药物分析-吩噻嗪类含量测定 PPT课件
合集下载
吩噻嗪类药物的分析精品PPT课件
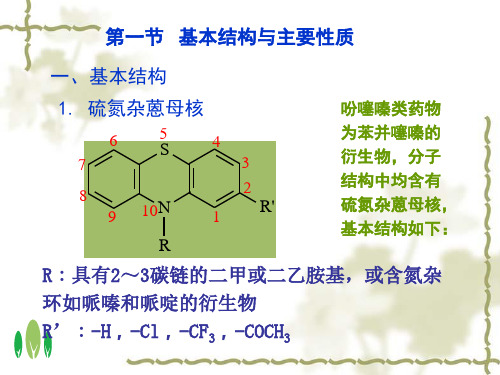
盐酸 (1→20) 乙醇
10 256
8
264与315
A 0.46
-
- 0.65
- - - -
915
-
883~93 7 - -
553~59 3
630 -
第三节有关物质的检查
❖ 合成工艺
COOH NH2 重氮化 NaNO2,HCl
COOH
N2Cl Cu2Cl2
COOH H2N
Cl
Cl
Cu,缩合
COOH NH
H3C N CH3 CH2
N
Cl
S O 3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺S-氧化物(Ⅴ)
O H3C N CH3
CH2
N
Cl
S
3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺N-氧化物(Ⅵ)
❖ 有关物质的检查方法: (ChP2010)
HPLC法 供试品:0.4 mg/ml
S
S
PdCl2
Pd2+ 2Cl-
pH2
N
R'
S
R
该反应被ChP(2010)用于吩噻嗪类药物及其制剂 的鉴别。不受氧化产物亚砜和砜的干扰
(四)含卤素取代基的反应 1.焰色反应 2.显色反应 吩噻嗪类药物2位的含氟取代基,可经 有机破坏后,分解后的氟化物,在酸性条件下,与 茜草锆试液反应呈色。
(五)氯化物的鉴别反应 1.硝酸银的沉淀反应 2.氧化还原反应
4.紫外和红外吸收:含S、N的三环共轭的大体系。S、 N与苯环形成p-共轭。紫外吸收主要由母核三环的π 系统所产生,一般具三个峰值。204~209nm(205nm附 近)、250~265nm(254nm附近)和300~325nm (300nm附近)。最强峰多在 250~265nm。
药物分析--吩噻嗪类含量测定

【示例11-32】盐酸氯丙嗪注射液测定JP(15)
• 提取后HClO4非水滴定法:
取本品约0.15g置 分液漏斗,加氨水碱化 水浴加热挥干乙醚 残渣 溴甲酚绿-结晶紫指示液 HClO4非水滴定法 紫红色→蓝紫色 乙醚提取6次,合并
丙酮溶解
每1ml高氯酸滴定液(0.05mol/L)相当于17.77mg 的 C17H19N2ClS.HCl。
四、液相色谱-质谱联用技术
液相色谱-质谱联用技术集HPLC的高分离效能和
MS的高专属性及高灵敏度于一体,已经成为目前测 定复杂生物样本中微量药物的首选方法,同时也是
进行药物及有关物质定性分析的常用方法。
色谱图:
图11-4 血浆中7种抗抑郁类和5种抗精神病类药物的LC-MS/MS检测典型色谱图
1.五氟利多;2.匹莫奇特;3.氯普噻吨;4.硫利达嗪;5.三氟拉嗪;6.阿 米替林;7.氯米帕明;8.氟西汀;9.丙米嗪;10.马普替林;11.地昔帕明; 12.舍曲林;IS.还阳酚
三氯甲烷提取 6次
浓缩提取液至5~10ml
蒸气浴 空气流蒸干
硫酸氯丙嗪 max =298测定A。
H 2SO4溶解定容
对照品:50 g / ml 同法测定As。
cs A x c(mg / ml ) 500 V As
萃取→除去抗氧剂的干扰
(三)提取后双波长分光光度法 提取后分光光度法能够除去一些干扰物
4、适用范围
主要用于Kb<10-8的有机碱盐的含量测定。对碱性较 弱的杂环类药物,只要选择合适的溶剂、滴定剂和终 点指示的方法,可使pKb为8~13的弱碱性药物采用本 法滴定。 Kb为10-8~10-10时,宜选冰醋酸作为溶剂; Kb为10-10~10-12时,宜选冰醋酸与醋酐的混合溶 液; Kb<10-12时,应用醋酐作为溶剂。 冰醋酸中加入不同量的甲酸,也能使滴定突跃显著 增大。
第11章吩噻嗪类药物分析PPT课件

第十一章 吩噻嗪类抗精神病类 药物的分析
1
概述
1
点击输入简要文字内容,文字内容需概括精炼,不用多余 的文字修饰,言简意赅的说明分项内容……
2
点击输入简要文字内容,文字内容需概括精炼,不用多余 的文字修饰,言简意赅的说明分项内容……
3
点击输入简要文字内容,文字内容需概括精炼,不用多余 的文字修饰,言简意赅的说明分项内容……
流动相A(%) 流动相B(%)
67
33
90
10
100
0
67
33
38
• 调灵敏度:对照品进样20μl,主峰高为满量程的 20%, 测定:供试品和对照品溶液各20μl,记录谱图的 保留时间是主成分出峰时间的4倍,供试品杂质峰 的面积不得大于对照品溶液主峰的面积0.5倍 (0.5%)。各杂质峰面积和不得大于对照品溶液 主峰面积2倍(2.0%)。
4. HClO4在冰醋酸溶液中,具强氧化性,可氧化吩
噻嗪类药物产生红色的氧化物,干扰指示剂终点的 观察。排除方法: (1)改用中性溶剂 (2)加抗坏血酸 (3)电位法指示终点
43
盐酸氯丙嗪测定ChP(2010)
取本品约0.2g ,精密称定,加冰醋酸10ml与醋 酐30ml,溶解后,照电位滴定法,用高氯酸滴 定液(0.1mol/L)滴定,并将滴定的结果用空白试 验校正。每1ml高氯酸滴定液(0.1mol/L)相当于 35.53mg 的C17H19N2ClS.HCl。
【鉴别】 取本品,加无水乙醇制成每1ml 中含
5g 的溶液,照分光光度法(附录ⅣA)测定, 最大吸收波长在 254nm和306 nm 本品的红外吸收图谱应与对照的图谱(光谱集 243 图)一致
28
三、色谱法
1
概述
1
点击输入简要文字内容,文字内容需概括精炼,不用多余 的文字修饰,言简意赅的说明分项内容……
2
点击输入简要文字内容,文字内容需概括精炼,不用多余 的文字修饰,言简意赅的说明分项内容……
3
点击输入简要文字内容,文字内容需概括精炼,不用多余 的文字修饰,言简意赅的说明分项内容……
流动相A(%) 流动相B(%)
67
33
90
10
100
0
67
33
38
• 调灵敏度:对照品进样20μl,主峰高为满量程的 20%, 测定:供试品和对照品溶液各20μl,记录谱图的 保留时间是主成分出峰时间的4倍,供试品杂质峰 的面积不得大于对照品溶液主峰的面积0.5倍 (0.5%)。各杂质峰面积和不得大于对照品溶液 主峰面积2倍(2.0%)。
4. HClO4在冰醋酸溶液中,具强氧化性,可氧化吩
噻嗪类药物产生红色的氧化物,干扰指示剂终点的 观察。排除方法: (1)改用中性溶剂 (2)加抗坏血酸 (3)电位法指示终点
43
盐酸氯丙嗪测定ChP(2010)
取本品约0.2g ,精密称定,加冰醋酸10ml与醋 酐30ml,溶解后,照电位滴定法,用高氯酸滴 定液(0.1mol/L)滴定,并将滴定的结果用空白试 验校正。每1ml高氯酸滴定液(0.1mol/L)相当于 35.53mg 的C17H19N2ClS.HCl。
【鉴别】 取本品,加无水乙醇制成每1ml 中含
5g 的溶液,照分光光度法(附录ⅣA)测定, 最大吸收波长在 254nm和306 nm 本品的红外吸收图谱应与对照的图谱(光谱集 243 图)一致
28
三、色谱法
吩噻嗪类抗精神病药物-精品医学课件

CH.P 2010
除去包衣, 研细,称取细粉适量 加水5ml,振摇, 滤过, 按盐酸氯丙嗪项下鉴别
常用氧化剂:
硫酸 硝酸 过氧化氢
三氯化铁
(三)*与钯离子呈色反应
重金属 专属性强 不受亚砜和砜的干扰
R
N
R'
2
+ PdCl 2
S
R
N
R'
S
Pd2+
2Cl -
S
N
R'
R
红色
(四)含卤素取代基的反应
(一)直接法 (二)提取后分光光度法 (三)提取后双波长分光光度法 (四)二阶导数法
P307示例11-35
规格:1ml:25mg V供:2ml A:0.452 求标示量% (98.7%)
(五)钯离子比色法
R
N
2
S
R'
+ PdCl 2
pH2±0.1
R
N
R'
S
Pd2+
S
N
R'
R
2Cl -
A500nm
N
Cl
S
Ⅳ
氧化物
CH 2CH 2CH 2N(CH 3)2
N
Cl
S
Ⅴ
o
o
CH 2CH 2CH 2N(CH 3)2
N
Cl
Ⅵ
S
(二)有关物质检查方法:
盐酸氯丙嗪
盐酸氯丙嗪(糖衣)片 避光操作
盐酸氯丙嗪注射液
HPLC
1.盐酸氯丙嗪的有关物质检查
供试品溶液:20mg,50ml量瓶定容 对照品溶液:精密量取适量,
药物 酸性
扫尾剂 冰醋酸,甲酸
除去包衣, 研细,称取细粉适量 加水5ml,振摇, 滤过, 按盐酸氯丙嗪项下鉴别
常用氧化剂:
硫酸 硝酸 过氧化氢
三氯化铁
(三)*与钯离子呈色反应
重金属 专属性强 不受亚砜和砜的干扰
R
N
R'
2
+ PdCl 2
S
R
N
R'
S
Pd2+
2Cl -
S
N
R'
R
红色
(四)含卤素取代基的反应
(一)直接法 (二)提取后分光光度法 (三)提取后双波长分光光度法 (四)二阶导数法
P307示例11-35
规格:1ml:25mg V供:2ml A:0.452 求标示量% (98.7%)
(五)钯离子比色法
R
N
2
S
R'
+ PdCl 2
pH2±0.1
R
N
R'
S
Pd2+
S
N
R'
R
2Cl -
A500nm
N
Cl
S
Ⅳ
氧化物
CH 2CH 2CH 2N(CH 3)2
N
Cl
S
Ⅴ
o
o
CH 2CH 2CH 2N(CH 3)2
N
Cl
Ⅵ
S
(二)有关物质检查方法:
盐酸氯丙嗪
盐酸氯丙嗪(糖衣)片 避光操作
盐酸氯丙嗪注射液
HPLC
1.盐酸氯丙嗪的有关物质检查
供试品溶液:20mg,50ml量瓶定容 对照品溶液:精密量取适量,
药物 酸性
扫尾剂 冰醋酸,甲酸
吩噻嗪类药物的分析PPT课件
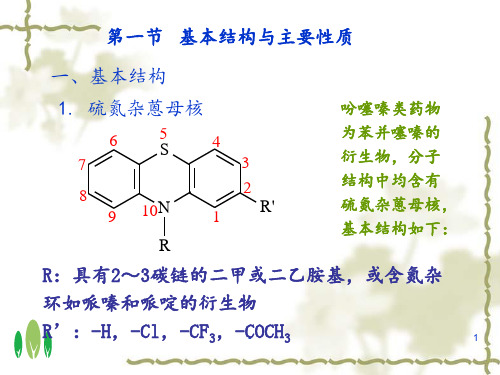
O H3C N CH3
CH2
N
Cl
S
3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺N-氧化物(Ⅵ)
-
23
❖ 有关物质的检查方法: (ChP2010)
HPLC法 供试品:0.4 mg/ml
主成分自身对照法
对照品:供试品稀释至2μg/ml
色谱柱:辛烷基硅烷键和硅胶
流动相:乙腈-0.5%三氟乙酸(50:50)
密称定.加冰醋酸10ml与醋酐30ml溶解后,照电位 滴定法,用高氯酸 (0.1mol/L)滴定液滴定,并将滴 定结果用空白试验校正。
每1ml高氯酸滴定液(0.1mol/L) 相当于35.53mg盐酸 氯丙嗪。
-
27
注意:
滴定剂的稳定性 非水溶液滴定法所用的溶剂为醋
酸,具有挥发性,膨胀系数较大,故高氯酸滴定液 的浓度受温度的影响。若滴定样品与标定HClO4溶液 时的温度不一致,温差超过10℃时,应重新标定;温 差未超过10℃时,应将高氯酸滴定液的浓度用下列 公式加以校正:
原理:吩噻嗪类药物可与一些金属离子(如Pd2+)在适 当pH值的溶液中形成有色的络合物,借以进行比色测定。
S
S
PdC2l
Pd2+ 2Cl-
pH2
N
R'
S
R
-
41
吩噻嗪类药 P物 d2 p H 2 0. 1 红色络合物
ma~ x 500nm 在max处测定 A,对照法定量
反应在 pH2±0.1 的缓冲液中进行
20
分 离 异 构 体 C 6H 5C l,C l
H N S
H3C N CH3 CH2
N
成盐 Cl HCl,C2H5OH
药物分析-吩噻嗪类抗精神病药物的鉴别试验21页PPT
▪
27、只有把抱怨环境的心情,化为上进的力量,才是成功的保证。——罗曼·罗兰
▪
28、知之者不如好之者,好之者不如乐之者。——孔子
▪
29、勇猛、大胆和坚定的决心能够抵得上武器的精良。——达·芬奇
▪
ห้องสมุดไป่ตู้
30、意志是一个强壮的盲人,倚靠在明眼的跛子肩上。——叔本华
谢谢!
21
药物分析-吩噻嗪类抗精神病药物的鉴 别试验
51、山气日夕佳,飞鸟相与还。 52、木欣欣以向荣,泉涓涓而始流。
53、富贵非吾愿,帝乡不可期。 54、雄发指危冠,猛气冲长缨。 55、土地平旷,屋舍俨然,有良田美 池桑竹 之属, 阡陌交 通,鸡 犬相闻 。
▪
26、要使整个人生都过得舒适、愉快,这是不可能的,因为人类必须具备一种能应付逆境的态度。——卢梭
药物分析--吩噻嗪类含量测定

第十页,共24页。
二、分光光度法
(一)直接分光光度法
在254nm的波长处,C17H19ClN2S.HCl的吸收系数
(
E
1 )% 为915计算。
1cm
第十一页,共24页。
(二)提取后分光光度法-排除辅料干扰
【示例11-36】盐酸氯丙嗪口服液USP32NF量 2取 7 盐酸氯丙嗪适量Vml氨 水 碱化 氯丙嗪
第十六页,共24页。
A
= E C 1 %
254
1 cm (2 5 4 )
sl
A 1 % E = C 1 c m ( 2 5 4 )
254 (s) s
A 1 % E = C 1 c m ( 2 7 7 )
2 7 7 (s) s
A
= (E
1 %
1 cm (1 )
E
) C 1 %
1 cm ( 2 )
xl
碱性溶液pKb>10,冰醋酸 中滴定突跃不明显,需加
入醋酐,使碱性增强, 突跃更明显
第四页,共24页。
2.一般方法
* 供试品:以消耗标准液体积计算 * 溶剂: HAc,一般用量10~30ml
* 滴定剂:0.1mol/L HClO4 无水HAc溶液 * 指示剂:电位法
第五页,共24页。
3. HClO4在冰醋酸溶液中,具强氧化性,可氧化吩噻 嗪类药物产生红色的氧化物,干扰指示剂终点的观 察。排除方法:
冰醋酸中加入不同量的甲酸,也能使滴定突跃显著增大。
第七页,共24页。
【示例11-31】盐酸氯丙嗪测定ChP(2010)
取本品约0.2g ,精密称定,加冰醋酸10ml与醋酐 30ml,溶解后,照电位滴定法,用高氯酸滴定液 (0.1mol/L)滴定,并将滴定的结果用空白试验校 正。每1ml高氯酸滴定液(0.1mol/L)相当于 35.53mg 的C17H19N2ClS.HCl。
药物分析11吩噻嗪类抗精神病药物的分析ppt课件

7 8
6 9
4 1
3 2 R'
Company Logo
第一节
H 3C CH
基本结构与性质
典型药物分子结构
N CH CH N S
2 3 3
CH
盐酸异丙嗪
Company Logo
第一节
H 3C
基本结构与性质
CH3
典型药物分子结构
Company Logo
显色反应
(如氟奋乃静等含氟有机药物)
氟化物 ZrF6
酸性茜素锆试液
红色 黄色
2
第一节
基本结构与性质
O O (C H 2 ) 8 C H 3
典型药物分子结构
N N CH2 N S CF3
氟奋乃静
Company Logo
第一节
基本结构与性质
CH N N CH N S
2 3
典型药物分子结构
CF3
盐酸三氟拉嗪
Company Logo
Company Logo
第二节
化学鉴别法
鉴别试验
吩噻嗪类药物具有弱碱性、 易氧化性、与金属离子配合 呈色的特性
可采用与生物碱沉淀剂、氧 化显色反应、与钯离子配合 呈色反应来鉴别本类药物
Company Logo
第二节
化学鉴别法
鉴别试验
JP15盐酸氯丙嗪的鉴别方法
Company Logo
四、紫外光吸收特性
主要理化性质
硫氮杂蒽母核为共轭三环的π系统,紫外区三个吸收峰值,约 205nm、254nm、300nm,最强多在254nm附近,根据2位、 10位取代基不同,可引起最大吸收峰的位移。当母核的二价硫被 氧化为砜或亚砜,则呈现四个峰值。
药物分析--吩噻嗪类含量测定

(五)钯离子比色法
吩噻嗪类药物在pH 2±0.1的缓冲溶液中,与 钯离子(Pd2+)形成红色配合物,在500nm波长 附近具有最大吸收
由于钯离子仅与未被氧化的硫元素配合显色, 消除了药物中氧化物对测定的干扰。
比色液的浓度:50 ~ 250 g/mL 显色稳定性:10分钟后可稳定约2小时
【示例11-38】盐酸异丙嗪注射液USP32-NF27
冰醋酸中加入不同量的甲酸,也能使滴定突跃显著 增大。
【示例11-31】盐酸氯丙嗪测定ChP(2010)
取本品约0.2g ,精密称定,加冰醋酸10ml与醋 酐30ml,溶解后,照电位滴定法,用高氯酸滴 定液(0.1mol/L)滴定,并将滴定的结果用空白试 验校正。每1ml高氯酸滴定液(0.1mol/L)相当于 35.53mg 的C17H19N2ClS.HCl。
二、分光光度法
(一)直接分光光度法
在254nm的波长处,C17H19ClN2S.HCl的吸收系
数(E
1 1
% cm
)为915计算。
(二)提取后分光光度法-排除辅料干扰
【示例11-36】盐酸氯丙嗪口服液USP32NF量 2取 7 盐酸氯丙嗪适量Vml氨 水 碱化 氯丙嗪
三 氯 甲烷 提取 6次 合并三氯甲烷提取液 蒸 气 浴浓缩提取液至5~10ml空 气 流蒸 干
2.一般方法
* 供试品:以消耗标准液体积计算 * 溶剂: HAc,一般用量10~30ml * 滴定剂:0.1mol/L HClO4 无水HAc溶液 * 指示剂:电位法
3. HClO4在冰醋酸溶液中,具强氧化性,可氧化吩噻 嗪类药物产生红色的氧化物,干扰指示剂终点的观 察。排除方法:
(1)改用中性溶剂 (2)加抗坏血酸 (3)电位法指示终点
杂环类药物的分析—吩噻嗪类药物(药物分析课件)

二、鉴别试验
(一)紫外特征吸收和红外吸收光谱
本类药物可用UV和IR鉴别,Ch.P中UV鉴别实例如下:
药物名称 溶剂
浓度(μg/ml) λmax(nm) A
E11%c
盐酸氯丙嗪 盐酸(9→1000) 5
254 0.46 915
m
306
——
盐酸异丙嗪 盐酸(0.01mol/L) 6
249
— 883-937
作为对照溶液(1)和(2)。 • 吸取上述三种溶液各10μl,分别点于同一硅胶GF254薄层板上,以己
烷-丙酮-二乙胺(8.5:1:0.5)为展开剂,展开后,晾干,置紫外光灯 (254nm)下检视
• 供试品溶液如显杂质斑点,不得多于3个; • 其杂质斑点与对照溶液(2)的主斑点比较,不得更深; • 如有一点超过,应不得深于对照溶液(1)的主斑点。
H C CH2OH
N(CH3)2
N(CH3)2
H3C CH3
使得最终合成产物变为:
-- 薄层色谱法 -- 高低浓度对比法
H3C N CH3 H3C
N
S
此异构体在丙酮中溶解度大,精制 也不易除去。
另外,异丙嗪不太稳定,易氧化, 贮藏过程可能分解。
盐酸异丙嗪【有关物质】检查方法:
• 取本品,加二氯甲烷制成每1ml中含10mg的溶液,作为供试品溶液; • 精密量取适量,加二氯甲烷制成每1ml中含0.15mg和0.05mg的溶液,
奋乃静 无水乙醇
7
258 0.65 —
癸氟奋乃静 乙醇
10
260
——
盐酸氟奋乃静 盐酸(9 →1000) 10
255
— 553-593
盐酸三氟拉嗪 盐酸(1 →20) 10
药物分析--吩噻嗪类含量测定25页PPT

药物分析--吩噻嗪类含量测定
11、用道德的示范来造就一个人,显然比用法律来约束他更有价值。—— 希腊
12、法律是无私的,对谁都一视同仁。在每件事上,她都不徇私情。—— 托马斯
13、公正的法律限制不了好的自由,因为好人不会去做法律不允许的事 情。——弗劳德
14、法律是为了保护无辜而制定的。——爱略特 15、像房子一样,法律和法律都是相互依存的。。——非洲 2、最困难的事情就是认识自己。——希腊 3、有勇气承担命运这才是英雄好汉。——黑塞 4、与肝胆人共事,无字句处读书。——周恩来 5、阅读使人充实,会谈使人敏捷,写作使人精确。——培根
11、用道德的示范来造就一个人,显然比用法律来约束他更有价值。—— 希腊
12、法律是无私的,对谁都一视同仁。在每件事上,她都不徇私情。—— 托马斯
13、公正的法律限制不了好的自由,因为好人不会去做法律不允许的事 情。——弗劳德
14、法律是为了保护无辜而制定的。——爱略特 15、像房子一样,法律和法律都是相互依存的。。——非洲 2、最困难的事情就是认识自己。——希腊 3、有勇气承担命运这才是英雄好汉。——黑塞 4、与肝胆人共事,无字句处读书。——周恩来 5、阅读使人充实,会谈使人敏捷,写作使人精确。——培根
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
碱性溶液pKb>10,冰 醋酸中滴定突跃不明显, 需加入醋酐,使碱性增 强,突跃更明显
2.一般方法
* 供试品:以消耗标准液体积计算 * 溶剂: HAc,一般用量10~30ml * 滴定剂:0.1mol/L HClO4 无水HAc溶液 * 指示剂:电位法
3. HClO4在冰醋酸溶液中,具强氧化性,可氧化吩噻 嗪类药物产生红色的氧化物,干扰指示剂终点的观 察。排除方法:
定量依据
样品在二波长下吸收度差值(A):
A
Aab 1
Aab 2
(
Aa 1
Ab 1
)
(
Aa 2
Ab 2
)
Aa 1
Aa 2
( 1
2 )Cal
=(E11%cm(1 )
E1% 1cm(2
)
)Ca
l
Ab 1
Ab 2
A Ca
即,吸收度差值(A)仅与待测组分的浓度有关,
而与干扰组分无关,干扰组分的干扰被消除。
BH++C7H15SO2O- pH3.5 [BH+C7H15SO2O-]
影响离子对形成的条件
反离子的性质与浓度 流动相的组成 pH 离子强度等
影响待测组分的保留
四、液相色谱-质谱联用技术
液相色谱-质谱联用技术集HPLC的高分离效能 和MS的高专属性及高灵敏度于一体,已经成为目 前测定复杂生物样本中微量药物的首选方法,同时 也是进行药物及有关物质定性分析的常用方法。
【示例11-32】盐酸氯丙嗪注射液测定JP • (提1取5)后HClO4非水滴定法:
取本品约0.15g置
乙醚提取6次,合并
分液漏斗,加氨水碱化
水浴加热挥干乙醚 HClO4非水滴定法
残渣
丙酮溶解 溴甲酚绿-结晶紫指示液
紫红色→蓝紫色
每1ml高氯酸滴定液(0.05mol/L)相当于17.77mg 的 C17H19N2ClS.HCl。
【示例11-37】盐 (酸U氯SP丙32嗪-注NF射2液7)的含量测定
测定波长: 254nm(最大吸收波长) 277nm(等吸收波长)
空白对照液:0.1mol/L盐酸溶液
计算公式: 12.5C(A254 A277 )U /V (A254 A277 )S
式中:C(mg/ml)为对照品溶液的浓度; V(ml)为所取供试品的体积; 括号内的公式分别为供试品溶液(U)和对照
A
(
A 254 Cs
A 277 Cs
)s
Cx
(A254 (A254
A277 )x A277 )s
Cs
D=500 250 =12500 10
含量(mg
/
ml)=
12500 V 1000
(A254 (A254
A277 )x A277 )s
Cs
(五)钯离子比色法
吩噻嗪类药物在pH 2±0.1的缓冲溶液中,与 钯离子(Pd2+)形成红色配合物,在500nm波长 附近具有最大吸收
色谱图:
图11-4 血浆中7种抗抑郁类和5种抗精神病类药物的LC-MS/MS检测典型色谱图
1.五氟利多;2.匹莫奇特;3.氯普噻吨;4.硫利达嗪;5.三氟拉嗪;6.阿 米替林;7.氯米帕明;8.氟西汀;9.丙米嗪;10.马普替林;11.地昔帕明; 12.舍曲林;IS.还阳酚
数(E11c%m )为915计算。
(二)提取后分光光度法-排除辅料干扰
【示例11-36】盐酸氯丙嗪口服液USP32NF量2取7 盐酸氯丙嗪适量Vml 氨水碱化氯丙嗪
三氯甲烷提取6次 合并三氯甲烷提取液 蒸气浴浓缩提取液至5~10ml 空气流蒸干
H2SO4溶解定容 硫酸氯丙嗪 max =298测定A。
合并盐酸提取液 并定容至50ml
称取标准品和对照品溶液各2ml +PdCl2显色,测A470
对照品0.124mg/ml
含量(mg
/
ml)=50
Ax As
Cs
三、高效液相色谱法
(一)反相高效液相色谱法(RP-HPLC)
固定相:十八烷基硅烷键合硅胶(ODS) 流动相:水-甲醇或水-乙腈系统,极性较强
冰醋酸中加入不同量的甲酸,也能使滴定突跃显著 增大。
【示例11-31】盐酸氯丙嗪测定ChP(2010)
取本品约0.2g ,精密称定,加冰醋酸10ml与醋 酐30ml,溶解后,照电位滴定法,用高氯酸滴 定液(0.1mol/L)滴定,并将滴定的结果用空白试 验校正。每1ml高氯酸滴定液(0.1mol/L)相当于 35.53mg 的C17H19N2ClS.HCl。
极性较强的组分在分离时先流出柱子,极性较弱 的组分后流出柱子,适合于共存组分极性差异较大样 品的分析。杂环类药物分析一般在流动相中加入含氮 碱性竞争试剂(扫尾剂:醋酸铵、三乙胺、二乙胺、乙 腈等),以抑制碱性药物与未硅烷化的硅醇基作用。
(二)离子对高效液相色谱法(Ion-RP-HPLC)
常用的离子对试剂:烷基磺酸盐阴离子对试剂, 如戊烷磺酸钠、庚烷磺酸钠、十二烷磺酸钠等。
BH+A- + HClO4
BH+ClO4 + HA
游离碱类盐
被置换出的弱酸
当HA酸性较强时,反应不能定量 完成,必须除去或降低HA的酸性, 使反应顺利地完成。
因此,要根据不同情况采用相应测定条件。
HClO4滴定
供
冰
试 +醋
品
酸
结 指示剂 果
空白试 验校正
10ml~30ml
若供试品为氢卤酸 盐再加5%醋酸汞的 冰醋酸液3ml~5ml
品溶液(S)在下标所示波长处的吸光度差。
A254 =E11c%m(254)Csl
A 1% E = C 1cm(254)
254 ( s ) s
A 1% E = C 1cm(277)
277(s) s
A=(E11%cm(1 )
E1% 1cm(2
)
)C
xLeabharlann lCxAx
(E E ) 1% 1cm(1 )
1% 1cm(2 )
对照品:50 g / ml 同法测定As。
c(mg / ml) 500 cs Ax V As
萃取→除去抗氧剂的干扰
(三)提取后双波长分光光度法
提取后分光光度法能够除去一些干扰物 质,但却不能除去吩噻嗪类药物的氧化物。 提取时,氧化物随游离碱进入有机相;测定 时,因结构相近,氧化物在吩噻嗪类药物的 测定波长处产生吸收,干扰测定。
(1)改用中性溶剂 (2)加抗坏血酸 (3)电位法指示终点
4、适用范围
主要用于Kb<10-8的有机碱盐的含量测定。对碱性较 弱的杂环类药物,只要选择合适的溶剂、滴定剂和终 点指示的方法,可使pKb为8~13的弱碱性药物采用 本法滴定。 ✓ Kb为10-8~10-10时,宜选冰醋酸作为溶剂; ✓ Kb为10-10~10-12时,宜选冰醋酸与醋酐的混合 溶液; ✓ Kb<10-12时,应用醋酐作为溶剂。
由于钯离子仅与未被氧化的硫元素配合显色, 消除了药物中氧化物对测定的干扰。
比色液的浓度:50 ~ 250 g/mL 显色稳定性:10分钟后可稳定约2小时
【示例11-38】盐酸异丙嗪注射液USP32-NF27
精密称取适量置 分液漏斗
依次加氯化钾 氢氧化钠碱化 甲醇
正庚烷提 取3次
合并正庚 烷提取液
盐酸酸化
第十一章 吩噻嗪类抗精神病药物 的分析
第四节 含 量 测 定
• 性质:弱碱性、紫外光吸收特性、金属离 子配合呈色
• 测定方法:酸碱滴定法、分光光度法、高 效液相色谱法和液相色谱-质谱联用技术
一、酸碱滴定法
(一)非水溶液滴定法 结构:母核上10位取代基的烃胺-NR2、哌嗪基及 哌啶基具有碱性 1. HClO4非水溶液滴定法基本原理
(二)乙醇水溶液的氢氧化钠滴定法
• 本类药物的盐酸盐的水溶液显酸性,在乙醇
-水溶液中,可用NaOH滴定剂滴定,电位法指示 滴定终点。
等当点
突跃体积
NaOH HCl NaCl H2 0
V1
B.HCl NaOH B NaCl H2 0
V2
滴定体积△V= V1-V2
二、分光光度法
(一)直接分光光度法 在254nm的波长处,C17H19ClN2S.HCl的吸收系