药典分析方法确认与验证
药物的分析方法与验证

• (4)对于易水解的酯类药物,可采用定量过量加入碱滴定液水解后,用酸 滴定液回滴的剩余滴定法。如氯贝丁酯、阿司匹林片的含量测定。
• 示例: 氯贝丁酯(Clofibrate,降血脂药)的含量测定
• 取本品2 g,精密称定,置锥形瓶中,加中性乙醇(对酚酞指示液显中 性)10mL与酚酞指示液数滴,滴加氢氧化钠滴定液(0.lmol/L)至显粉红色,再 精密加氢氧化钠滴定液(0.5mol/L)20mL,加热回流1小时至油珠完全消失, 放冷,用新沸过的冷水洗涤冷凝管,洗液并入锥形瓶中,加酚酞指示液数滴, 用盐酸滴定液(0.5mol/L)滴定,并将滴定的结果用空白试验校正。每1mL氢 氧化钠滴定液(0.5mol/L)相当于121.4mg的C12H15ClO3。
第二法:测定有机弱酸及其盐类(异维A酸、苄氟噻嗪、环吡酮等)
• 第二法:除另有规定外,精密称取供试品适量 [约消耗碱滴定液(0.1 mol/L)8 mL],加各品种 项下规定的溶剂使溶解,再加规定的指示液 1~2滴,用规定的碱滴定液(0.1 mol/L)滴定。 终点颜色应以电位滴定时的突跃点为准,并将 滴定的结果用空白试验校正。
• 为消除这些酸性物质的干扰,采用两步滴定法:
第一步—中和供试品中的各种酸性杂质,消耗的氢氧化钠量较少,故采用较 稀的碱滴定液(0.1mol/L)
第二步—水解后返滴定,加入定量过量较浓的碱滴定液(0.5mol/L),进行加热 回流1h,以保证水解完全,并可以准确滴定剩余量。
(三)氧化还原滴定法
• 该法是以氧化还原反应为基础的滴定分析法,既可直接测定具有氧化性或还 原性的物质,又可间接测定不具有氧化性或还原性的物质。
药典分析方法确认与验证

通过分析含有高于和低于必需的检测水平的、已知待分析物浓度的样品,以显示检测限度足够低。例如,如果必需检测浓度在0.1%的杂质,则应当证明该分析规程将可靠地检测在这个水平的杂质。
药典分析方法确认与验证
D
精密度、线性等内容的验证,证明修订后的分析方法的合理可行。
当变更达到一定程度时,则需要完整的验证。如分析方法完全改变,则应按新方法进行完整的验证。
方法验证的一般原则:
通常情况下,分析方法需进行方法验证。对于仅需按照实验室日常测试操作步骤即可测定的检验项目不需要进行验证,如外观、崩解时限、密度、重量、pH值、灰分、装量等。
对于展示出背景噪音的仪器分析规程,ICH文件描述了一个通用方法,用来比较从以下样品测得的信号,这些样品分别为含已知低浓度被分析物的样品和空白样品。这样就确立了能够可靠检测的待分析物的最低浓度。可接受的典型信噪比是2:1或3:1。其他方法取决于校正曲线斜率的测定和响应值的标准差。
5QUANTITATION LIMIT定量限度
第一类:对成药中原料药或活性成分(包括防腐剂)的主要组分进行定量测定的分析规程。
第二类:对成药中原料药或降解物质中杂质进行测定的分析规程。这些规程包括定量测定和限度测定。
第三类:对工作特性(例如,溶出度、药物释放)进行测定的分析规程。
第四类:鉴别测试。
每一个类别均需要不同的分析信息。每个种类通常所需要的数据要素在表2中列出。
分析工作特性
第二类
第一类
定量测试
限度测试
第三类
第四类
准确度பைடு நூலகம்
药典方法学验证指导原则

药典方法学验证指导原则药典方法学验证指导原则是指用于确认药品分析方法准确性、可靠性和适用性的规范化指南。
这些原则旨在确保药品分析方法在生产和质量控制中的可靠性,保证药品质量的稳定和一致性。
以下是一般性的药典方法学验证指导原则:1.准确性(Accuracy):方法应能够提供与真实值或已知标准接近的结果。
验证过程通常涉及对已知浓度样品的测试,以评估方法的测量准确度。
2.精密度(Precision):方法应在重复测试中产生一致的结果。
通过进行重复性测试来评估方法的精密度,这有助于确定方法在变异性方面的表现。
3.线性度(Linearity):方法应能够准确测量不同浓度范围内的样品,并且结果应该与样品的浓度成正比。
验证线性通常通过测试不同浓度水平的样品来进行。
4.灵敏度(Sensitivity):方法应能够检测到样品中非常小的变化,并产生可靠的测量结果。
灵敏度验证包括检测限(LOD)和定量限(LOQ)的评估。
5.特异性(Specificity):方法应能够准确地识别和量化目标化合物,而不受其他可能存在的干扰物质的影响。
这通常通过特异性测试来确认。
6.稳定性(Stability):方法在一定时间内应保持稳定,不会因为时间、环境变化或存储条件的变化而失效。
验证方法的稳定性可以确保其长期可靠性。
7.重复性(Repeatability)和再现性(Reproducibility):重复性指同一实验室、同一操作者、同一设备和条件下重复测试的结果一致性;再现性指不同实验室、不同操作者、不同设备和条件下重复测试的结果一致性。
这些是验证方法可靠性和可复制性的重要指标。
8.系统性误差(Systematic Errors)和随机误差(Random Errors):确保方法验证过程中能够识别和控制可能出现的系统性和随机误差,以确保结果的准确性和精确性。
这些原则构成了验证药品分析方法的基本指导,有助于确保分析方法的可靠性、精确性和适用性,并且是保障药品质量和安全的重要步骤。
分析方法确认与验证管理规程

一、目的:规范药品检验方法确认与验证的管理,证明采用的方法适合相应的检测要求及在实验室条件下的适用性,保证检验结果准确、可靠。
二、适用范围:适用于中药和化学药品(包括物料和产品)的理化分析方法和仪器分析方法的确认与验证。
不适用于中药和化学药品(包括物料和产品)微生物分析方法的确认与验证。
三、相关职责:QC班组长:QC班组长根据检验方法的来源确定开展方法确认或验证。
由其本人或者其他有经验的检验人员起草方案;负责确认或验证方案的培训;安排有经验的人员参与方法确认或验证过程实施;对确认或验证过程中出现的偏差要严格按照相关管理规程的程序执行,如需要提出变更申请;对确认或验证工作中出现的问题及时纠正并记录;总结确认或验证报告。
QC主管:负责审核检验方法确认或验证方案,并对方案的执行过程进行追踪;负责组织偏差的调查,变更的审核;负责总结报告的审核。
化验室QA:监督各项目按照已制定的方案进行;参与确认或验证过程中的偏差调查;对提出的变更进行评估,确认变更的是否成立,跟踪变更实施。
QA主管:负责审核检验方法确认或验证方案,确保其法规符合性;参与偏差的调查,变更的审核;负责总结报告的审核。
验证专员:审核检验方法确认或验证方案;审核总结报告;负责验证证书的发放;负责方法确认或验证方案、记录和报告的整理、存档。
质量管理负责人:批准检验方法确认或验证方案;批准方案实施过程中出现的偏差和变更;批准总结报告。
四、制定依据:《药品GMP指南》(质量控制实验室与物料系统)、《药品生产质量管理规范》(2010年修订)、《中国药典》(2020年版)、《化学药物质量控制分析方法验证技术指导原则》、ICH分析方法验证:正文和方法学Q2(R1 )、中国药品检验标准操作规范(2010年版)(原子吸收分光光度法)。
五、内容:1、术语1.1方法验证方法验证系指根据检验项目的要求,预先设置一定的验证内容和验证标准要求,并通过设计合理的实验来验证所采用的分析方法是否符合检验项目的要求。
分析方法的验证和确认与转移ppt实用课件

此外,含有不同辅料、抗氧化剂、缓冲剂或包装浸出物的制剂产品可能会对药典 方法产生潜在干扰。
在这些情况下,需要进行更加ቤተ መጻሕፍቲ ባይዱ尽的评价,证明方法适用于特定的原料药或制剂 产品。其它分析性能参数,比如杂质方法的检测限/定量限和精密性评价,对证 明药典方法在实际使用条件下适用非常有用。
当分析方法转移顺利完成后,分析人员应起草分析方法转移报告,其中应描述转移的结果,是否有符合标准规定,并应有结论确定接收方是可以使用转移的分析方法。
经过讨论并获得U各S方P一3致9同<1意225> Verification of Compendial Procedure
4法规规定的其他需要验证的方法
--CP2021<9101>USP 39<1226> Validation of Compendial Procedure
方法转移的步骤--USP39<1224>
阶段 分析方法
负责人 接收方
工作
1.分析方法应起草成书面文件,其中应对细节详细说明并有明确指 示,以便经过培训的人员可以顺利的进行检验。2.如果使用的是液 相或气相进行检验,那么进样的数量以及进样的顺序应明确规定; 如果是溶出试验,进行溶出试验的样品数量也应明确规定。
1采用新的检验方 法;2检验方法需 要变更的;3采用 CP及其他法定标准 未收载的方法;4 法规规定的其他需 要验证的方法
1不需要进行验证 的检验方法; 2药典方法和其他 法定方法
分析方法由A实验 室转移到B实验室
按照方法的用途, 对方法学验证参数 进行全部或部分验 证
(仅供参考)分析方法转移、确认和验证

分析方法验证 (GMP 实验室)
• 按已批准的分析方法验证方案,进行方法验证实验 • 计算、审核验证结果 • 编写、审核、批准分析方法验证报告包括相关的典型色谱图 • 完善、审核、批准并放行最终的分析测试方法
• 空白溶剂 / 辅料溶液 Diluent / Placebo Solution • 原料药 / 制剂中掺入杂质 • 强力降解试验 Stress Studies (∼10% degradation)
溶液:热、酸及碱水解、氧化、光照 固态:高温、高湿、光照 • API 主峰不受溶剂、辅料以及任何杂质峰干扰 • API 主峰纯度分析 (purity: NLT 990 or purity angle < purity threshold)
HPLC含量测定方法验证 (续)
耐用性 Robustness
• HPLC 系统参数变化
流动相比例 (± 10%) 缓冲液的 pH 值 (± 0.2) 流速 (± 0.2 mL/min) 柱温 (± 5°C)
Meet SS Criteria
• 样品制备的参数变化
振摇/超声时间 (± 5 min) 萃取溶剂的比例 (± 10%) 样品过滤的影响 对照品及样品溶液的稳定性 (0, 12, 24, 48 hrs)
使用药典专论 HPLC 杂质分析方法 药典制剂专论 Compendial DP Monograph
• 系统适用性 System Suitability • 方法重现性 Method Reproducibility • 方法准确性 Method Accuracy • 溶液稳定性 Solution Stability
2024年版《中国药典》通则调整—9101药品质量标准分析方法验证指导原则

2024年版《中国药典》通则对9101号章节进行了调整,主要涉及药品质量标准分析方法验证指导原则。
该调整旨在提高药品质量标准分析方法的可靠性和准确性,为药品质量控制提供更可靠的依据。
以下是对该调整的详细内容的介绍。
第一部分:引言和目的该部分对该章节的目的和重要性进行了介绍。
主要包括以下几个方面:1.引言:对药品质量标准分析方法验证的定义和重要性进行了说明,强调该过程对药品质量控制的作用。
2.目的:明确了药品质量标准分析方法验证的目的是为了验证和确认该方法可适用于特定药品的质量标准控制。
第二部分:验证原则该部分对药品质量标准分析方法验证的基本原则进行了详细说明。
主要包括以下几个方面:1.可行性研究:强调在进行药品质量标准分析方法验证之前,应进行可行性研究,评估该方法是否可以适用于特定药品的质量标准控制。
2.验证参数:明确了药品质量标准分析方法验证应包括的验证参数,如准确性、精密度、线性范围、检测限、定量限等。
3.验证方案:对药品质量标准分析方法验证的具体方案进行了要求。
包括验证规程的编制、实验条件的确定、验证样品的选择等。
4.验证结果的评估:对药品质量标准分析方法验证结果的评估进行了说明。
强调验证结果应能够满足药品质量标准的要求。
5.验证文件的管理:对药品质量标准分析方法验证所涉及的文件、记录和资料的管理要求进行了详细说明。
第三部分:验证程序该部分对药品质量标准分析方法验证的具体程序进行了详细说明。
主要包括以下几个步骤:1.验证计划的编制:明确了编制药品质量标准分析方法验证计划的内容和要求。
2.验证方案的制定:对药品质量标准分析方法验证方案的制定进行了详细说明,包括实验设计、验证参数的选择、实验方法的确定等。
3.验证实验的进行:对药品质量标准分析方法验证实验的具体步骤和要求进行了详细说明。
4.验证结果的评估和分析:对药品质量标准分析方法验证结果的评估和分析进行了说明。
5.验证报告的编制:明确了编制药品质量标准分析方法验证报告的内容和要求。
分析方法验证与确认管理规程

分析方法验证与确认管理规程(总10页)--本页仅作为文档封面,使用时请直接删除即可----内页可以根据需求调整合适字体及大小--3 定义检验方法验证:证明采用的方法适用于相应检测要求。
检验方法确认:证明使用法定方法在目前实验室条件下是否能获得可靠结果,是否适用于相应的检测工作。
在本质上和验证一样,但不一定是验证项目的全部。
药典方法:经过国家药监部门批准的药典收载的质量标准和检验方法法定方法:法定方法包括药典方法、国标方法等。
准确度:是指用该方法测定的结果与真实值或参考值接近的程度,一般用回收率表示。
精密度:是指在规定的测试条件下同一个均匀供试品经多次取样测定所得结果之间的接近程度。
重复性:在相同条件下,由同一个分析人员测定所得结果的精密度称为重复性。
中间精密度:在同一个试验室,不同时间由不同分析人员用不同设备测定结果之间的精密度称为中间精密度。
重现性:在不同实验室由不同分析人员测定结果之间的精密度称为重现性。
专属性:是指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的方法能正确测定出被测物质的特性。
检测限:是指供试品中被测物能被检出的最低量。
定量限:是指供试品中被测物能被定量测定的最低量。
线性:是指在设计范围内,测试结果与试样中被测物浓度直接成正比关系的程度。
范围:是指能达到一定精密度、准确度和线性,测试方法适用的高低浓度或量的区间。
耐用性:是指在测定条件有小的变动时,测定结果不受影响的承受程度。
4 职责标准验证岗提升现行质量标准工作时,对研究后确定的标准草案进行检验方法验证工作,以确保检验方法的适用性、科学性。
对技术部移交的新品质量标准草案进行确认,以确保检验方法适用性、科学性。
对技术部移交的新品应研究建立设备清洁验证残留物检验方法,并进行方法学验证。
理化检验岗对首次采用的药典或国标检验方法,进行检验方法确认工作,以证明检验方法的适用性,确保检验数据准确、可靠。
对标准验证岗研究后确定的质量标准草案中变更后的检验方法进行确认。
中国药典版生物样品定量分析方法验证指导原则

二、生物分析方法验证
为了验证批内准确度,应 取一个分析批的定量下限及低、中、高浓度质控样品,每个浓 度至少用5个样品 浓度水平覆盖标准曲线范围:定量下限,在不高于定量下限浓度3倍的低浓度质控样品, 标准曲线范围中部附近的中浓度质控样品,以及标准曲线范围上限约75 %处的高浓度质控 样品
准确度均值一般应在质控样品标示值的±15%之内,定量下限准确度应在标示值的±20%范围 内
除正常基质外,还应关 注其他样品的基质效应, 例如溶血的或髙血脂的 血浆样品等
二、生物分析方法验证
9.稳定性
必须在分析方法的每一步骤确保稳定性,用于检查稳定性的条件,例如样品基质、抗凝剂 、容器材料、储存和分析条件,都应该与实际试验样品的条件相似 用文献报道的数据证明稳定性是不够的 采用低和髙浓度质控样品(空白基质加人分析物至定量下限浓度3倍以内以及接近定量上限 ),在预处理后以及在所评价的条件储存后立即分析 由新鲜制备的校正标样获得标准曲线,根据标准曲线分析质控样品,将测得浓度与标示浓 度相比较,每一浓度的均值与标示浓度的偏差应在±15%范围内 应通过适当稀释,考虑到检测器的线性和测定范围,检验储备液和工作溶液的稳定性
中国药典(2020版)9012 生物样品定量分析方法验
证指导原则
中国药典(2020版)9012生物样品定量分析方法验证指导原则
二、生物分析方法验证 三、试验样品分析
目录
中国药典(2020版)9012生物样品定量分析方法验证指导原则
9012生物样品定量分析方法验证指导原则 —、范围 准确测定生物基质(如全血、血清、血浆、尿)中的药物浓度,对于药物和制剂研发非常重 要 这些数据可被用于支持药品的安全性和有效性,或根据毒动学、药动学和生物等效性试验 的结果做出关键性决定 因此,必须完整地验证和记录应用的生物分析方法,以获得可靠的结果 本指导原则提供生物分析方法验证的要求,也涉及非临床或临床试验样品实际分析的基本 要求,以及何时可以使用部分验证或交叉验证,来替代完整验证 本指导原则二和三主要针对色谱分析方法,四针对配体结合分析方法 生物样品定量分析方法验证和试验样品分析应符合本指导原则的技术要求
分析方法的验证ppt课件

1
内容:
一、介绍 二、分析方法验证 三、常见问题
2
一、介绍
方法分析:是为完成检验项目而设定和建立的测试方法,它 详细描述每一个分析试验所必须的步骤。 一般包括分析方法原理、仪器及仪器参数、试剂、供试品溶 液与对照品溶液等的制备、测定、计算公式及限度要求等。
3
一、介绍 分析方法确认或验证相关指南
通过实验室之间的检测来评价;
如果方法需要标准化如收载到药典中的 方法,则应考虑重现性。这些资料不是 申请上市文档的三部分。
协作标定 能力验证 药品标准的实验 复核
24
5)精密度
系统重复性:相同条件下,同一分析人员对同一样品溶液多 次测试结果的接近程度。 同一均匀标准样品溶液,连续重复测定六次,计算各组分响 应值与平均值之间的相对标准偏差。 方法重复性:相同条件下,同一分析人员对同一个均匀样品 ,多次取样测定所得结果的接近程度。 同一样品,至少称取六份,再测定每一份样品含量,计算每 一次测定的含量与平均含量间的相对标准偏差。
中其他成分(如防腐剂等)的含量,溶出度与释放度等检 查中的溶出量,以及含量均匀度。 化学药品/中药制剂中其他需控制成分(如残留物、添加剂 等)的测定
7
二、分析方法验证
5、分析方法验证的具体内容
1)专属性 2)线性 3)范围 4)准确性 5)精密度 6)检测限 7)定量限 8)耐用性 9)系统适用性
(完全辨别),在此情况下,建议采用两种或两种以上的分 析方法以达到所需的辨别水平。
9
1)专属性
含量测定目的是得到样品中被分析物的含量或效价的准确 结果。
对于色谱法和其他分离方法来说,应当用具有代表性的色 谱图来证明专属性,并在图上适当地标出每一种成分。色谱 法的分离度应符合要求。
药典生物样品定量分析方法验证指导原则

在对已被验证的分析方法进行小幅改变情况下,根据改变的实质内容,可能需要部分方法验证。可能的改变包括:生物分析方法转移到另一个实验室,改变仪器、校正浓度范围、样品体积,其他基质或物种,改变抗凝剂、样品处理步骤、储存条件等。应报告所有的改变,并对重新验证或部分验证的范围说明理由。
(三)交叉验证
3. 定量下限
定量下限是能够被可靠定量的样品中分析物的最低浓度,具有可接受的准确度和精密度。定量下限是标准曲线的最低点,应适用于预期的浓度和试验目的。
4. 标准曲线
应该在指定的浓度范围内评价仪器对分析物的响应,获得标准曲线。通过加入已知浓度的分析物(和内标)到空白基质中,制备各浓度的校正标样,其基质应该与目标试验样品基质相同。方法验证中研究的每种分析物和每一分析批,都应该有一条标准曲线。
生物样品定量分析方法验证指导原则
一、范围
准确测定生物基质(如全血、血清、血浆、尿)中的药物 浓度,对于药物和制剂研发非常重要。这些数据可被用于支持药品的安全性和有效性,或根据毒动学、药动学和生物等效性试验的结果做出关键性决定。因此,必须完整地验证和 记录应用的生物分析方法,以获得可靠的结果。
本指导原则提供生物分析方法验证的要求,也涉及非临床或临床试验样品实际分析的基本要求,以及何时可以使用部分验证或交叉验证,来替代完整验证。本指导原则二和三主要针对色谱分析方法,四针对配体结合分析方法。
批间准确度
通过至少3个分析批,且至少两天进行,每批用定量下 限以及低、中、高浓度质控样品,每个浓度至少5个测定值来评价。准确度均值一般应在质控样品标示值的±15%范围内,对于定量下限,应在标示值的±20%范围内。
报告的准确度和精密度的验证数据应该包括所有获得的测定结果,但是已经记录明显失误的情况除外。
药典方法确认规程
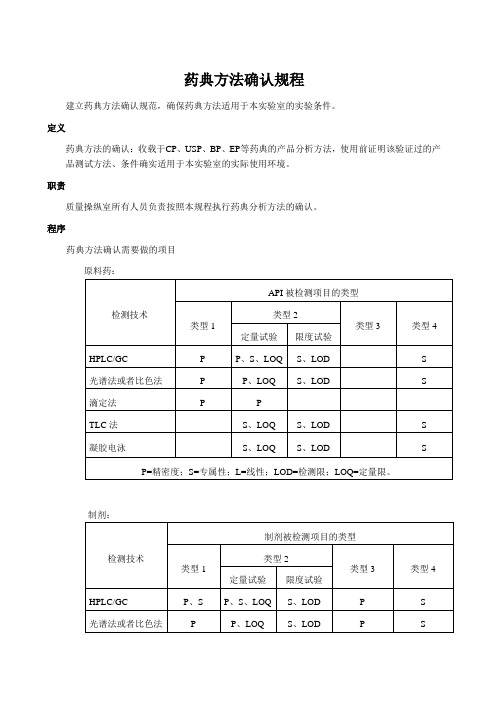
药典方法确认规程建立药典方法确认规范,确保药典方法适用于本实验室的实验条件。
定义药典方法的确认:收载于CP、USP、BP、EP等药典的产品分析方法,使用前证明该验证过的产品测试方法、条件确实适用于本实验室的实际使用环境。
职责质量操纵室所有人员负责按照本规程执行药典分析方法的确认。
程序药典方法确认需要做的项目制剂:被检测项目类型的说明:系统适用性实验按药典正文要求的内容进行;正文没有要求的内容,如RSD%,按药典附录的要求进行。
专属性是指在其他成分(如杂质、降解物、辅料等)可能存在下,使用的分析方法能够正确鉴定、检出被分析物质的特性。
通常,鉴别、杂质检查、含量测定方法中均应考察其专属性。
如使用的方法不够专属,应使用多个方法予以补充。
鉴别反应专属性鉴别试验应确证被分析物符合其特征。
专属性试验要求证明能与可能共存的物质或者结构相似化合物区分,需确证含被分析物的供试品呈正反应,而不含被测成分的阴性参照呈负反应,结构相似或者组分中的有关化合物也应呈负反应。
杂质检测专属性在杂质可获得的情况下,可向供试品中加入一定量的杂质,证明杂质与共存物质能得到分离与检出,并具适当的准确度与精密度。
在杂质或者降解产物不能获得的情况下,专属性可通过与另一种已证明合理但分离或者检测原理不一致、或者具较强分辨能力的方法进行结果比较来确定。
或者将供试品用强光照射,高温,高湿,酸、碱水解及氧化的方法进行破坏(制剂应考虑辅料的影响),比较破坏前后检出的杂质个数与量。
必要时可使用二极管阵列检测与质谱检测,进行色谱峰纯度检查。
含量检测专属性➢关于API主成分含量测定可在供试品中加入杂质,考察测定结果是否受干扰,并与未加杂质的供试品比较测定结果;在杂质或者降解产物不能获得的情况下,可使用另一个经验证了的或者药典方法进行比较,对比两种方法测定的结果。
也可使用破坏性试验(强光照射,高温,高湿,酸、碱水解及氧化),得到含有杂质或者降解产物的试样,用两种方法进行含量测定,比较测定结果。
分析方法的验证确认和转移

- -1 目的明确分析方法的验证、确认和转移的管理制度,确保所采用的分析方法适合于相应检测要求和目的,被测样品质量可控,保证得到一致、可靠和准确的测定结果,同时也证明检验人员有能力操作分析方法。
2 适用围分析方法的验证:在建立药品质量标准时,分析方法需经历证;在药品生产工艺变更、制剂的组分变更、原分析方法进展修订时,质量标准分析方法也需进展验证。
分析方法确实认:在采用药典分析方法或法定分析方法进展检验时,不需要再对方法进展验证,但是需要进展方法确认,以证明承检实验室能够正确地操作药典方法。
分析方法的转移:分析方法由公司的研发实验室转移到质控实验室;由于生产线转移使分析方法从A生产地点转移到B生产地点;分析方法由*公司转移到合同公司;由于*公司购置了Y公司的产品,方法由Y公司转移到*公司。
3 职责及责任者3.1分析方法验证及确认的职责及责任者3.1.1 检验员负责方案的起草、工作具体实施以及完成记录、起草报告,负责报告验证/确认中发生的任何偏差。
3.1.2 化验室主任负责方案、记录、报告的审核,负责对发生的偏差组织调查,确保方案的正确实施。
3.1.3 质量部负责人负责方案、报告的审核,监视工作的实施,对验证/确认工作中出现的问题提出改进意见并监视落实。
3.1.4 质量受权人负责方案及报告的批准。
3.2分析方法转移的职责及责任者3.2.1 研发员或转移方人员负责方案的起草、工作具体实施以及完成记录、起草报告,负责报告验证/确认中发生的任何偏差。
3.2.2如需要可邀请转移方人员作为验证团队的一员,参加接收方的实验室方法转移。
3.2.2 化验室主任负责方案、记录、报告的审核,负责对发生的偏差组织调查,确保方案的正确实施。
3.2.3 质量部负责人负责方案、报告的审核,监视工作的实施,对验证/确认工作中出现的问题提出改进意见并监视落实。
3.2.4 质量受权人负责方案及报告的批准。
4 定义4.1 分析方法:分析方法是为完成检验工程而设定和建立的测试方法,它详细描述了完成分析检验的每一步骤。
分析方法确认指导原则

分析方法确认指导原则2020年版《药典》四部通则9099分析方法确认(analytical method verification)是指首次使用法定分析方法时,由现有的分析人员或实验室对分析方法中关键的验证指标进行有选择性的考察,以证明方法对所分析样品的适用性,同时证明分析人员有能力使用该法定分析方法。
指导原则9101《分析方法验证指导原则》中提供了建立分析方法需要验证的指标,分析方法的确认并不是重复验证过程。
本指导原则不涉及微生物分析方法的确认。
—、确认过程(verification process)分析方法的确认过程,是指应用法定方法对药物及其制剂进行测定时,评价该方法能否达到预期的分析目的。
分析人员应具备一定的药物分析经验和知识,经培训后能够理解和执行法定方法。
分析方法确认应当由上述分析人员开展,以确保法定方法能够按预期顺利实施。
如果法定方法确认失败,并且相关工作人员(或起草人员)未能协助解决失败的问题,也可能是该方法不适用于在该实验室测定待分析的样品。
二、确认要求(verification requirements)1 . 确认原则分析方法确认一般无需对法定方法进行完整的再验证,但是需要将指导原则9101《分析方法验证指导原则》表1 中列出的分析方法验证的指标用于方法的确认。
分析方法确认的范围和指标取决于实验人员的培训和经验水平、分析方法种类、相关设备或仪器、具体的操作步骤和分析对象等。
分析方法确认的指标和检验项目(鉴别、杂质分析、含量测定等)有关,不同的检验项目,方法确认所需的指标也不同。
2 . 考察指标分析方法确认应包含对影响方法的必要因素的评估。
对于化学药,方法确认应考虑原料药的合成路线和制剂的生产工艺等因素;对于中药,方法确认应考虑中药材种类、来源、饮片制法和制剂的生产工艺等因素,从而评价法定方法是否适用于原料药和制剂基质。
在原料药和制剂含量测定时,方法专属性是确认法定分析方法是否适用的关键指标。
药物的分析方法验证、转移和确认

生物检定方法(biological assay,or bioassay)
生物制品(包括一秒、抗体、细胞因子和酶等)的分 析方法通常被称作“生物检定方法”根据ICH的定义,
1. 分析方法验证的指导原则
美国、欧盟、日本、澳大利亚、中国等国家和地区的 药品监管机构以及ICH、WHO、ISO等国际组织都有 专门的关于方法学验证的指导原则。其中FDA在 2000年和2001年分别发布了“化学药品分析步骤和
分析方法验证指导原则”和“生物分析方法验证指导 原则”;EMEA于2004年发布的GMP指导原则对
生物检定的内容包括:测定细胞水平的生化或生理学 反应、免疫学作用下的酶反应速率或者能够生物学反 应以及配体或者受体结合反应。由于生物制品的复杂 性,生物检定方法的范围很广,包括:理化分析方法、 效价测定方法、细胞活性测定、酶活性测定、免疫测 定等,随着新技术和新生物制品的不断出现,生物检 定的范围仍在继续不断扩大。
临床前和临床试验效果,包括:生物等效性、生物利 用度、药代动力学和毒代动力学等,者对于药品研发 来说至关重要,在药品注册时,这些研究材料都是提 交给药品监管当局进行审评。
单从分析方法手段和操作过程来说,生物分析方法和 化学药品分析方法属于同一范畴,都是采用分析仪器 (HPLC、GC、MS等等)和手段对被测物进行定性
和定量分析。但是,由于样品的特殊性,相对于化学 药品分析方法来说,生物分析方法更加复杂和困难。
由于采用的分析手段和试验操作基本上一致,因此两 者的方法验证、转移和确认的基本原则和验证项目总 体上是一致的。
药典方法的确认与验证

There may be additional elements which should be considered (e.g. filters)
for its intended purpose under conditions of actual use Has been widely adopted by the industry
Future Steps
Encourage use of good scientific judgment and appropriate risk assessment
The complexity of the procedure The complexity of the material to which the procedure is applied The training and experience of the user
Differences in drug substance impurity profile or drug product composition may increase risks, and a more thorough assessment may be required
Need to demonstrate the procedure will yield acceptable results utilizing the personnel, equipment, and reagents available
药典方法确认规程

药典方法确认规程一、概述药典方法确认是指对涉及药品质量控制的方法进行确认,以确保其能够准确、可靠地用于药品质量控制的目的。
方法确认的目的是为了评价和验证该方法的适用性、灵敏度、精密度、准确度等指标,以提高方法的可靠性和准确性。
药典方法确认规程是对药典方法进行确认的标准化程序和要求的文件,以保证方法的确认工作可以科学、规范地进行。
二、流程和要求1.确认方法选择选择确认的方法应根据药品的特性和质量的要求进行选择,确认方法的选择应考虑以下因素:(1)方法的适用范围和原理;(2)方法的灵敏度;(3)方法的准确性和精密度;(4)方法的稳定性和可重复性。
2.确认方法验证确认方法的验证是确认方法准确性和可靠性的一个重要步骤。
验证的内容主要包括以下几个方面:(1)方法的线性度验证,即浓度与响应的关系;(2)方法的精密度验证,即方法的重复性和中间精密度的验证;(3)方法的准确度验证,即方法的稳定性、回收率和加标回收率等的验证;(4)方法的特异性验证,即方法对其他干扰物质的选择性。
3.确认方法的验证结果和评价对验证结果进行评价,评价的主要依据是验证参数的符合药典规定的要求。
4.确认方法的执行对经过确认的方法进行标准化操作,包括以下几个方面:(1)方法的标识和记录,包括方法的名称、编号、版本、适用范围等信息;(2)方法的操作规程和记录,包括关键步骤、操作条件、操作记录等;(3)方法的仪器和试剂的管理和校准;(4)方法的数据处理和验证,包括数据的计算、处理和记录。
5.确认方法的周期性评价和更新对已确认的方法进行周期性评价,包括对每次分析结果的比对、统计和验证等,以确保方法的可靠性和准确性。
三、注意事项1.确认方法的选择应根据药品特性和质量要求进行选择,并符合药典规定的方法。
2.确认方法的验证内容应全面、科学地进行验证,确保方法的准确性和可靠性。
3.确认方法的执行应严格按照规程进行,确保方法操作的标准化和可靠性。
4.确认方法的周期性评价和更新应及时进行,确保方法的持续性和可靠性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分析方法的验证和确认主要内容:分析方法的验证分析方法的确认分析方法的转移方法验证、方法确认和方法转移的文件管理【背景介绍】只有经过验证或确认的分析方法,才可以用于物料和产品的检验以及清洁验证。
检验方法必须经过验证或确认是物料和产品放行的前提之一。
因为只有经过验证或确认的分析方法才可以可靠有效地用于控制药品的内在质量。
分析方法:分析方法是为完成检验项目而设定和建立的测试方法,它详细描述了完成分析检验的每一步骤。
一般包括分析方法原理、仪器及仪器参数、试剂、供试品溶液与对照品溶液等的制备、测定、计算公式及限度要求等。
不同分析方法各有特点,同一分析方法可用于不同的检验项目,但验证内容会有不同。
例如,采用高效液相色谱法进行药品的鉴别和杂质定量检验,两检验项目在方法验证时要求不同。
前者重点要求验证专属性,而后者则需要重点验证专属性、准确度和定量限。
【适用范围】适用于:化学药品(包括物料和产品)的理化分析方法和仪器分析方法的验证与确认。
清洁验证方法的验证。
不适用于:化学药品的微生物方法,生物制品分析方法验证。
【方法验证】方法验证就是根据检验项目的要求,预先设置一定的验证内容和验证标准要求,并通过设计合理的实验来验证所采用的分析方法是否符合检验项目的要求。
建立质量标准时,应对分析方法中的各检验项目进行完整的验证。
当药品生产工艺变更、制剂的组分变更、原分析方法修订时,可根据变更的内容决定对分析方法进行部分验证还是完整的验证。
当原料药合成工艺发生变更时,可能引入新的杂质,杂质检查方法和含量测定方法的专属性就需要进行验证,以证明有关物质检查方法能够检测新引入的杂质,且新引入的杂质对主成分的含量测定应无干扰。
当质量标准中某一项目分析方法发生部分改变时,如采用高效液相色谱法测定含量时,检测波长发生改变,则需要重新进行检测限、定量限、专属性、准确认、精密度、线性等内容的验证,证明修订后的分析方法的合理可行。
当变更达到一定程度时,则需要完整的验证。
如分析方法完全改变,则应按新方法进行完整的验证。
方法验证的一般原则:通常情况下,分析方法需进行方法验证。
对于仅需按照实验室日常测试操作步骤即可测定的检验项目不需要进行验证,如外观、崩解时限、密度、重量、pH值、灰分、装量等。
方法验证的内容应根据检验项目的要求,结合所采用分析方法的特点确定。
同一分析方法用于不同的检验项目会有不同的验证要求。
需要验证的检验项目检验项目是为控制药品质量,保证药品安全有效而设定的测试项目。
要所检验项目的设定目的和验证内容的不同要求,本指南将需验证和检验项目分为四类:鉴别试验杂质的限度检查杂质的定量测定含量测定,包括原料药或制剂中有效成分的含量,制剂中其他成分(如防腐剂等)的含量,溶出度与释放度等检查中的溶出量,以及含量均匀度。
除此这外还有一些物理项目的检测如粒径分布、旋光度、熔点和硬度,其要求与其他检验项目有所不同,通常其分析方法验证应有不同的要求。
鉴别的目的在于判定被分析物是目标化合物,而非其他物质。
用于鉴别的分析方法要求具有较强的专属性和耐用性。
杂质检查主要用于控制主成分以外的杂质,如无机杂质、有机杂质等。
杂质检查分限度检查法和定量测定两部分。
用于限度检查的分析方法验证侧重专属性、检测限和耐用性。
用于定量测定的分析方法验证强调专属性、准确度、精密度、线性、范围、定量限和耐用性。
含量测定对准确度要求较高,因此所采用的分析方法要求具有一定的专属性、准确度和线性等要求。
《中国药典》2010年版中规定了不同的检验项目需要验证不同的内容。
方法验证内容:Accuracy准确度Precision精密度Specificity专属性Detection Limit检测限度Quantitation Limit定量限度Linearity线性Range范围Robustness耐用性1准确度定义---- 系指用该方法测定的结果与真实值或参考值接近的程度。
一般用回收率表示。
准确度应在规定的范围内测试。
测定---- 当对一种药物进行分析时,准确度可以通过该分析规程来分析一个已知纯度的物质(例如,某个标准物质)来进行测定,或者通过比较运用这个分析方法所得的结果与另一个已经鉴定的、其准确度已被说明或被解释过的方法所得的结果来进行测定。
对于杂质的定量分析,应使用以已知数量杂质增敏的样品来评估准确度。
当不能获得特定杂质或降解产物的样品时,应将结果与另一独立方法获得的结果进行比较。
通过测定被加入到样品中的已知数量的被分析物来计算准确度,得到回收百分比,或得到平均值与可接受的真实值之间的差异,同置信区间一起。
准确度的评估可以通过多种不同的方式完成,包括评价在含量测定的整个范围内被分析物的回收率,或评价估计浓度与实际浓度之间关系的线性。
2精密度定义---- 分析规程的精密度是当该分析规程单独分析均质样品的多个样本时,若干检验结果的一致程度。
分析规程的精密度通常以一系列测量数值的标准差或相对标准差(变异系数)来表示。
精密度可以是分析规程在普通操作条件下可重现性或可重复性程度的度量单位。
在这样的背景下,重现性指的是该分析规程在不同实验室的应用,例如在一个协作实验室里进行研究。
中间精密度(也称为“耐用性”)体现了在实验室内的差异,如在相同的实验室,但在不同的日期,或使用不同的分析员或设备。
可重复性指的是在同一个实验室内,一段较短的时间内,相同的分析员使用相同的设备、同一个分析方法的应用情况。
测定----一个分析方法的精密度是通过对足够的同一样品的分析来计算有统计学意义的标准偏差或相对标准偏差来测定的。
在这个背景下的分析是从样品准备到最终实验结果的完整分析程序的对样品的独立分析。
ICH文件建议可重复性的评估应该使用最少九次检测,覆盖该分析规程所规定的范围(例如,三个浓度和每个浓度三次重复进样,或在100%测试浓度上进行最少六次测定)。
3专属性定义----当待分析物含有预期会有的其他组分(例如,杂质、降解产物、矩阵组分)时,准确可靠地评估待分析物的能力。
某个分析规程缺乏专属性可以通过其他辅助性分析规程进行补偿。
纯度检测:确保执行的所有分析规程能够令对于待分析物各杂质含量的准确陈述得以做出。
含量检测:提供准确的结果,令对样品中待分析物的含量或效力的准确陈述得以做出。
测定---- 对于定性分析(鉴别检验),应当论证其在可能存在的、结构密切相关的物质中进行选择的能力。
从含有待分析物的样品中得到阳性结果(可能通过与已知标准物质的比较),而从不含待分析物的样品得到阴性结果,以对其选择能力加以确认,并还要确认阳性响应不是来自与待分析物结构相似或密切相关的物质。
对于检测杂质的分析规程,专属性可以通过以适当水平的杂质将原料药或成药增敏,并论证这些杂质的测定达到了适当的准确度和精密度。
对于含量检测,对专属性的论证要求能够显示出该分析规程不受各杂质或辅料的影响。
在实际操作中,通过以适当水平的杂质或辅料将原料药或成药增敏,并证明含量检验结果不受这些外来物质的影响,来完成论证。
4DETECTION LIMIT检测限度定义---- 检测限度是限度检测的特性。
它是指在规定的试验条件下,样品中可被检测到的待分析物的最小数量,但是无需定量。
因此,限度检测仅仅说明了待分析物的数量高于或低于某个特定水平。
检测限度通常以在样品中的待分析物浓度(例如,百分比、十亿分率)表示。
测定---- 对于非仪器分析规程,检测限度的测定方法通常为,对含有已知浓度待分析物的样品进行分析,并确立能够可靠地被检测出来的待分析物的最低水平。
通过分析含有高于和低于必需的检测水平的、已知待分析物浓度的样品,以显示检测限度足够低。
例如,如果必需检测浓度在0.1%的杂质,则应当证明该分析规程将可靠地检测在这个水平的杂质。
对于展示出背景噪音的仪器分析规程,ICH文件描述了一个通用方法,用来比较从以下样品测得的信号,这些样品分别为含已知低浓度被分析物的样品和空白样品。
这样就确立了能够可靠检测的待分析物的最低浓度。
可接受的典型信噪比是2:1或3:1。
其他方法取决于校正曲线斜率的测定和响应值的标准差。
5QUANTITATION LIMIT定量限度定义---- 定量限度是样品矩阵中低含量物质的定量分析的特性,例如在原料药中的杂质和成品药物中的降解产物。
它是在规定试验条件下,能够以可接受的精密度和精确度进行测定的样品中待分析物的最小量。
定量限度以样品中待分析物的浓度(例如,百分比、十亿分率)来表示。
测定---- 对于非仪器分析规程,定量限度的测定方法通常为对含有已知浓度待分析物的样品进行分析,并确立能够以可接受的准确度和精密度被检测出来的待分析物的最低水平。
对于展示出背景噪音的仪器分析规程,ICH文件描述了一个通用方法,用来比较从以下样品测得的信号,这些样品分别为含已知低浓度被分析物的样品和空白样品。
这样就确立了能够可靠地定量待分析物的最低浓度。
可接受的典型信噪比是10:1。
其他方法取决于校正曲线斜率的测定和响应值的标准差。
无论用什么方法,均应该在随后通过分析适当数量的、已知接近或制备于定量限度的样品,来验证定量限度。
6 7 LINEARITY AND RANGE线性和范围线性的定义---- 分析规程的线性是该规程直接地、或通过明确给出的数学转换而间接地,得出与特定范围内的样品中待分析物浓度呈比例关系的测试结果的能力。
因此,在此部分,“线性”是指浓度与检验结果之间的线性关系。
在某些情况下,为了实现线性,浓度和/或测量数据可以进行转换。
范围的定义---- 分析规程的范围是分析物的较高浓度和较低浓度(含)之间的区间,已经证实在此区间内,使用该规程进行测定具有适当水平的精密度、准确度、和线性。
该范围通常以与该分析规程得到的测试结果相同的单位表示,例如百分比,百万分率等。
线性和范围的确定---- 应在分析规程整个范围内确立线性。
线性的确立最开始应该通过目测代表内容物中待分析物浓度的多个信号构成的图表来进行。
如果其呈现线性关系,测试结果应该通过适当的统计学方法(例如,以最小二乘方来计算回归线)来确立。
来自回归线自身的数据可以用于提供线性程度的数学评估。
应该提交其相关系数、Y轴截距、回归线斜率、残差平方和。
通过用规程分析含有待分析物的、浓度在范围的极限值上和在范围中的样品,确认该分析规程提供了可接受的精密度、准确度、线性,以进行该规程范围的验证。
ICH建议,为了确立线性,通常使用最少五个浓度。
8 ROBUSTNESS耐用性定义---- 分析规程的耐用性是规程文件中列出的操作参数在微小、故意的变更中不受影响的能力的衡量单位,并在日常使用中提供了其适用性的指标。
耐用性可以在分析规程的研发中确定。
第一类:对成药中原料药或活性成分(包括防腐剂)的主要组分进行定量测定的分析规程。