实时荧光定量PCR—— qMan探针法及设计原则
实时定量PCR试验设计原则及应用

[12][13]merase chain reaction [J ].Am J Trop Med Hyg ,1991(45):688-694.马良,陈雅棠,刘约翰.聚合酶链反应检测粪便中微小隐孢子虫[J ].中国寄生虫学与寄生虫病杂志,1996,14(2):111-114.吴良,陈盛霞,曹建平.隐孢子虫病的流行与诊断研究进展[J ].国际医学寄生虫病杂志,2006,33(5):277-281.□收稿日期:2008-12-29作者简介:黎松庆(1982—),男,助理兽医师,主要从事畜牧兽医技术推广工作。
实时定量PCR 试验设计原则及应用黎松庆,叶朗光,黎旭宇,邓树轩(广东省东莞市大岭山镇农业技术服务中心,广东东莞523820)摘要:笔者对实时定量PCR 试验设计原则及其在医学、分子生物学、食品检测的应用上进行了概述。
关键词:实时定量PCR ;试验设计;应用中图分类号:R446.61文献标识码:A 文章顺序编号:1672-5190(2009)01-0043-02The Design and Application of The Quantitative Real-time PCRLI Song-qing,et al(Dalingshan Service Center of Agricultural Technology of Dongguan City in Guangdong Province ,Dongguan 523820,China )Abstract :The design principles and application in medicine ,molecular biology,food testing of the quantitative real-time PCR were re -viewed in this paper.Keywords :quantitative real-time PCR;design;application 普通PCR 反应能够对模板链进行指数似的拷贝,但由于反应过程中模板、反应物的限制,或焦磷酸分子产物对于DNA 聚合酶的抑制作用,以至PCR 反应最后不能够指数似的拷贝模板,所以PCR 反应终止时候的产量总是难以确定,也无法知道起始的模板的数量[1]。
实时荧光pcr法优选全文

可编辑修改精选全文完整版实时荧光pcr法实时荧光PCR法是一项重要的分子遗传学技术,能够获得准确、可靠而快速的结果。
它是一种用于监测和调节基因表达的技术,在研究生物活动和生物学过程中发挥重要作用。
本文简要介绍了实时荧光PCR的原理,其对各种生物活动的应用,同时也讨论了实时荧光PCR 未来发展的趋势。
一、实时荧光PCR的基本原理实时荧光PCR是一种高灵敏的PCR技术,它主要是利用荧光标记探针来检测模板的扩增过程,从而得出DNA片段扩增的数量和速度。
实时荧光PCR通常利用5’和3’端有荧光活性的探针对模板DNA进行检测,以高精度确定模板DNA的数量和种类。
与传统PCR技术不同,实时荧光PCR技术可以实时跟踪和检测生物序列的复制,而不需要将样本放置于催化剂,模板DNA可以通过反应体系中的荧光探针产生荧光信号,根据荧光信号的强弱进行实时调节复制过程。
二、实时荧光PCR适用的生物活动实时荧光PCR技术可应用于许多已知的生物活动,用于多种数据收集,包括但不限于:筛选细菌,鉴定病毒,检测微生物,检测基因表达,提取和组装基因组,测定突变状态,预测可变位点,鉴定病原体,杂合状态分析,发育生物学研究,柔性检测变体等。
例如,实时荧光PCR可用于研究癌症相关基因的表达和转录状态,还可用于检测家禽禽流感病毒,研究家禽的流行特性,以及检测芽胞杆菌抗性基因多态性。
三、实时荧光PCR未来发展趋势随着现代科技的发展,实时荧光PCR技术也发生了巨大变化,一系列新技术已经应用于现代实时荧光PCR技术中,大大提高了这种技术的准确性和快速性。
例如,超高通量实时荧光PCR技术,使研究者可以以更高的效率来检测和分析生物序列;多重PCR技术,可以有效提高检测敏感度和追踪多个位点的表达;实时荧光PCR技术的循环法,可以使检测更准确,但是耗时较长。
此外,基于活性水平的荧光定量PCR技术,也被广泛应用于实时荧光PCR中,可以以可视化方式监测和调节基因表达。
定量PCR Taqman探针设计要领-2

定量PCR Taqman探针设计要领-2第三步:寻找一家信赖的公司合成引物和探针,一般引物合成大家比较熟悉,而且价格也比较便宜(特别是这两年便宜了许多),而探针则相对来说贵了许多,一般Taqman探针合成在1000到5000元不等(不同的合成要求价钱不同)——而这只是标记价钱,序列合成基本上和引物合成价钱相似。
第四、五、六步:一般的定量PCR反应体系与普通PCR其实也差不了多少,只是要加入Taqman探针,另外不同就是分步法的不同。
其中需要注意的是:* 扩增酶最好选用热启动酶* 引物和探针的浓度需要进行优化,有人建议从50nM开始,在50nM—900nM之间优化,一般为200nM(注意探针需要避光保存。
* 同样Mg+和酶量也需要进行优化,酶的推荐反应浓度是1.25-1.5U(50ul)* DNA模板的添加量通常在100 ng以下,因不同种类的DNA模板中含有的靶基因的拷贝数不同,必要时可进行梯度稀释,确定最佳的DNA模板添加量。
如果欲进行2 Ste p RT-PCR反应的第二步PCR扩增反应,第一步的RT反应液作为DNA模板时的添加量不要超过PCR反应液总体积的10%。
另外循环参数虽然在引物和探针设计完之后也就确定了,但是有时也需要进行优化。
第七步:在进行数据分析的时候,通常用不同浓度的标准样品的Ct值来产生标准曲线,然后计算相对方程式。
方程式的斜度可以用来检查PCR的效率,对于100%PCR效率来说,一个理想的斜率是3.32。
最佳的标准曲线是建立在PCR的扩增效率为90%-100 %(100%意味着在每个循环之后,模板的总数将增加为前一次的2倍)的基础上。
所有标准曲线的线性回归分析需要存在一个高相关系数(R2≥0.99) ,这样才能认为实验的过程和数据是可信的。
使用这个方程式我们可以计算出未知样本的初始模板量。
大多数定量PCR仪都有这样一个软件,它可以从标准曲线中自动地计算出未知样本的初始模板量。
荧光定量PCR应用指南

荧光定量PCR应用指南荧光定量PCR(Fluorescent quantitative PCR)是依靠荧光信号的强度来定量PCR产物的一种PCR技术。
该技术通过在PCR反应体系中添加特异性荧光探针,利用荧光信号的强度来反映PCR产物的数量。
荧光定量PCR在生物医学研究、基因表达分析、致病微生物检测等领域有着广泛的应用。
下面将介绍荧光定量PCR的原理、实验步骤和一些注意事项。
一、原理:荧光定量PCR最常用的荧光探针是TaqMan探针,TaqMan探针是由两个DNA引物和一个荧光标记的探针组成。
当PCR反应进行到延伸阶段时,引物和探针结合到靶序列上形成荧光信号。
荧光信号的强度与PCR产物的数量成正比,通过检测荧光信号的强度可以定量PCR产物的数量。
二、实验步骤:1.设计引物和探针:选择特异性的引物和探针对目标序列进行扩增。
引物和探针的设计需要遵循一些原则,如避免引物和探针之间的自互补性、探针需选择合适的荧光染料等。
2.准备PCR反应体系:根据PCR反应需要,准备PCR反应体系。
一般包括模板DNA、引物、探针、聚合酶、缓冲液等。
3.荧光定量PCR反应:将PCR反应体系加入到荧光定量PCR仪中,进行PCR反应。
PCR反应的条件需要根据目标序列的特性进行优化,包括温度、延伸时间等。
4.数据分析:通过荧光定量PCR仪记录荧光信号的强度,根据荧光信号的强度可以反应PCR产物的数量。
通过数据分析软件对荧光信号进行定量计算,获得PCR产物的数量。
三、注意事项:1.引物和探针的设计需要严格控制,确保其特异性,避免与非靶序列结合。
2.PCR反应的条件需要进行优化,不同的目标序列可能需要不同的温度、延伸时间等。
3.控制实验中的阳性对照和阴性对照,确保实验结果的可靠性。
4.严格遵守无菌操作,避免PCR反应体系受到外源性污染。
5.选择合适的荧光定量PCR仪进行实验,确保获得准确的荧光信号强度数据。
6.实验过程中需严格按照操作规程进行,避免操作失误对实验结果产生影响。
实时定量PCR引物和探针设计操作步骤PrimerExpress软件

实时定量PCR引物和探针设计操作步骤Primer Express软件Primer Express 是实时定量PCR引物和探针设计的专用软件。
遵守以下三个原则有助于快速建立定量PCR反应体系:1.所有扩增按照同样的原则设计 (Primer Express);2.所有PCR反应在ABI PRISM ?7000/7900上使用同样的热循环条件;3.所有反应使用相同的PCR试剂。
引物和探针的设计原则下述原则的重要程度由上往下越来越低,请尽量满足编号靠前的条件。
它们中有的已经在Primer Expre软件中设置成缺省值,有的则需要在选择引物和探针时由设计者加以运用。
如果是设计SYBRGreen 引物,也要选择TaqMan Primer and Probe design并遵守这些规则,但是只需要合成引物就可以了。
TaqMan 探针:1. 保持G-C含量在30-80%之间。
2. 避免同一碱基重复过多。
特别是G,不可超过4个及以上。
3. 5' end不能是G。
4. 尽量使探针中的Cs多于Gs。
如果不能满足,则使用互补链上的探针。
5. 对于单探针反应,用Primer Express?软件计算出来的Tm值应当在68-70 °C 之间。
引物:1. 在探针确定以后再选择引物。
2. 引物要尽可能地接近探针,但是不要重叠。
3. 保持G-C含量在30-80%之间。
4. 避免同一碱基重复过多。
特别是G,不可超过4个及以上。
5. 用Primer Express?软件计算出来的Tm值应当在58-60 °C之间。
6. 3' end 的5个碱基中G and/or C碱基的总数不能超过2个。
实时TaqMan 引物和探针设计Begin by opening Primer Express and selecting "File", "New", and "TaqMan? Primer & Probe Design". The following screen will appear. You can close the TaqMan? Primer & Probe Data box as shown.输入或插入序列Import or paste a sequence into the window (Import shown). To paste a sequence from a Word or text file, first copy it to the clipboard. Be sure to only select the sequence (including numbers or annotations is OK); do not include extraneous information such as accession numbers etc. Next, select "Edit" and "Paste". The sequence will appear in the Sequence screen of Primer Express. Or, to Import a Sequence, click the "Import DNA File" button as shown. The software will then ask you to locate the sequence file. Select it from a folder, hard drive, disk, or desktop. Again, no annotations should be present in this sequence.A file is then imported after selecting the file location.保存输入的序列Select "File" and "Save" to give the sequence a name. This will be displayed in the File Name Box and will save the sequence in the Archive Folder.引物和探针设计参数Click the "Parameters" tab. This displays the Universal default parameters used to search for suitable TaqMan? primer & probe sets for real-time assays. It is strongly recommended that you do not adjust any of the parameters.引物和探针的排序及选择Primer Express is now ready to find Primers and Probes. Click the "Primers" tab, select "Options" and "Find Primers/Probes Now". The software will display the progress in the small window below the sequence.** Please disregard the "Optimal Primer Pairs Only" checkbox and the "Penalty" heading. By checking the Optimal Primer Pairs Only box, you will be severely limiting the range of your search, since the parameters it employs are not based on TaqMan? design guidelines. The Penalty score assigned to your Primer & Probe set is based on factors such as amplicon length. Since the default TaqMan? design parameters keep amplicons under 150 bp, this can be disregarded as well.Primer/probe sets will be listed when the search is complete. Scroll to the right to view the Probes. Click on the "Start" heading under probes to sort probes by sequence. This will group similar probes, simplifying the search.探针的选择Select a probe that is less than 30 bp in length and contains more C's than G's. The probes displayed are on the sense strand only. If the probes displayed do not have more C's than G's, then you will need to use the complement probe (as illustrated in this example). If you need to use the complement, make sure that the probe selected here does not have a C at the 3' end of the probe (otherwise, the complement will have a G at the 5' end ? which is not allowed).The probe selected meets the first criteria above, but not the second (9 G's, 5 C's). Highlight this probe.Return to the sequence by clicking the "Sequence" tab.Lock in the probe sequence by clicking the Probe Button on the Tool Bar and highlight the probe sequence. The probe will turn green and be displayed in lower case when it is locked.引物选择Find compatible primers by returning to the "Primers" tab, selecting "Options" and "Find Primers & Probes Now". This will find new primer sets that will work with the probe you have selected. You can click on "Start" under Forward Primer to sort the displayed sequences. Search for a primer from the list displayed the meets the following criteria:1.No more than 2 G's and/or C's within the last 5 bases on the 3' end of the primer; and2.No runs of identical nucleotides, especially 4 or more G's.From the list of forward primers displayed, select a primer that has no more than 2 G's and/or C's within the last 5 bases on the 3' end of the primer. Highlight one of the primers that matches this criteria. If no forward primer matches this criteria then select a primerwith 3 G's and/or C's. The example shown below matches the criteria and will serve as a suitable forward primer. Once you have selected the appropriate primer click on the "Sequence" tab to return to the Sequence window.Lock the forward primer by clicking the "Forward Primer" button on the toolbar, then highlighting the forward primer sequence. A blue arrow will be displayed under the forward primer showing that it is locked.Click on the "Primers" tab and perform a new search. Scroll to the Reverse Primers displayed and select a reverse primer following the same criteria for forward primer selection (G/C rule on the 3' end of primer).Return to the Sequence page and lock in on the Reverse Primer using the Reverse Primer Tool.This now displays the primers and probe you have selected. Return to the Primers tab and perform one final search to display your results.保存搜索结果Click on "Save List" at the bottom of the screen to save your selection in a tab delimited format. Click "Order" to generate an editable/printable text file of your sequences:互补探针的选择In the example above, you must use the complementary probe so as to insure that the probe has more C's than G's. Remember, the probe you use cannot have a G at the 5' end, thus the sense probe used for this search cannot have a C at the 3' end.In order to generate the probe complement, return to the Sequence screen. Highlight the probe sequence, select "Edit", and "Copy Complement". You will not see the complementary sequence at this point; it is copied to the clipboard:Return to the Order window and "Paste" the complement in this window, overwriting the probe displayed. You have the option of editing the primer/probe names, and adding the reporter/quencher dyes to the probe sequence.This document can now be saved and put into a Word document or attached to an e-mail message.在Results Archive中保存搜索结果Your search can also be saved in the Results Archive Folder. Click on the "Results" tab. The forward and reverse primers are displayed in their respective boxes, and the probe sequence is displayed in the "Cycle Params" box The probe sequence displayed is the original strand. To view/save the complementary strand, highlight the probe from the Sequence and select "Copy Complement". "Paste" the complement probe into the "Cycle Params". The complementary probe strand is now displayed. It is important to note that if you leave the Results page, the probe sequence will default back to the original. Each time you returnto the Results page you will need to re-paste the complementary probe strand. Note: The information displayed below the selected primer and probe sequences should be ignored when performing TaqMan Assays. The Universal TaqMan? Guidelines do not require you to perform optimizations, thus, the cycling/concentration, etc. information displayed here can be ignored. Save the Results by selecting "Save Results". A message will display showing the results were saved.打印结果 To print the Results, select "Open Results" from the "File" menu. The last (newest) results file will be the last one in the list (at the bottom of the list): Highlight and click "Open".This is the relevant information needed to order your primer/probe set. To print, click and drag, highlighting the information you want and selecting "Copy" from the "Edit" menu, placing it on the clipboard. This should be everything from the Sequence name through the TaqMan? probe annealing information.This is the relevant information needed to order your primer/probe set. To print, click and drag, highlighting the information you want and selecting "Copy" from the "Edit" menu, placing it on the clipboard. This should be everything from the Sequence name through the TaqMan? probe annealing information.You can then paste your sequence information in to a Word document; from here you can print a copy for your records.订购信息Be sure to include information on your needed synthesis scale and the corresponding part number, your reporter dye(s), your quencher (TAMRA), and your personal information (name, institution, address, phone fax etc.).。
实时荧光定量PCR的原理、操作及其应用PPT课件

THANKS FOR WATCHING
感谢您的观看
它利用荧光染料或荧光探针标记特异性引物,在PCR反应过 程中,随着DNA或RNA的扩增,荧光信号被逐渐释放,通过 检测荧光信号的强度,可以实时监测DNA或RNA的扩增量。
实时荧光定量PCR的原理概述
实时荧光定量PCR的基本原理是在PCR反应体系中加入荧光染料或荧光探针,这些荧光物质 与DNA或RNA结合后,在特定波长光的激发下产生荧光信号。
环介导等温扩增技术
在恒温条件下进行核酸扩增,具有快速、特异性强和灵敏度高等优 点。
纳米材料与PCR的结合
利用纳米材料的特性,提高PCR的检测效率和灵敏度。
提高检测灵敏度与特异性
信号放大技术
通过使用信号放大系统,提高检测信号,从而提高检测灵 敏度。
特异性引物和探针的设计
针对目标基因设计特异性引物和探针,降低非特异性扩增 和交叉污染的风险。
选择合适的内参基因
选择稳定的内参基因
01
内参基因应具有稳定的表达水平,不受实验处理的影响。
验证内参基因的稳定性
02
通过实时荧光定量PCR技术,对候选内参基因进行稳定性评估。
使用多个内参基因参基因进行数据校正。
数据解读与报告
标准化处理
对原始数据进行标准化处理,消 除不同样本间的差异。
样本处理
对样本进行破碎、离心、 提取核酸等处理,以获得 待测的DNA或RNA。
浓度和纯度测定
使用紫外分光光度计等设 备测定核酸浓度和纯度, 确保符合实验要求。
引物设计与选择
引物设计
根据目标基因序列,利用 引物设计软件进行引物设 计。
引物筛选
根据实验需求,筛选出特 异性好、扩增效率高的引 物。
实时荧光Taqman 探针设计

一、实时荧光Taqman 探针设计总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。
即real-time PCR的扩增片段是50bp----150bp。
当找不到150bp的保守片段时,必须确保探针的片段是保守的。
在设计探针和引物时,要同时考虑在两条链上设计引物与探针。
但要注意的是:在那条链上设计探针时,就应靠近在同一条链上设计的引物(即上游引物)。
这样,可保证在将来扩增时,即便没有完全扩增,也有荧光信号报告出来。
两者的距离最好是探针的5’端离上游引物的3’有一个碱基,但也可以重叠。
若在原序列中找不到合适的探针与引物(1主要是探针和上游引物的距离太远,而离下游引物的距离却较近时;2突变位点要求在探针的5’ 端也能检测到荧光信号,但却是在3’端),可在互补的序列中设计引物与探针。
另real-time PCR中的探针和引物的Tm值,均要高于平常PCR的引物和杂交的探针的Tm 值。
二、探针的设计探针设计的基本原则:1.保守:探针要绝对的保守,有时分型就单独依靠探针来决定。
理论上有一个碱基不配对,就可能检测不出来。
若找不到完全保守的片段,也只能选取有一个碱基不同的片段。
且这个不同的碱基最好在探针的中间,对探针与目的片段的杂交影响不大,不相同的碱基最好不要在两端,因为两端不利于探针的杂交。
且最好为A或T,而不能为G或A,因为A、T为双键,而G、A为三键。
2.探针长度Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,最好为70℃,确保探针的Tm值要比引物的Tm值高出10℃,这样可保证探针在煺火时先于引物与目的片段结合。
因此探针最好是富含GC的保守片段,保证其的Tm值较高。
现在有Taqman MGB探针,在TAMER之后再标记一个MGB,可使探针的Tm值较高,即使探针片段较短,也可达到Taqman探针的Tm值要求(68-70℃)。
实时荧光定量PCR方法简介

实时荧光定量PCR方法简介一.实时荧光定量PCR的基本原理理论上,PCR过程是按照2n(n代表PCR循环的次数)指数的方式进行模板的扩增。
但在实际的PCR反应过程中,随着反应的进行由于体系中各成分的消耗(主要是由于聚合酶活力的衰减)使得靶序列并非按指数方式扩增,而是按线性的方式增长进入平台期。
因此在起始模板量与终点的荧光信号强度间没有可靠的相关性。
如采用常规的终点检测法(利用EB染色来判断扩增产物的多少,从而间接的判断起始拷贝量),即使起始模板量相同经PCR 扩增、EB染色后也完全有可能得到不同的终点荧光信号强度。
为了能准确判断样品中某基因转录产物(mRNA)的起始拷贝数,实时荧光定量PCR采用新的参数——Ct值,定量的根本原理是Ct值与样品中起始模板的拷贝数的对数成线性反比关系。
Ct值是如何得到的在实时荧光定量PCR的过程中,靶序列的扩增与荧光信号的检测同时进行,定量PCR仪全程采集荧光信号,实验结束后分析软件自动按数学算法扣除荧光本底信号并设定阈值从而得到每个样品的Ct值。
Ct值的定义Ct值中的“C”代表Cycle(循环),“t”代表检测threshhold(阈值),其含义是PCR扩增过程中荧光信号强度达到阈值所需要的循环数;也可以理解为扩增曲线与阈值线交点所对应的横坐标。
Ct值与样品中模板的对应关系Ct值与样品中起始模板的拷贝数的对数成线性反比关系(y=ax+b,x代表起始模板拷贝数的对数,y代表Ct值)。
与终点法相比利用Ct值的优势由于Ct值是反映实际PCR反应过程中扩增即将进入指数期的参数,该参数几乎不受试剂消耗等因素的影响,因此利用Ct值判断的起始模板拷贝数更加精确,重复性也更好。
传统的终点检测法是在PCR扩增经历了指数扩增期进入平台期后利用EB等染料染色来判断扩增产物的多少,从而间接的判断起始拷贝量,这种方法的精确度不高、重复性也不好。
下图中是96个复孔的实时扩增曲线(完全相同的反应体系、相同的反应protocol、相同的样品起始浓度),可以看到Ct值具有很好的重复性,而终点的荧光信号强度差异达到300个单位。
定量PCR Taqman探针设计要领

定量PCR+Taqman探针设计要领自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR 荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR要求。
当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:目的基因查找比对→探针与引物设计→探针与引物合成→配置反应体系→反应参数→重复实验,优化条件→获得曲线数据,比对标准曲线→再重复验证。
第一步:在第一步目的基因查找比对过程中可以利用NCBI genbank序列以及DNAstar等软件完成目的DNA 或者RNA的查找与比对——这在分析测序报告的时候相信很多人操作过,这一步需要注意的就是要保证所分析的序列在一个contig(重叠群,即染色体的一些区域中毗邻DN***段重叠的情况)内。
第二步:如果其它条件一致,那么这个第二步——引物探针的设计就可以说是定量PCR成败的关键了,通过各方面经验的总结有以下几个基本的原则:总体原则* 先选择好探针,然后设计引物使其尽可能的靠近探针。
* 所选序列应该高度特异,尽量选择具有最小二级结构的扩增片段——这是因为二级结构会影响反应效率,而且还会阻碍酶的扩增。
建议先进行二级结构检测,如果不能避免二级结构,那么就要相应提高退火温度。
* 扩增长度应不超过400bp,理想的最好能在100-150bp内,扩增片段越短,有效的扩增反应就越容易获得。
较短的扩增片段也容易保证分析的一致性。
* 保持GC含量在20%和80%之间,GC富含区容易产生非特异反应,从而会导致扩增效率的降低,以及出现在荧光染料分析中非特异信号。
定量PCR引物探针设计原则完整版

定量P C R引物探针设计原则Document serial number【NL89WT-NY98YT-NC8CB-NNUUT-NUT108】定量PCR引物、探针设计原则自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR 要求。
当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:目的基因查找比对→探针与引物设计→探针与引物合成→配置反应体系→反应参数→重复实验,优化条件→获得曲线数据,比对标准曲线→再重复验证。
第一步:在第一步目的基因查找比对过程中可以利用NCBIgenbank序列以及DNAstar等软件完成目的DNA或者RNA的查找与比对——这在分析测序报告的时候相信很多人操作过,这一步需要注意的就是要保证所分析的序列在一个contig(重叠群,即染色体的一些区域中毗邻DNA片段重叠的情况)内。
第二步:如果其它条件一致,那么这个第二步——引物探针的设计就可以说是定量PCR成败的关键了,通过各方面经验的总结有以下几个基本的原则:总体原则先选择好探针,然后设计引物使其尽可能的靠近探针。
所选序列应该高度特异,尽量选择具有最小二级结构的扩增片段——这是因为二级结构会影响反应效率,而且还会阻碍酶的扩增。
建议先进行二级结构检测,如果不能避免二级结构,那么就要相应提高退火温度。
扩增长度应不超过400bp,理想的最好能在100-150bp内,扩增片段越短,有效的扩增反应就越容易获得。
qPCR实时荧光定量PCR可编辑全文

1.1 基本原理
在实时荧光定量PCR中,对整个PCR反应扩增过程进行 了实时的监测和连续地分析扩增相关的荧光信号,随着反应时 间的进行,监测到的荧光信号的变化可以绘制成一条曲线。
在PCR反应早期,产生荧光的水平不能与背景明显地区别 ,而后荧光的产生进入指数期、线性期和最终的平台期。FQPCR可行性定量是在PCR扩增的指数期进行的。首先需设定 一定荧光信号的域值,一般这个域值是以PCR反应的前15个 循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是 3~15个循环的荧光信号的标准偏差的10倍。
1.2 化学反应原理
依据反应中所选荧光物质的不同,实时荧光定量PCR的 化学反应原理通常分为荧光染料和荧光探针两种:
(1) SYBR荧光染料:SYBR是一种与双链DNA小沟结合的染料 。在PCR反应体系中,加入过量SYBR荧光染料, SYBR荧光 染料特异性掺入DNA双链后,发射荧光信号,而不掺入链中的 SYBR染料分子不会发射任何荧光信号,从而保证荧光信号 的增加与PCR产物的增加完全同步。
(1)特异性好。使用针对靶序列设计的特异性探针对定量分子 进行识别,具有很高的准确性。同时,靶序列由引物和探针 双重控制,特异性好、假阳性低。
(2)灵敏度高。荧光定量PCR检测技术是综合了PCR测灵敏度。
(3)线性关系好、线性范围宽。由于荧光信号的产生和每次扩 增产物成一一对应的关系,通过荧光信号的检测可以直接 对产物进行定量。
1.实时荧光定量PCR技术原理及特点
实时荧光定量PCR是将荧光能量传递技术 (fluorescenceresonanceenergytransfer,FRET)应用于常规 PCR仪中,指在PCR反应体系中加入荧光基团或荧光染料,利用 荧光信号积累,从而实时监测整个PCR过程,最后通过标准曲线 对未知模板浓度进行定量分析的方法 。
实时荧光定量PCR-TaqMan探针法及设计原则

Taqman MGB 探针设计
探针的5’端避免出现G,即使探针水解为单个碱基,与报告 基团相相连的G碱基仍可淬灭基团的荧光信号。
05 04 03
Tm值应为65-67℃。 尽量缩短Taqman MGB探针,但探针长度不少于碱基,应避免出现4个或 4个以上的G重复出现。
性; • 探针的DNA折叠和二级结构;尽量避开二级结构; • Tm值在65-70℃,通常比引物TM值高5-10℃(至少要5℃),
以确保在退火过程中探针先于引物与目的片段结合,GC含量在 40%-70%; • 探针的5’端应避免使用G鸟嘌呤——因为5'G会有淬灭作用, 而且即使是被切割下来还会存在淬灭作用; • 整条探针中,碱基C的含量要明显高于G的含量——G含量高会 降低反应效率,这时就应选择配对的另一条链作为探针; • 为确保引物探针的特异性,最好将设计好的序列在blast中核实 一次,如果发现有非特异性互补区,建议重新设计引物探针。
实时荧光定量PCR——TaqMan探针法
TaqMan技术引物设计原则
• 序列选取应在基因的保守区段; • 避免引物自身或与引物之间形成4个或4个以上连续配对,避免引
物自身形成环状发卡结构; • 典型的引物18到24个核苷长。引物需要足够长,保证序列独特性,
并降低序列存在于非目的序列位点的可能性。 • Tm值在55-65℃,GC含量在40%-60%; • 引物之间的TM相差避免超过2℃; • 引物的3’端避免使用碱基A,引物的3’端避免出现3个或3个以
上连续相同的碱基; • 为避免基因组的扩增,引物设计最好能跨两个外显子; • Taqman探针技术要求片段长度在50bp-150bp; • 引物末端(最后5个核苷酸)不能有超过2个的G和C。
两种定量分析方法的比较及Taqman探针引物设计原则

两种定量分析⽅法的⽐较及Taqman探针引物设计原则两种定量分析⽅法的⽐较及Taqman 探针、引物设计原则遗传物质DNA ⾸先要把所携带的遗传信息转录成为信使RNA (mRNA ),携带遗传信息的mRNA 从细胞核进⼊到细胞质中与核糖体结合,在核糖体中mRNA 携带的遗传信息被翻译成为多肽,多肽经过进⼀步加⼯后变成蛋⽩质,⾄此遗传物质DNA 完成了表达过程。
期间的转录过程是基因表达中⾮常重要的调节步骤,所转录的mRNA 的多少直接影响着相关最终蛋⽩质的多少,所以通过对细胞内某条基因mRNA 含量多少的分析,就能⼤致判断出该条基因的表达是否活跃。
定量PCR 仪是在普通PCR 仪的基础上加装了荧光激发装臵和荧光检测装臵,PCR 扩增和检测同时进⾏;在PCR 反应体系中加⼊荧光基团,利⽤荧光信号的积累实时监测整个PCR 进程,最后通过标准曲线对未知模板进⾏定量分析。
该技术于1996年由美国Applied Biosystems 公司推出,由于该技术不仅实现了PCR 从定性到定量的飞跃,⽽且与常规PCR 相⽐,它具有特异性更强、有效解决PCR 污染问题、⾃动化程度⾼等特点,⽬前已得到⼴泛应⽤。
定量PCR 常⽤的三个常⽤概念扩增曲线、荧光阈值、Ct 值扩增曲线:反映PCR 循环次数和荧光强度的曲线,定量PCR 仪每次轮PCR 扩增都会⾃动记录荧光强度的变化荧光阈值:样本的荧光背景值和阴性对照的荧光值,⼿动设臵的原则要⼤于样本的荧光背景值和阴性对照的荧光最⾼值,同时要尽量选择进⼊指数期的最初阶段,并且保证回归系数⼤于0.99。
CT 值: PCR 扩增过程中,扩增产物的荧光信号达到设定的阈值时所经过的扩增循环次数。
扩增曲线阈值及CT 值荧光定量PCR 的数学原理理想的PCR 反应:X=X0*2n⾮理想的PCR 反应:X=X0* (1+Ex)n(n :扩增反应的循环次数;X :第n 次循环后的产物量;X0:初始模板量;Ex :扩增效率)在扩增产物达到阈值线时: C(t) valueXCt=X0 (1+Ex)Ct =M (1)XCt:荧光扩增信号达到阈值强度时扩增产物的量,在阈值线设定以后,它是⼀个常数,我们设为M⽅程式(1)两边同时取对数得:log M=log X0 (1+Ex)Ct (2)整理⽅程式(2)得:log X0= - log(1+Ex) *Ct+ log M (3)由此可见,log X0浓度与循环数呈线性关系,根据样品扩增达到域值的循环数即Ct值就可计算出样品中所含的该基因的初始模板量。
实时荧光定量pcr技术原理与应用

实时荧光定量pcr技术原理与应用实时荧光定量PCR技术原理与应用一、引言实时荧光定量PCR技术(Real-time quantitative PCR,qPCR)是一种基于荧光信号的PCR方法,可以实时监测PCR反应的进程。
相比传统的末端定量PCR方法,qPCR具有更高的灵敏性、准确性和可靠性。
本文将介绍qPCR的原理和应用。
二、原理qPCR利用荧光探针在PCR过程中发出的荧光信号来定量PCR产物的数量。
其原理基于PCR技术,但在反应过程中引入了荧光探针。
在PCR反应的扩增过程中,荧光探针与靶DNA序列特异性结合,通过荧光信号的增强来检测靶DNA的数量。
qPCR的主要荧光探针有两种:TaqMan探针和SYBR Green探针。
TaqMan探针是一种双标记探针,它包含一个荧光染料和一个荧光淬灭剂。
在PCR反应中,TaqMan探针与靶DNA序列结合后,酶的活性将荧光染料释放出来,产生荧光信号。
SYBR Green探针是一种非特异性结合荧光染料,其荧光信号与PCR产物的数量成正比。
三、应用1. 基因表达分析qPCR广泛应用于基因表达分析。
通过检测靶基因的mRNA水平,可以评估基因的表达情况。
qPCR可以检测低表达基因和高表达基因之间的差异,并对基因调控进行定量研究。
这对于研究基因功能、诊断疾病以及药物开发具有重要意义。
2. 病原体检测qPCR在病原体的检测和诊断中起着重要作用。
通过设计特异性引物和荧光探针,可以快速、准确地检测病原体的DNA或RNA。
例如,在临床诊断中,qPCR被广泛用于检测病毒、细菌、真菌等致病微生物。
3. 遗传疾病筛查qPCR也被用于遗传疾病的筛查。
通过检测患者的DNA样本,可以准确判断某些遗传疾病的患病风险。
例如,qPCR可以检测染色体异常、突变等遗传变异,为遗传咨询和疾病预防提供重要依据。
4. 肿瘤标记物检测qPCR在肿瘤标记物检测中也有广泛应用。
通过检测肿瘤相关基因的表达水平,可以评估肿瘤的发生和发展情况。
定量PCR 引物、探针设计原则

定量PCR 引物、探针设计原则自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR要求。
当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:目的基因查找比对→探针与引物设计→探针与引物合成→目的DNA第二步:保持GC)?典型的引物18到24个核苷长。
引物需要足够长,保证序列独特性,并降低序列存在于非目的序列位点的可能性。
但是长度大于24核苷的引物并不意味着更高的特异性。
较长的序列可能会与错误配对序列杂交,降低了特异性,而且比短序列杂交慢,从而降低了产量。
Tm值在55-65℃(因为60℃核酸外切酶活性最高),GC含量在40%-60%引物之间的TM相差避免超过2℃引物的3’端避免使用碱基A,引物的3’端避免出现3个或3个以上连续相同的碱基为避免基因组的扩增,引物设计最好能跨两个外显子。
Taqman探针技术要求片段长度在50bp-150bp?引物末端(最后5个核苷酸)不能有超过2个的G和C。
?探针设计原则探针位置尽可能地靠近上游引物探针长度应在15-45bp(最好是20-30bp),以保证结合特异性检测探针的DNA折叠和二级结构Tm值在65-70℃,通常比引物TM值高5-10℃(至少要5℃),GC含量在40%-70%探针的5’端应避免使用G鸟嘌呤——因为5'G会有淬灭作用,而且即使是被切割下来还会存在淬灭作用。
整条探针中,碱基C的含量要明显高于G的含量——G含量高会降低反应效率,这时就应选择配对的另一条链作为探针。
实时荧光定量PCR技术的原理及应用-注意事项-适合菜鸟入门

实时荧光定量PCR技术的原理及应用-注意事项-适 合菜鸟入门
实验中污染的防控
实时荧光定量PCR技术的原理及应用-注意事项-适 合菜鸟入门
1、荧光定量PCR实验中污染源的分析
(1)、PCR产物引起的污染 在检测过程中,荧光定量PCR产物都是封闭在 PCR管中的,不存在开管检测,所以有污染的 话,最有可能是因为电泳检测荧光定量PCR产 物时引起的污染,只要将电泳室与PCR加样室 隔离开,就能有效的避免PCR产物的污染。
实时荧光定量PCR技术的原理及应用-注意事项-适 合菜鸟入门
• 优点 − 特异性高,可准确定量 − 灵敏度高 − 设计不同标记的探针,可进 行多重检测
• 缺点 − 一个探针只适用于一个目标 − 价格较高 − 探针设计较繁琐
实时荧光定量PCR技术的原理及应用-注意事项-适 合菜鸟入门
实时荧光定量PCR技术的原理及应用-注意事项-适 合菜鸟入门
TaqMan作用机理
SYBR Green I法溶解曲线分析
实时荧光定量PCR技术的原理及应用-注意事项-适 合菜鸟入门
荧光染料的特点及应用
(1)、能够与所有的核酸双链结合,受激后产生荧光,其荧光的强度与双链核 酸的含量及长度成正比,因此本底较高;
(2)、可做双链核酸的熔解曲线分析;
(3)、SYBR GreenⅠ染料本身价格便宜,但是做荧光定量PCR时对引物设计 的要求很高;对Taq酶要求较高,最好是HotStar Taq酶,或者操作时需要 严格的冷启动,冰上操作,否则引物二聚体及非特异性扩增会严重干扰结 果的准确性,尤其是模板含量较低时;
实时荧光定量PCR —一科学准确的定量方法
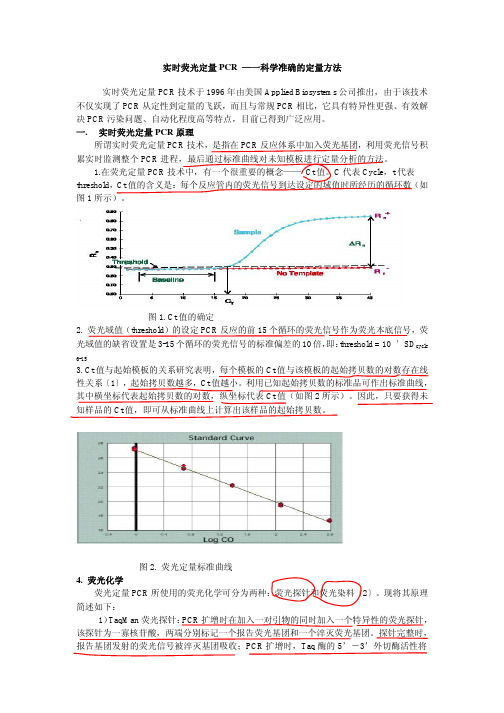
实时荧光定量PCR —一科学准确的定量方法实时荧光定量PCR技术于1996年由美国Applied Biosystems公司推出,由于该技术不仅实现了PCR从定性到定量的飞跃,而且与常规PCR相比,它具有特异性更强、有效解决PCR污染问题、自动化程度高等特点,目前已得到广泛应用。
一. 实时荧光定量PCR原理所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
1.在荧光定量PCR技术中,有一个很重要的概念—— Ct值。
C代表Cycle,t代表threshold,Ct值的含义是:每个反应管内的荧光信号到达设定的域值时所经历的循环数(如图1所示)。
图1. Ct值的确定2. 荧光域值(threshold)的设定PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3-15个循环的荧光信号的标准偏差的10倍,即:threshold = 10 ′ SD cycle6-153. Ct值与起始模板的关系研究表明,每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系〔1〕,起始拷贝数越多,Ct值越小。
利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代表Ct值(如图2所示)。
因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。
图2. 荧光定量标准曲线4. 荧光化学荧光定量PCR所使用的荧光化学可分为两种:荧光探针和荧光染料〔2〕。
现将其原理简述如下:1)TaqMan荧光探针:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。
探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR 产物形成完全同步。
实时荧光定量PCR-TaqMan探针法及设计原则

03
Taqman探针的合成与制备
探针的合成方法
化学合成法
通过化学反应将荧光基团和淬灭基团分别连接到DNA或RNA的5'和3'末端,形成 Taqman探针。
酶促合成法
利用DNA聚合酶将荧光基团和淬灭基团分别添加到DNA或RNA的特定位置,形成 Taqman探针。
探针的质量检测与纯化
质量检测
通过电泳、质谱和光谱分析等方法检测探针 的长度、荧光基团和淬灭基团的数量和位置 ,以及探针的纯度。
定义与原理
定义
实时荧光定量PCR-Taqman探针法是 一种基于荧光信号的实时监测技术, 用于定量分析DNA或RNA的拷贝数。
原理
通过在Taq酶催化下,利用荧光染料 标记的特异性探针与待测核酸进行特 异性结合,在PCR循环过程中实时监 测荧光信号的增强,从而实现对核酸 的定量分析。
发展历程与现状
02
Taqman探针的设计原则
探针的长度与结构
长度
通常为20-30bp,确保特异性并减少非特异性扩增。
结构
由报告基团、淬灭基团和连接臂组成,连接臂长度一般为5-6个脱氧核糖核苷酸。
探针的特异性
针对目标序列
确保探针与目标序列完全匹配,避免 交叉反应。
序列选择
选择基因特异性区域,避免基因组中 的高变区。
04
Taqman探针在实时荧光定量 PCR中的应用
样本处理与PCR反应体系建立
样本处理
确保样本质量,去除杂质和抑制剂,提 取高质量的DNA或RNA。
Taqman探针设计
根据目标基因序列设计Taqman探针, 确保探针的特异性和荧光信号的稳定
性。
引物设计
根据目标基因序列设计特异性引物, 确保引物与模板的结合效率和特异性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
• 整条探针中,碱基C的含量要明显高于G的含量——G含量高会 降低反应效率,这时就应选择配对的另一条链作为探针;
• 为确保引物探针的特异性,最好将设计好的序列在blast中核实 一次,如果发现有非特异性互补区,建议重新设计引物探针。
个或4个以上的G重复出现。 • 原则上MGB探针只要有一个碱基突变,MGB探针就会检
测到(MGB探针将不会与目的片段杂交,不产生荧光信号)。
Zhejiang Provincial Key Lab of Medical Genetics
4
Zhejiang Provincial Key Lab of Medical Genetics
2
TaqMan探针设计原则
• 先选择好探针,然后设计引物使其尽可能的靠近探针;
• 探针长度应在15-45bp(最好是20-30bp),以保证结合特异性;
• 探针的DNA折叠和二级结构;尽量避开二级结构;
• Tm值在65-70℃,通常比引物TM值高5-10℃(至少要5℃), 以确保在退火过程中探针先于引物与目的片段结合,GC含量在 40%-70%;
• Tm值在55-65℃,GC含量在40%-60%;
• 引物之间的TM相差避免超过2℃;
• 引物的3’端避免使用碱基A,引物的3’端避免出现3个或3 个以上连续相同的碱基;
• 为避免基因组的扩增,引物设计最好能跨两个外显子;
• Taqman探针技术要求片段长度在50bp-150bp;• 引物末端(最后5个苷酸)不能有超过2个的G和C。
Zhejiang Provincial Key Lab of Medical Genetics
3
Taqman MGB 探针设计
• 探针的5’端避免出现G,即使探针水解为单个碱基,与报 告基团相相连的G碱基仍可淬灭基团的荧光信号。
• Tm值应为65-67℃。 • 尽量缩短Taqman MGB探针,但探针长度不少于13bp。 • 尽量避免出现重复的碱基,尤其是G碱基,应避免出现4
实时荧光定量PCR——TaqMan探针法 •
Zhejiang Provincial Key Lab of Medical Genetics
1
TaqMan技术引物设计原则
• 序列选取应在基因的保守区段;
• 避免引物自身或与引物之间形成4个或4个以上连续配对, 避免引物自身形成环状发卡结构;
• 典型的引物18到24个核苷长。引物需要足够长,保证序 列独特性,并降低序列存在于非目的序列位点的可能性。