VASP自旋轨道耦合计算错误汇总
VASP自旋轨道耦合计算错误汇总

VASP自旋轨道耦合计算错误汇总静态计算时,报错:VERY BAD NEWS!Internal内部error in subroutine子程序IBZKPT:Reciprocal倒数的lattice and k-lattice belong to different class of lattices.Often results are still useful (48)INCAR参数设置:对策:根据所用集群,修改INCAR中NPAR。
将NPAR=4变成NPAR=1,已解决!错误:sub space matrix类错误报错:静态和能带计算中出现警告:WARNING:Sub-Space-Matrix is not hermitian共轭in DAV结构优化出现错误:WARNING:Sub-Space-Matrix is not hermitian in DAV4-4.681828688433112E-002对策:通过将默认AMIX=0.4,修改成AMIX=0.2(或0.3),问题得以解决。
以下是类似的错误:WARNING:Sub-Space-Matrix is not hermitian in rmm-3.00000000000000RMM:22-0.167633596124E+02-0.57393E+00-0.44312E-0113260.221E+00BRMIX:very serious problems the old and the new charge density differ old charge density:28.00003new28.060930.111E+00错误:WARNING:Sub-Space-Matrix is not hermitian in rmm-42.5000000000000ERROR FEXCP:supplied Exchange-correletion table is too small,maximal index:4794错误:结构优化Bi2Te3时,log文件:WARNING in EDDIAG:sub space matrix is not hermitian1-0.199E+01RMM:2000.179366581305E+01-0.10588E-01-0.14220E+007180.261E-01BRMIX:very serious problems the old and the new charge density differ old charge density:56.00230new124.70394 66F=0.17936658E+01E0=0.18295246E+01d E=0.557217E-02curvature:0.00expect dE=0.000E+00dE for cont linesearch0.000E+00ZBRENT:fatal error in bracketingplease rerun with smaller EDIFF,or copy CONTCAR to POSCAR and continue但是,将CONTCAR拷贝成POSCAR,接着算静态没有报错,这样算出来的结果有问题吗?对策1:用这个CONTCAR拷贝成POSCAR重新做一次结构优化,看是否达到优化精度!对策2:用这个CONTCAR拷贝成POSCAR,并且修改EDIFF(目前参数EDIFF=1E-6),默认为10-4错误:WARNING:Sub-Space-Matrix is not hermitian in DAV1-7.626640664998020E-003网上参考解决方案:对策1:减小POTIM:IBRION=0,标准分子动力学模拟。
报错解决

设置:初始值收敛值结果AMIX =0.0100;BMIX =0.0001 AMIX = 0.01; BMIX = 0.00 计算无误AMIX = 0.1000;BMIX = 0.0010 AMIX = 0.10; BMIX = 0.00 计算无误AMIX =0.20; BMIX = 0.01 AMIX =0.20; BMIX = 0.01 计算无误AMIX=0.2、BMIX=0.001 AMIX=0.2、BMIX=0.001 计算无误AMIX=0.3、BMIX=0.1 AMIX=0.3、BMIX=0.1 计算无误AMIX=0.4 AMIX = 0.40; BMIX = 1.00 静态log: WARNING in EDDRMM: call toZHEGV failed, returncode = 6 3 **,能带一样AMIX=0.02 AMIX = 0.02; BMIX = 1.00 计算无误AMIX=0.1 AMIX = 0.10; BMIX = 1.00 静态log: WARNING in EDDRMM: call toZHEGV failed, returncode = 6 3 **,能带一样AMIX=0.3 AMIX = 0.30; BMIX = 1.00 静态log: WARNING in EDDRMM: call toZHEGV failed, returncode = 6 3 **,能带一样BMIX=0.0001 AMIX = 0.40; BMIX = 0.00 计算无误以上参数设置,得到的能带图都一样,如下图:综上:设置AMIX=0.2(或0.3),BMIX默认(省事,等于1.0),可以保证计算过程无误。
还需进一步调整其他参数,算出正确的能带。
警告:算1QL弛豫、静态、能带时,都有这个提示:ADVICE TO THIS USER RUNNING 'V ASP/V AMP' (HEAR YOUR MASTER'S VOICE ...): You have a (more or less)'small supercell' and for smaller cells it is recommended to use the reciprocal-space projection scheme! The real space optimization is not efficient for small cells and it is also less accurate ... Therefore set LREAL=.FALSE. in the INCAR file对策:对于较小的晶胞(原子数小于20),设置LREAL=.FALSE.,计算结果比较精确。
VASP自旋轨道耦合计算错误汇总

VASP自旋轨道耦合计算错误汇总自旋轨道耦合是描述电子自旋和轨道运动之间相互作用的物理概念。
在VASP计算中,自旋轨道耦合是通过GGA+U方法处理的,但在实际计算中可能会出现一些错误。
下面是一些可能导致VASP自旋轨道耦合计算错误的原因及解决方案的汇总。
1.参数设置错误:在VASP计算中,轨道耦合的计算需要将INCAR文件中的参数设置正确。
首先,需要将ISPIN参数设置为2,以便考虑自旋极化。
其次,需要通过LDAU参数将自旋-轨道耦合效应的影响引入计算中。
在计算过程中,可以尝试不同的U值,并观察计算结果的变化。
2.缺乏足够的k点网格:自旋轨道耦合计算需要在倒空间中计算,因此需要足够高的k点网格密度。
如果k点网格密度过低,可能会导致计算结果不准确。
解决方法是增加k点网格密度,可以通过增加KPOINTS文件中的MP或MONKHORST参数来实现。
3.收敛条件设定不合理:VASP计算中,自旋轨道耦合的计算需要满足一定的收敛条件。
如果计算结果不收敛,则可能需要调整计算过程中的一些参数。
可以尝试增加ENCUT参数来提高计算精度,减小EDIFF参数来提高计算收敛性。
同时,还可以尝试改变电荷密度和波函数的混合策略,选择更合适的算法来解决计算问题。
4.初始结构选择不合理:不合理的初始结构选择可能导致计算结果不准确。
建议根据实验已知的结构或先前的计算结果来选择初始结构,并合理设置INCAR文件中的ISIF参数来优化结构。
5.系统对称性的处理错误:自旋轨道耦合计算过程中,VASP通常假设系统具有一定的对称性,因此在计算中会利用结构的对称性进行优化。
如果对称性处理错误,可能会导致计算结果的不准确。
建议在计算前进行空间群和点群对称性的分析,并在INCAR文件中正确设置ISYM参数来处理对称性。
总之,VASP自旋轨道耦合计算错误的原因有很多,可能是参数设置错误、收敛条件设定不合理、初始结构选择不合理、系统对称性处理错误等。
Vasp出错信息及解决方法

Vasp出错信息及解决方法使用vasp的过程中难免会出现一些警告、报错信息,现在将这样的信息和一些解决办法列出来。
欢迎大家一起讨论,把解决问题的办法记录下来,让我们在一起解决问题中前行。
欢迎补充~讨论~1.warning:the distance between some ions is very small一种可能的错误是因为:在poscat中的坐标类型(Direct 或者 car)没有顶格写(也就是说开头空格),如果你是笛卡尔坐标的话,它会识别为direct坐标,从而出现这样的警告。
2.WARNING: CHECK: NIOND is too smalldyna.F中的NIOND 默认值是 256,如果体系中的原子数大于256时将出现这个警告信息。
所以解决办法就是:将 NIOND 的值改成一个大于你计算体系的原子数,然后重新编译一下。
3.ERROR: there must be 1 or 3 items on line 2 of POSCARFORTRAN STOP造成这个错误的原因是上传POSCAR文件的时候是windows的格式,传到unix 系统下造成回车符不对。
最简单的解决方法为:试着在unix下直接写一个。
或者dos2unix filename4.VASP计算出现Segmentation Fault而终止运算在makefile中 FFLAGS 后面添加-heap-arrays,例如:FFLAGS = -FR -lowercase -assume byterecl -heap-arrays5.WARNING: aliasing errors must be expected set NGX to 154 to avoid them WARNING: aliasing errors must be expected set NGY to 158 to avoid them WARNING: aliasing errors must be expected set NGZ to 126 to avoid themaliasing errors are usually negligible using standard VASP settingsand one can safely disregard these warnings当设置PREC=LOW,NORMAL时会出现这样的警告信息,当PREC=high和accurate时就没有了,或者直接将NGX,NGY,NGZ设置成警告信息中给出的数值即可。
VASP磁性计算总结篇

在线说明书整理出来的非线性磁矩和自旋轨道耦以下是从VASP合的计算说明。
非线性磁矩计算:和CHGCAR文件。
1)计算非磁性基态产生WAVECAR)然后INCAR中加上2ISPIN=2文件和CHGCAR11 !读取WAVECAR ICHARG=1 或LNONCOLLINEAR=.TRUE. MAGMOM=注意:①对于非线性磁矩计算,要在x, y 和 z方向分别加上磁矩,如MAGMOM = 1 0 0 0 1 0 !表示第一个原子在x方向,第二个原子的y方向有磁矩②在任何时候,指定MAGMOM值的前提是ICHARG=2(没有WAVECAR和CHGCAR文件)或者ICHARG=1 或11(有WAVECAR和CHGCAR文件),但是前一步的计算是非磁性的(ISPIN=1)。
磁各向异性能(自旋轨道耦合)计算:注意: LSORBIT=.TRUE. 会自动打开LNONCOLLINEAR= .TRUE.选项,且自旋轨道计算只适用于PAW赝势,不适于超软赝势。
.自旋轨道耦合效应就意味着能量对磁矩的方向存在依赖,即存在磁各向异性能(MAE),所以要定义初始磁矩的方向。
如下:LSORBIT = .TRUE.SAXIS = s_x s_y s_z (quantisation axis for spin)默认值: SAXIS=(0+,0,1),即x方向有正的无限小的磁矩,Z方向有磁矩。
要使初始的磁矩方向平行于选定方向,有以下两种方法:MAGMOM = x y z ! local magnetic moment in x,y,zSAXIS = 0 0 1 ! quantisation axis parallel to zorMAGMOM = 0 0 total_magnetic_moment ! local magnetic moment parallel to SAXIS (注意每个原子分别指定)SAXIS = x y z ! quantisation axis parallel to vector (x,y,z),如 0 0 1两种方法原则上应该是等价的,但是实际上第二种方法更精确。
自旋轨道耦合计算探索过程分析
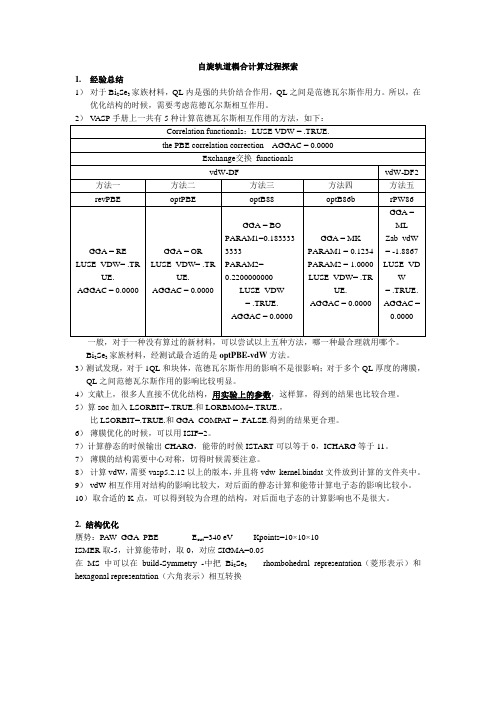
自旋轨道耦合计算过程探索1.经验总结1)对于Bi2Se3家族材料,QL内是强的共价结合作用,QL之间是范德瓦尔斯作用力。
所以,在优化结构的时候,需要考虑范德瓦尔斯相互作用。
一般,对于一种没有算过的新材料,可以尝试以上五种方法,哪一种最合理就用哪个。
Bi2Se3家族材料,经测试最合适的是optPBE-vdW方法。
3)测试发现,对于1QL和块体,范德瓦尔斯作用的影响不是很影响;对于多个QL厚度的薄膜,QL之间范德瓦尔斯作用的影响比较明显。
5)算soc加入LSORBIT=.TRUE.和LORBMOM=.TRUE.,比LSORBIT=.TRUE.和GGA_COMPAT = .FALSE.得到的结果更合理。
6)薄膜优化的时候,可以用ISIF=2。
7)计算静态的时候输出CHARG,能带的时候ISTART可以等于0,ICHARG等于11。
7)薄膜的结构需要中心对称,切得时候需要注意。
8)计算vdW,需要vasp5.2.12以上的版本,并且将vdw_kernel.bindat文件放到计算的文件夹中。
9)vdW相互作用对结构的影响比较大,对后面的静态计算和能带计算电子态的影响比较小。
10)取合适的K点,可以得到较为合理的结构,对后面电子态的计算影响也不是很大。
2. 结构优化赝势:PAW_GGA_PBE E cut=340 eV Kpoints=10×10×10ISMER取-5,计算能带时,取0,对应SIGMA=0.05在MS中可以在build-Symmetry -中把Bi2Se3 rhombohedral representation(菱形表示)和hexagonal representation(六角表示)相互转换图中黑色t 1、t 2、t 3基矢围成菱形原胞,用于计算块体,红色方框包含一个五元层 计算能带的布里渊区高对称点:块体:文献中倒空间高对称点坐标Г(0 0 0)-Z(π π π)-F(π π 0)-Г(0 0 0)-L(π 0 0), 根据正空间和倒空间坐标的转换关系,得到正空间中高对称点的坐标: Г(0 0 0)-Z(0.5 0.5 0.5)-F(0.5 0.5 0)-Г(0 0 0)-L(0 0 -0.5) KPOINTS 20 Line-mode Rec0.0 0.0 0.0 !Г 0.5 0.5 0.5 ! Z 0.5 0.5 0.5 ! Z 0.5 0.5 0.0 ! F 0.5 0.5 0.0 ! F 0.0 0.0 0.0 !Г 0.0 0.0 0.0 !Г 0.0 0.0 -0.5 ! L[通过比较结构,发现Ecut=580,KPOINTS=151515,得到的结构比较靠谱]3. 块体soc 的计算 文献能带结构图:块体(Bi 2Se 3-VASP-GGA-PAW-PBE )我们的结果(未考虑vdW+静态和能带都加soc计算结果与文献基本符合):4.薄膜的计算薄膜:Kpoints=10×10×1计算能带的K点和石墨烯(六角晶胞的)的K点一样:KPOINTS20Lone-modeRec0.66666667 0.33333333 0.0 !K0.0 0.0 0.0 !Г0.0 0.0 0.0 !Г0.5 0.0 0.0 !M考虑薄膜的对称性由MS六角结构,沿(001)方向切割,可以得到两种以Se原子作为表面原子的薄膜,如下图,分别为1QL和3QL的两种切法,右图比左图对称性要更好一些,这一区别在计算过程中会导致巨大的区别,我们通过比较,发现,只有右图的结果,才可以得到合理的结果,尤其是在多个QL的情况。
VASP自旋轨道耦合计算

VASP 自旋轨道耦合计算已有4532 次阅读2011-9-13 20:37|个人分类:VASP|系统分类:科研笔记将VASP 的makefile 文件中的 CPP 选项中的 -DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf 这4个选项去掉重新编译VASP才能计算自旋轨道耦合效应。
以下是从VASP在线说明书整理出来的非线性磁矩和自旋轨道耦合的计算说明。
非线性磁矩计算:1)计算非磁性基态产生WAVECAR和CHGCAR文件。
2)然后INCAR中加上ISPIN=2ICHARG=1 或 11 !读取WAVECAR和CHGCAR文件LNONCOLLINEAR=.TRUE.MAGMOM=注意:①对于非线性磁矩计算,要在x, y 和 z方向分别加上磁矩,如MAGMOM = 1 0 0 0 1 0 !表示第一个原子在x方向,第二个原子的y方向有磁矩②在任何时候,指定MAGMOM值的前提是ICHARG=2(没有WAVECAR和CHGCAR文件)或者ICHARG=1 或11(有WAVECAR和CHGCAR文件),但是前一步的计算是非磁性的(ISPIN=1)。
磁各向异性能(自旋轨道耦合)计算:注意: LSORBIT=.TRUE. 会自动打开LNONCOLLINEAR= .TRUE.选项,且自旋轨道计算只适用于PAW赝势,不适于超软赝势。
自旋轨道耦合效应就意味着能量对磁矩的方向存在依赖,即存在磁各向异性能(MAE),所以要定义初始磁矩的方向。
如下:LSORBIT = .TRUE.SAXIS = s_x s_y s_z (quantisation axis for spin)默认值: SAXIS=(0+,0,1),即x方向有正的无限小的磁矩,Z方向有磁矩。
要使初始的磁矩方向平行于选定方向,有以下两种方法:MAGMOM = x y z ! local magnetic moment in x,y,zSAXIS = 0 0 1 ! quantisation axis parallel to zorMAGMOM = 0 0 total_magnetic_moment ! local magnetic moment parallel to SAXIS (注意每个原子分别指定)SAXIS = x y z !quantisation axis parallel to vector (x,y,z),如 0 0 1两种方法原则上应该是等价的,但是实际上第二种方法更精确。
vasp计算报错解决

报错Error EDDDAV: Call to ZHEGV failed. Returncode = 7 1 8Serial version problems:Problem 1 : Error EDDDAV: Call to ZHEGV failed.Problem 2 : lib-4201 : UNRECOVERABLE library errorProblem 3 : WARNING in EDDRMM: call to ZHEGV failed, returncode = 6 3 9Parallel version problems:Will be added at some later dateProblem 1)The test calculation for copper system (benchmark.tar.gz, download from the VASP ftp-server) stops with an error message:"Error EDDDAV: Call to ZHEGV failed. Returncode = 9 1 8".The actual numbers change at the end of the error, but the message means that a LAPACK library call failed.Solution:The subroutine davidson.F must be handled with lower optimization settingAdd the following lines to the end of the VASP Makefile:davidson.o : davidson.F$(CPP)$(FC) $(FFLAGS) -O1 -c $*$(SUFFIX)(You remembered to use the TAB key instead of spaces with the second and the third line, right?)Problem 2)After lowering the davidson subroutine optimization level the calculation ends with another error:"lib-4201 : UNRECOVERABLE library error: Unable to find error message (check NLSPATH, file lib.cat)Encountered during a direct access unformatted READ from unit 21. Fortran unit 21 is connected to a direct unformatted unblocked file: "TMPCAR"/opt/gridengine/default/spool//compute-0-4/job_scripts/10170: line 31: 16997 Aborted (core dumped) ./vasp_path_serial >vasp_path_serial.out" Solution:Change/add the IWAVPR=10 line to your INCAR file. This is from the VASP manual, FAQ section, page 149"Question: I am running VASP on a SGI Origin, and the simple benchmark (benchmark.tar.gz) fails with lib-4201 : UNRECOVERABLE library error READ operation tried to read past theend-of-record.Encountered during a direct access unformatted READ from unit 21 Fortran unit 21 is connected to a direct unformatted unblocked file: "TMPCAR" IOT TrapAbort (core dumped)Answer: VASP extrapolates the wave functions between molecular dynamics time steps. To store the wave functions of the previous time steps either a temporary scratch file (TMPCAR) is used (IWAVPR=1-9) or large work arrays are allocated (IWAVPR=11-19).On the SGI, the version that uses a temporary scratch file does not compile correctly, and hence the user has to set IWAVPR to 10."Problem 3)When running the Hg benchmark (bench.Hg.tar.gz), the OUTCAR file has numerous lines saying:"WARNING in EDDRMM: call to ZHEGV failed, returncode = 6 3 9" Solution:This issue is addressed in the VASP support forum(http://cms.mpi.univie.ac.at/vasp-forum/forum_viewtopic.php?3.214) Short summary is given here. Possible reasons (this may, once again, be connected to failures calling LAPACK routines):1) The diagonalization algorithm is not stable for your system-> Change ALGO = Normal or ALGO = Fast in your INCAR file2) Your geometry is not reasonable. Maybe your initial structure or the algorithm handling ion relaxation is giving a bad structure-> Switch to a different ion relaxation scheme (IBRION line in your INCAR)-> Reduce the step size of the first step by reducing the POTIM value in your INCAR你的几何学不合理。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
VASP自旋轨道耦合计算错误汇总静态计算时,报错:VERY BAD NEWS!Internal内部error in subroutine子程序IBZKPT:Reciprocal倒数的lattice and k-lattice belong to different class of lattices.Often results are still useful (48)INCAR参数设置:对策:根据所用集群,修改INCAR中NPAR。
将NPAR=4变成NPAR=1,已解决!错误:sub space matrix类错误报错:静态和能带计算中出现警告:WARNING:Sub-Space-Matrix is not hermitian共轭in DAV结构优化出现错误:WARNING:Sub-Space-Matrix is not hermitian in DAV4-4.681828688433112E-002对策:通过将默认AMIX=0.4,修改成AMIX=0.2(或0.3),问题得以解决。
以下是类似的错误:WARNING:Sub-Space-Matrix is not hermitian in rmm-3.00000000000000RMM:22-0.167633596124E+02-0.57393E+00-0.44312E-0113260.221E+00BRMIX:very serious problems the old and the new charge density differ old charge density:28.00003new28.060930.111E+00错误:WARNING:Sub-Space-Matrix is not hermitian in rmm-42.5000000000000ERROR FEXCP:supplied Exchange-correletion table is too small,maximal index:4794错误:结构优化Bi2Te3时,log文件:WARNING in EDDIAG:sub space matrix is not hermitian1-0.199E+01RMM:2000.179366581305E+01-0.10588E-01-0.14220E+007180.261E-01BRMIX:very serious problems the old and the new charge density differ old charge density:56.00230new124.70394 66F=0.17936658E+01E0=0.18295246E+01d E=0.557217E-02curvature:0.00expect dE=0.000E+00dE for cont linesearch0.000E+00ZBRENT:fatal error in bracketingplease rerun with smaller EDIFF,or copy CONTCAR to POSCAR and continue但是,将CONTCAR拷贝成POSCAR,接着算静态没有报错,这样算出来的结果有问题吗?对策1:用这个CONTCAR拷贝成POSCAR重新做一次结构优化,看是否达到优化精度!对策2:用这个CONTCAR拷贝成POSCAR,并且修改EDIFF(目前参数EDIFF=1E-6),默认为10-4错误:WARNING:Sub-Space-Matrix is not hermitian in DAV1-7.626640664998020E-003网上参考解决方案:对策1:减小POTIM:IBRION=0,标准分子动力学模拟。
通过POTIM控制步长。
POTIM:当IBRION=1,2或3时,是力的一个缩放常数(相当于确定原子每步移动的大小),默认值为0.5。
对策2:改IBRION=1,采用准牛顿算法来优化原子的位置。
原IBRION=2,采用共轭梯度算法来优化原子的位置对策3:修改ISMEAR对策4:换成CG弛豫(共轭梯度算法)IBRION=2(决定结构优化过程中,原子如何移动或弛豫)IBRION=2离子是否运动,1不运动但做NSW外循环。
0动力学模拟,1准牛顿法离子弛豫2CG法离子弛豫,3采用衰减二阶运动方程离子弛豫,INCARrelax中设置IBRION=2,未解决!对策5:用的CG算符,出现的错误是CG算符不能算,在INCAR中加上IALG=Fast(电子优化采用blocked Davidson 方法[IALGO=38:IALG=Normal]和RMM-DIIS算法[IALGO=48:IALG=Very_Fast]混合)试一试IALG=Fast(两种方法混用)IALG=Very_Fast(等价于IALGO=48)IALG=Normal(等价于IALGO=38)INCAR中加上IALG=Fast已解决!(1QL、2QL已解决,3QL以上未解决)VASP FORUM:the error is due to a LAPCK call(ZHEGV):ZHEGV computes all the eigenvalues本征值,and optionally随意地,the eigenvectors of a complex generalized Hermitian-definite eigenproblem.there may be several reasons for that error:1)the RMM-DIIS diagonalisation algorithm is not stable for your specific setup of the calculation.-->use ALGO=Normal (blocked Davidson)or ALGO=Fast(5steps blocked Davidson,RMM-DIIS)用ALGO=Normal IALGO=48或者ALGO=Fast2)a)maybe your input geometry was not reasonable(error occurs at the very first ionic step,please have a look for the geometry data of your run in OUTCAR)orb)the last ionic relaxation step lead to an unreasonable geometry(compare the input and output geometries of the last ionic relaxation steps in XDATCAR).In that case(2b)it can be helpful to-->switch to a different relaxation algorithm(IBRION-tag)-->reduce the step size of the first step by setting POTIM smaller than the default value改变IBRION,减少步长POTIM3)The installation of the LAPACK on your machine was not done properly:use the LAPACK which is delivered with the code (vasp.4.lib/lapack_double.o)4)If the error persist although you switched to the Davidson algorithm:on some architectures(especially SGI)some LAPACK routines are not working properly.However,it is possible to avoid the usage of the ZHEGV subroutine by commenting the line #define USE_ZHEEVX in davidson.F,subrot.F,and wavpre_noio.F and recompiling VASP.关于Mixing方法的调试:针对这类错误:DAV:13-0.242323773333E+030.98155E+02-0.87140E+01488320.949E+01BRMIX:very serious problems the old and the new charge density differ old charge density:252.00012new252.299790.809E+01WARNING:Sub-Space-Matrix is not hermitian in DAV90.133520549894753.....解决办法只需调整AMIX,BMIX的值,把他们设置小一些。
Mixing方法:IMIX=type of mixing混合、混频,AMIX=linear mixing parameter,AMIN=minimal mixing parameter,BMIX=cutoff wave vector for Kerker mixing scheme,AMIX_MAG=linear mixing parameter for magnetization,BMIX_MAG=cutoff wave vector for Kerker mixing scheme for mag,WC=weight factor for each step in Broyden mixing scheme,INIMIX=type of initial for each step in Broyden mixing scheme,MIXPRE=type of preconditioning in Broyden mixing scheme,MAXMIX=maximum number steps stored in Broyden mixer.一般采用其默认值,除非在电子迭代难以收敛的情况,才手动设置AMIX和BMIX等参数值。
】对策:grep AMIX OUTCARAMIX=0.40;BMIX= 1.00AMIX_MAG= 1.60;BMIX_MAG= 1.00initial mixing is a Kerker type mixing with AMIX=0.4000and BMIX=1.0000设置:初始值收敛值结果AMIX=0.0100;BMIX=0.0001AMIX=0.01;BMIX=0.00计算无误AMIX=0.1000;BMIX=0.0010AMIX=0.10;BMIX=0.00计算无误AMIX=0.20;BMIX=0.01AMIX=0.20;BMIX=0.01计算无误AMIX=0.2、BMIX=0.001AMIX=0.2、BMIX=0.001计算无误AMIX=0.3、BMIX=0.1AMIX=0.3、BMIX=0.1计算无误AMIX=0.4AMIX=0.40;BMIX=1.00静态log:WARNING in EDDRMM:call toZHEGV failed,returncode=63**,能带一样AMIX=0.02AMIX=0.02;BMIX=1.00计算无误AMIX=0.1AMIX=0.10;BMIX=1.00静态log:WARNING in EDDRMM:call toZHEGV failed,returncode=63**,能带一样AMIX=0.3AMIX=0.30;BMIX=1.00静态log:WARNING in EDDRMM:call toZHEGV failed,returncode=63**,能带一样BMIX=0.0001AMIX=0.40;BMIX=0.00计算无误以上参数设置,得到的能带图都一样,如下图:综上:设置AMIX=0.2(或0.3),BMIX默认(省事,等于1.0),可以保证计算过程无误。