – $ bwa aln ref/ref1.fa reads/example1.R.fq > aln_example1.R.sai
执行路径建成以后, cd 回到工作目录,输入 fastqc -h 按回车能够
看到以下信息则表示安装成功
2.2 安装 BWA
● 解压缩
– $ cd ~/tools/bwa/ – $ tar -jxvf bwa-0.7.3a.tar.bz2
● 编译
பைடு நூலகம்– $ cd bwa-0.7.3a/ – $ make
2.1 安装 FastQC
● 解压缩
– $ cd ~/tools/fastqc/ – $ unzip fastqc_v0.10.1.zip
● 激活执行命令
– $ cd FastQC – $ chmod +x fastqc
● 建立执行路径
– $ cd ~/bin/ – $ ln -s ~/tools/fastqc/FastQC/fastqc ./
– $ cd ~/tools/samtools/ – $ tar -jxvf samtools-0.1.19.tar.bz2
● 编译
– $ cd samtools-0.1.19/ – $ make
● 建立执行路径
– $ cp samtools ~/bin/ – $ cp bcftools/vcfutils.pl ~/bin/ – $ cp bcftools/bcftools ~/bin/
二代测序变异位点解读 -回复

二代测序变异位点解读-回复如何解读二代测序变异位点。
引言:近年来,随着二代测序技术的快速发展,我们能够获得大规模的基因组测序数据,从而揭示出许多与人类健康和疾病相关的重要信息。
而在这些基因组数据中,变异位点是研究者们关注的一个重要研究对象。
本文将介绍如何解读二代测序的变异位点,包括变异位点的定义、检测方法以及进一步解读的方法和应用。
一、什么是变异位点?1.定义:变异位点指的是一个个体的基因组序列与参考基因组序列存在差异的位置。
变异位点可以分为单核苷酸变异(Single Nucleotide Variant, SNV)、小片段插入缺失(Small Insertion and Deletion, Indel)和结构变异(Structural Variation, SV)等多种类型。
2.分类:(1)单核苷酸变异(SNV)是最常见的类型,包括单核苷酸多态性(Single Nucleotide Polymorphism, SNP)和单核苷酸变异(Single Nucleotide Mutation, SNM)。
SNP是指在一个位置上两种以上的碱基频率超过1的变异,而SNM指的是在一个位置上只有一种碱基的变异。
(2)小片段插入缺失(Indel)是指在一个基因或基因组中,相邻的一段序列插入或缺失。
(3)结构变异(SV)是指在基因组中发生的较大的片段插入、缺失、重复、倒位、转座等。
二、如何检测变异位点?1.二代测序方法:目前,二代测序方法主要包括Illumina HiSeq、Ion Torrent、PacBio SMRT 等。
这些技术能够以较低的成本高通量地获得个体的基因组序列。
2.数据分析流程:(1)数据质控:对测序数据进行质量控制,去除低质量序列和接头序列等。
(2)比对参考:将质控过的测序数据与参考基因组进行比对,得到每个位点的碱基信息。
(3)变异检测:利用比对结果,采用各种算法和工具进行变异检测,包括单样本变异检测、群体组学变异检测等。
二代测序概念

二代测序概念二代测序概念一、引言随着生物学研究的不断深入,对于DNA序列的解读和分析需求越来越大。
传统的Sanger测序技术虽然被广泛应用,但由于其低通量、高成本等缺点,不能满足现代生物学研究的需求。
因此,二代测序技术应运而生。
二、二代测序技术简介二代测序技术是指将DNA分子通过PCR扩增或文库构建等方法转化为大量的片段,并在高通量平台上进行并行测序。
与传统Sanger测序技术相比,二代测序技术具有高通量、高准确性、低成本等优势,因此被广泛应用于基因组学、转录组学、表观基因组学等领域。
三、二代测序技术原理1. PCR扩增法PCR扩增法是将DNA模板通过PCR反应扩增成短片段,并在高通量平台上进行并行测序。
PCR反应过程中需要引物和酶的参与,其中引物是指能够特异性结合到待扩增区域的两个小分子,酶则是指能够催化DNA链的合成。
2. 文库构建法文库构建法是将DNA分子通过酶切或化学方法转化为小片段,并将这些片段连接到适当的载体上,形成文库。
在高通量平台上进行并行测序时,需要将文库中的DNA片段解离,使其单独存在,并进行测序。
四、二代测序技术流程二代测序技术流程包括样品准备、DNA提取、文库构建或PCR扩增、高通量测序和数据分析等步骤。
其中,样品准备是指从生物体中提取出所需的组织或细胞;DNA提取是指将样品中的DNA分子提取出来;文库构建或PCR扩增是将DNA分子转化为小片段,并连接到载体上;高通量测序是对文库或PCR产物进行并行测序;数据分析则是对测序结果进行处理和分析。
五、二代测序技术应用二代测序技术已经广泛应用于基因组学、转录组学、表观基因组学等领域。
其中,基因组学研究主要关注染色体结构和功能;转录组学研究主要关注基因表达水平和调控机制;表观基因组学研究主要关注DNA甲基化和组蛋白修饰等影响基因表达的因素。
六、二代测序技术发展趋势随着二代测序技术的不断发展,其应用领域也在不断扩大。
未来,二代测序技术有望应用于个性化医疗、环境监测、食品安全等领域,为人类健康和社会发展做出更大的贡献。
二代测序原理及报告解读

测序质量
度
深度
目标区平均深度 >20X比例
99.8%
274.67
99.7%
解决方案:
对于未覆盖的基因,或基因的部分外显子,尤其在与患者症状相符的目 的基因范围内,通过计算分析,排除大片段缺失后,实验室需使用一代测 序补齐未覆盖部分。
感谢观看
二代测序 报告详细解读
二代测序原理
边 复 制 边 测 序
扩
复
增
制
二代测序原理
制复
增扩
边 复
制
边测序DNA的建立捕获目标片段二代测序
安捷伦捕 获试剂盒
illumima 测序平台
二代测序流程复杂,参数繁多
需要检测的基因序列 所有碱基数量之和
现在报告中使用的参数
目标区覆 目标区平
测序质量
盖度
均深度
核苷酸 氨基酸 染色体 测序 变化 变化 位置 深度
Hom/ Het
正常人 群 携带率
c.2266 p.R75 C>G 6G
chr3:18 406981 8
75/82 (0.52)
het
-
对应第 对应第 该基因 该位点正常型 属于纯和 反映突
几位点 几位点 所在染 测序深度/突变 (hom)还 变型在
2.3 总结突变与患病的关系
• 从遗传方式看有没有患病的可能性 • 从突变类型看有没有患病的可能性
ACMG突变解析指南
未报道致病位点,致病可能性的评估指南,节选其中可能致病性很强列表如下:
3.基因与疾病背景介绍
• 基因介绍来自于Gene数据库 • 疾病介绍来自于OMIM、罕见病数据库或其他外文权威
数据库
4.附表
1. 检测基因包基因列表 2. 可疑阳性报告,附不明突变致病性预测指南附表 3. 检测到的其他突变位点(已去掉正常多态位点)
第二代测序数据分析原理ppt课件

组间差异基因上调与下调个数统计,可以通过此图观察上调与下调的一个总体趋势
45
差异基因火山图,可以观察到差异基因总体分布
46
GO功能分类
• 目的:利用数据库注释信息将 UniGene进行 GO 功能分类。 原理:利用数据库的注释结果,应用blast2GO算 法进行GO功能分类,得到所有序列在Gene Ontology 的三大类:molecular function, cellular component, biological process 的各个层次所占 数目,一般取到14层。 结果:MF,BP,CC三大分类结果文件以及 UniGene2GO 关系列表,三大类别中第二层次上 的柱状分布图和饼图,GO功能的层次分布图。
15
第三代测序技术:单分子测序
Helicos Biosciences VisiGen
Pacific Biosciences Mobious Nexus I三 代测序技术中,序列都是在荧光或者化学发光物质的协助 下,通过读取DNA 聚合酶或DNA 连接酶将碱基连接到DNA 链上过程中
释放出的光学信号而间接确定的。 除了需要昂贵的光学监测系统,还要记录、存储并分析大量的光学图像 ,这都使仪器的复杂性和成本增加。依赖生物化学反应读取碱基序列更
增加了试剂、耗材的使用,在目前测序成本中比例相当大。 直接读取序列信息,不使用化学试剂,对于进一步降低测序成本是非常 可取的。为了实现这样的目标,目前就有很多人在研究纳米物理技术。 在全球,许多公司和组织,如Agilent,DNA Electronics,IBM, NabSys, Oxford Nanopore Technologies,Sequenom 等都在进行纳米孔测序的开发
57
二三代测序技术的介绍和比较

目录
I. 测序技术发展 II. 二代测序 III. 三代测序 IV. 应用
二代和三代测序技术的比较
测序技术发展
1925
DNA是 遗传物
质
1953
DNA双 螺旋
1966
遗传密 码
1977
化学降解 和
Sanger 测序
1986
第一台 自动测 序仪
1998
第一台 全自动 测序仪 3700
二代和三代测序技术的比较
三代测序—Ion Torrent
Ion Torrent是一种基于半导体芯片的新一代革命性测序技术。
基本特 原点 理
当➢ D不N需A要聚昂合贵酶的把物核理苷成酸像聚等合设到备延,伸成 中的本D相N对A较链低上,时体,积会小释,放操出作一简个单氢; 离➢ 子快,速反,应除池了中2天的文PH库发制生作改时变间,,位整 于池个下上的机离测子序感可受在器2-感3.受5小到时H内+离完子成; 信➢ 号不,过H整+个离芯子片信的号通再量直并接不转高化,为目数前 字信是号10,G从左而右读,出适D合N小A基序因列组。和外显
因组的试剂成本最低
困难;仪器昂贵
太平洋生物科 学公司
PacBio
SMRT
实时单分子DNA测 序
荧光/光学
~1000
并不能高效地将DNA聚合酶加到
高平均读长,比第一代的测序时间 测序阵列中;准确性一次性达标的
降低;不需要扩增;最长单个读长 机会低(81-83%);DNA聚合酶
接近3000碱基
在阵列中降解;总体上每个碱基测
2005
2006
Roche4 54
Illumin a
Solexa GA测序
学习时间丨测序技术简介及对比——二代测序
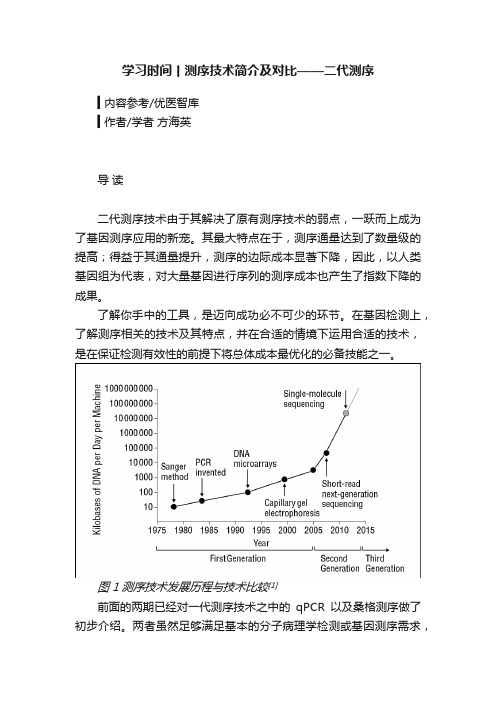
学习时间丨测序技术简介及对比——二代测序▍内容参考/优医智库▍作者/学者方海英导读二代测序技术由于其解决了原有测序技术的弱点,一跃而上成为了基因测序应用的新宠。
其最大特点在于,测序通量达到了数量级的提高;得益于其通量提升,测序的边际成本显著下降,因此,以人类基因组为代表,对大量基因进行序列的测序成本也产生了指数下降的成果。
了解你手中的工具,是迈向成功必不可少的环节。
在基因检测上,了解测序相关的技术及其特点,并在合适的情境下运用合适的技术,是在保证检测有效性的前提下将总体成本最优化的必备技能之一。
图 1测序技术发展历程与技术比较[1]前面的两期已经对一代测序技术之中的qPCR以及桑格测序做了初步介绍。
两者虽然足够满足基本的分子病理学检测或基因测序需求,但是对于多基因的综合测序分析就开始力不从心;并且如果规模进一步扩大,成本与时间的指数增长就成为了一代测序的阿喀琉斯之踵。
根据美国国立卫生研究院(NIH)国家人类基因组研究院(NHGRI)的数据,人类全基因组测序计划于1990年立项,耗时13年才得以初步完成,耗资达到27亿美元[2]。
在该项计划初步完成之后,如何缩短测序所需时间,降低测序成本,就成了一个重要的发展方向。
在NHGRI发起的降低测序成本的计划刺激下,发展出了一系列的二代测序技术(NGS)。
图 2每百万碱基DNA序列测序成本演变[3]对于大规模的基因组测序来说,2011年时间点上,桑格测序法的平均成本为每百万碱基约500美元,而二代测序已经将平均测序成本压到了每百万碱基0.5美元以下[4]。
目前,二代测序技术已经将人类基因组测序成本压到了1000美元,上机测序时间48小时以下[5]。
二代测序技术相比于一代测序而言,总体上有数个共通的,大的改进。
首先,比起一代测序主流上通过细菌质粒克隆的方法对DNA碎片进行前期克隆处理,二代测序普遍使用了无细胞系统进行NGS建库的前期准备;这样可以排除质粒中克隆载体序列的干扰[6]。
高通量测序:第二代测序技术详细介绍

在过去几年里,新一代DNA 测序技术平台在那些大型测序实验室中迅猛发展,各种新技术犹如雨后春笋般涌现。
之所以将它们称之为新一代测序技术(next-generation sequencing),是相对于传统Sanger 测序而言的。
Sanger 测序法一直以来因可靠、准确,可以产生长的读长而被广泛应用,但是它的致命缺陷是相当慢。
十三年,一个人类基因组,这显然不是理想的速度,我们需要更高通量的测序平台。
此时,新一代测序技术应运而生,它们利用大量并行处理的能力读取多个短DNA 片段,然后拼接成一幅完整的图画。
Sanger 测序大家都比较了解,是先将基因组DNA 片断化,然后克隆到质粒载体上,再转化大肠杆菌。
对于每个测序反应,挑出单克隆,并纯化质粒DNA。
每个循环测序反应产生以ddNTP 终止的,荧光标记的产物梯度,在测序仪的96 或384 毛细管中进行高分辨率的电泳分离。
当不同分子量的荧光标记片断通过检测器时,四通道发射光谱就构成了测序轨迹。
在新一代测序技术中,片断化的基因组DNA 两侧连上接头,随后运用不同的步骤来产生几百万个空间固定的PCR 克隆阵列(polony)。
每个克隆由单个文库片段的多个拷贝组成。
之后进行引物杂交和酶延伸反应。
由于所有的克隆都是系在同一平面上,这些反应就能够大规模平行进行。
同样地,每个延伸所掺入的荧光标记的成像检测也能同时进行,来获取测序数据。
酶拷问和成像的持续反复构成了相邻的测序阅读片段。
Solexa 高通量测序原理--采用大规模并行合成测序法(SBS, Sequencing-By-Synthesis)和可逆性末端终结技术(Reversible Terminator Chemistry)--可减少因二级结构造成的一段区域的缺失。
--具有高精确度、高通量、高灵敏度和低成本等突出优势--可以同时完成传统基因组学研究(测序和注释)以及功能基因组学(基因表达及调控,基因功能,蛋白/核酸相互作用)研究----将接头连接到片段上,经PCR 扩增后制成Library 。
quality score 二代测序

Quality Score 二代测序1. 什么是Quality Score?Quality Score(质量分数)是在二代测序中用来评估测序数据质量的指标。
在二代测序中,DNA或RNA样本会被分解成短片段,并通过高通量测序技术进行测序。
每个片段都会被测序仪读取多次,形成一个序列数据集。
Quality Score是对每个测序片段的测序质量进行评估的数值。
它反映了测序片段的可靠性和准确性,对于后续的生物信息学分析和数据解读至关重要。
2. Quality Score的计算方法Quality Score是通过测序仪读取测序片段时,对每个碱基进行质量评估得出的。
在二代测序中,常用的Quality Score计算方法有两种:Phred Score和Solexa Score。
2.1 Phred ScorePhred Score是最常用的Quality Score计算方法之一。
它是基于碱基的测序错误概率计算得出的质量分数。
Phred Score的计算公式如下:Q = -10 * log10(P)其中,Q表示Quality Score,P表示碱基的测序错误概率。
Phred Score的取值范围是0到40,数值越高表示测序质量越高,错误概率越低。
2.2 Solexa ScoreSolexa Score是Illumina公司独有的Quality Score计算方法。
它也是基于碱基的测序错误概率计算得出的质量分数。
Solexa Score的计算公式如下:Q = -10 * log10(P / (1 - P))其中,Q表示Quality Score,P表示碱基的测序错误概率。
Solexa Score的取值范围是-5到62,数值越高表示测序质量越高,错误概率越低。
与Phred Score相比,Solexa Score在测序质量较低时能够提供更高的分辨率。
3. Quality Score的应用Quality Score是二代测序中非常重要的指标,它在以下几个方面都有重要的应用:3.1 数据筛选Quality Score可以用于筛选测序数据,去除质量较低的片段。
二代测序 重组基因组

二代测序重组基因组
二代测序技术是一种高通量测序技术,能够快速、准确地测定DNA或RNA序列。
在重组基因组研究中,二代测序技术发挥着重要作用。
重组基因组是指通过DNA重组或转移,使得原本不同来源的DNA片段在新的组合中出现。
这种重组可以发生在同一染色体上,也可以发生在不同染色体之间。
二代测序技术在重组基因组研究中的应用有以下几个方面:
1. 检测重组事件,通过对重组基因组进行二代测序,可以帮助科研人员检测和确定基因组中发生的重组事件。
这有助于理解重组事件对基因组结构和功能的影响,同时也有助于研究重组事件与遗传疾病之间的关联。
2. 研究种群遗传多样性,通过对不同个体的重组基因组进行二代测序,可以揭示不同种群之间的遗传多样性和基因流动情况,有助于理解种群演化和遗传变异的机制。
3. 基因组编辑和转基因研究,二代测序技术可以帮助科研人员对重组基因组进行全面、高通量的测序,从而为基因组编辑和转基
因研究提供重要数据支持。
通过对编辑后的重组基因组进行测序,
可以验证编辑效果并评估可能的副作用。
4. 疾病研究,重组基因组与一些遗传性疾病的发生密切相关。
利用二代测序技术对重组基因组进行测序,有助于发现与疾病发生
相关的重组事件和突变,为疾病的诊断和治疗提供重要信息。
总之,二代测序技术在重组基因组研究中具有重要的应用前景,可以帮助科研人员深入理解基因组的结构和功能,揭示遗传变异与
生物学特征之间的关系,为人类健康和生物多样性保护提供重要支持。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
With Casava 1.8 the format of the '@' line has changed
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
Quality
• A quality value Q is an integer mapping of p (i.e., the probability that the corresponding base call is incorrect). • Phred quality score:
Per Base Sequence Content
•This module issues a warning if the difference between A and T, or G and C is greater than 10% in any position. • This module will fail if the difference between A and T, or G and C is greater than 20% in any position.
Illumina sequence identifiers
@HWUSI-EAS100R:6:73:941:1973#0/1
Versions of the Illumina pipeline since 1.4 appear to use #NNNNNN instead of #0 for the multiplex ID, where NNNNNN is the sequence of the multiplex tag.
A FASTQ file conte this
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Overrepresented Sequences
AATTAGATCGGAAGAGCACACGTCTGAACTCCAGTCACTCGAAGATCTCG 65311 1.636 TruSeq Adapter, Index 10 (97% over 36bp) ATTAGATCGGAAGAGCACACGTCTGAACTCCAGTCACTCGAAGATCTCGT 6464 0.162 TruSeq Adapter, Index 10 (97% over 36bp) AATAGATCGGAAGAGCACACGTCTGAACTCCAGTCACTCGAAGATCTCGT 4633 0.116 TruSeq Adapter, Index 10 (97% over 36bp) AATTAGTCGGAAGAGCACACGTCTGAACTCCAGTCACTCGAAGATCTCGT 4463 0.112 TruSeq Adapter, Index 10 (97% over 34bp) AATTATGGATAATTAAAGTATTCCCCCCTTTTTTTTATGATATTTTTGAC3994 0.100 No Hit
Basic Statistics
Filename File type Encoding Total Sequences NHS066-47_L4_1.fq.gz Conventional base calls Sanger / Illumina 1.9 3992798
Filtered Sequences
Per Sequence Quality Scores
•A warning is raised if the most frequently observed mean quality is below 27 - this equates to a 0.2% error rate. • An error is raised if the most frequently observed mean quality is below 20 - this equates to a 1% error rate.
Duplicate Sequences
•This module will issue a warning if non-unique sequences make up more than 20% of the total •This module will issue a error if non-unique sequences make up more than 50% of the total
Warning: >0.1% Failure: >1%
Overrepresented Kmers
•This module will issue a warning if any k-mer is enriched more than 3 fold overall, or more than 5 fold at any individual position •This module will issue a error if any k-mer is enriched more than 10 fold at any individual base position
American Standard Code for Information Interchange (ASCII)
FastQC
• / projects/fastqc/ • Double click “run_fastqc.bat” to run FastQC • The analysis results for 11 modules • Green tick for normal • Orange triangle for slightly abnormal • Red cross for very unusual
Sequence length
0
100
%GC
37
Per Base Sequence Quality
•The central red line is the median value •The yellow box represents the inter-quartile range (25-75%) •The upper and lower whiskers represent the 10% and 90% points •The blue line represents the mean quality
• The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used
Quality
Encoding
• Sanger format can encode a Phred quality score from 0 to 93 using ASCII 33 to 126 • Illumina's newest version (1.8) of their pipeline CASAVA will directly produce fastq in Sanger format • Solexa/Illumina 1.0 format can encode a Solexa/Illumina quality score from -5 to 62 using ASCII 59 to 126 • Starting with Illumina 1.3 and before Illumina 1.8, the format encoded a Phred quality score from 0 to 62 using ASCII 64 to 126 • Starting in Illumina 1.5 and before Illumina 1.8, the Phred scores 0 to 2 have a slightly different meaning
Saving a Report
NHS066-47_L4_1.fq_fastqc.zip
SRA数据库及数据获取
SRA数据库及数据获取
SRA数据库及数据获取
SRA数据库及数据获取
查看和下载SRR576183
Fastq-dum将SRA文件转化成 FASTQ格式
• fastq-dump --split-files -DQ “+” ./SRR576183.sra • fastq-dump --split-files -DQ “+” -gzip ./SRR576183.sra
Per Sequence GC Content
•A warning is raised if the sum of the deviations from the normal distribution represents more than 15% of the reads •This module will indicate a failure if the sum of the deviations from the normal distribution represents more than 30% of the reads
Per Base N Content
•This module raises a warning if any position shows an N content of >5% •This module will raise an error if any position shows an N content of >20%