新药设计与合成——中国药科大学
化学生物学中的化学药物设计与合成

化学生物学中的化学药物设计与合成引言化学药物作为治疗疾病的重要工具已经成为当代医学的重要组成部分。
化学生物学作为化学与生物学之间的交叉学科,致力于发展新的化学药物,通过合成方法的研究和生物活性评价的方法,提高药物的疗效和选择性。
本文将分别从化学药物设计和化学药物合成两个方面阐述化学生物学在药物发展中的重要性。
一、化学药物设计化学药物设计是将化学数据和生物学数据相结合,通过计算机辅助设计等方法来进行药物分子的合理改造和修饰的过程。
其中主要包括药物基因组学、药物代谢学、药物传递以及药物分子的计算模拟。
1. 药物基因组学药物基因组学研究药物对基因表达的影响,并以此为基础设计新的药物。
现代高通量测序技术的发展,使得我们能够了解不同基因变异对药物反应的影响。
通过药物基因组学的研究,可以预测患者对药物的反应,从而进行个体化药物治疗。
2.药物代谢学药物代谢学研究药物在体内的代谢途径、代谢产物和代谢酶的作用。
药物代谢的了解可以预测药物的药代动力学特性,进一步指导药物设计。
例如,通过设计抑制药物代谢的药物,延长药物在体内的作用时间。
3.药物传递药物传递是指药物在体内到达靶区的过程。
化学生物学通过研究纳米材料的设计和功能化,实现药物的定向传递。
例如,通过修饰纳米颗粒的化学性质和形状,可以使药物在体内具有靶向性,降低剂量和毒副作用。
4.药物分子的计算模拟药物分子的计算模拟是通过分子动力学模拟和计算化学方法,预测药物与生物分子的相互作用。
通过计算模拟,可以优化药物分子的结构和性质,提高药物的活性和选择性。
二、化学药物合成化学药物合成是在药物设计的基础上,通过合成方法制备具有特定生物活性的化合物。
合成方法的选择和反应的策略在合成过程中起着至关重要的作用。
1.合成方法的选择合成方法的选择是根据目标化合物的结构和性质来决定的。
合成方法一般包括碳碳键形成反应、碳氮键形成反应、氧化反应等。
合成方法的选择要求高效、环保,并且能够产生高纯度的目标化合物。
2014年中国药科大学硕士研究生招生专业目录

_ 02制药过程及设备
100701药物化学
82
101思想政治理论
201英语一
710药学基础综合(一)
复试:药物化学-有机合成综合(笔试),比重各占50%。
_ 01新药分子设计与合成研究
_ 02新药设计与结构生物学研究
_ 03天然产物的合成、优化与评价研究
_ 04化学生物学与创新药物研究
中药学院复试科目:天然药物化学;
生科院复试科目:生物工程。
_ 02临床药学
临床药学方向——临床医学概论和临床药物治疗学综合(各占50%)(笔试)
_ 03管理药学
管理药学方向——管理学原理及西方经济学(微观部分)(各占1/2)
105600中药学(专业学位)
35
101思想政治理论
204英语二
350中药专业基础综合
同等学力加试:人力资源管理,财务管理
_ 01医药技术经济与管理研究
_ 02医药产业经济与管理研究
_ 03企业经营管理与诊断咨询研究
_ 04药品生产运作与注册管理
_ 05医药营销战略
_ 06物流与供应链管理
005理学院
14
070300化学
7
101思想政治理论
201英语一
713有机化学
811分析化学
01-05方向复试科目:现代仪器分析(笔试);
本专业只接收2名少数民族骨干计划考生。
_ 01医药政策与法规研究
_ 02医药知识产权研究
_ 03药品质量监督与管理
_ 04医药产业经济及政策研究
_ 05药物资源的合理利用
_ 06医药国际商务
_ 07医疗保险研究
1007Z8★药物经济学
医药化学中的药物合成与设计

医药化学中的药物合成与设计医药化学是一门综合性强的学科,其研究对象主要是药物的合成、性质、结构和作用等方面。
在医药领域中,药物的合成与设计显得尤为关键。
药物合成的目的是生产出高效、低毒、高选择性的化合物,而药物设计则是针对特定的靶点进行合理的结构设计。
本文将阐述医药化学中药物合成与设计的相关内容。
一、药物合成药物合成的目标是在尽可能短的时间内合成目标药物,并尽可能提高药物的产量。
药物的合成可以通过天然产物合成、半合成和全合成三种方式进行。
全合成指从非生物来源或简单的化学物质出发,通过化学合成方法制备目标化合物。
半合成则是在天然产物的基础上进行所需结构的修饰,以改善其疗效或生物利用度。
在药物合成的过程中,选择合适的合成路径和反应条件至关重要。
常见的反应有亲核取代反应、加成反应、氧化还原反应、羟化反应等。
此外,在合成过程中,需要严格控制反应条件,如温度、催化剂、溶剂种类和比例等。
二、药物设计药物设计是指根据疾病的特点和药理学机制,选择最佳的分子结构,以达到预期的疗效。
药物设计的过程中,需要对靶标分子进行深入的研究,确定活性位和分子的关键部分,结构分析和计算机模拟技术也成了不可缺少的手段。
药物设计的成功在很大程度上取决于药物分子与靶标分子之间的相互作用。
药物设计主要分为配体设计和基础设计两种。
配体设计根据药物分子与靶标分子间非共价力的特点,以小分子有机化合物为药物分子。
而基础设计则是以靶标分子的生物大分子如蛋白质、DNA作为药物分子。
这两种方法各有优劣,应根据具体情况酌情选择。
三、药物合成与设计的结合药物合成和设计并不完全独立,两者相互作用,共同推动药物科学的发展。
药物合成的相关研究和发展,为药物设计提供了更多实际应用的资源,药物设计则进一步推动了药物合成技术的发展和提升。
药物合成和设计的相互作用,深刻影响到药物领域的发展。
总之,药物合成与设计是医药化学学科的核心内容之一,也是研究药物的关键工作。
药物的合成需要控制不同反应的条件,最终得到高效、低毒的化合物;药物的设计则是对靶标分子的深入研究后,寻找合适的分子结构,以达到理想的疗效。
新型药物分子设计与合成研究

新型药物分子设计与合成研究第一章:引言在当今医学领域,新型药物的研发一直是一个热门话题。
随着科技的不断发展,药物分子设计与合成研究变得尤为关键。
本文将介绍新型药物分子设计与合成研究的意义和挑战,以及当前研究的新进展。
第二章:药物分子设计2.1 分子设计基础药物分子设计是指通过对分子结构进行合理的设计,以达到特定药理活性的一种方法。
在药物分子设计中,我们需要考虑到分子的性质、结构和活性。
2.2 药物分子设计方法目前,常用的药物分子设计方法有结构同源性建模、配体药效关系、基于药物靶点的设计以及计算机辅助分子设计等。
这些方法在不同的研究领域中都得到了广泛的应用。
第三章:药物分子合成3.1 药物合成基础药物合成是指通过一系列的化学反应,将目标分子合成出来的过程。
药物分子的合成需要考虑到合成路线的选择、合成方法和合成步骤的优化等。
3.2 药物合成方法目前,常用的药物合成方法包括有机合成、绿色合成和多步合成等。
这些方法在药物分子的合成过程中发挥着重要的作用。
第四章:新型药物分子设计与合成研究的意义4.1 治疗慢性疾病的需求随着人口老龄化的加剧,慢性疾病的发病率也在不断增加。
传统的药物已经无法满足对慢性疾病治疗的需求,因此需要开展新型药物的研究与合成。
4.2 高效低毒的药物研发传统药物研发过程中,往往需要大量的试验和研究,耗时耗力。
而新型药物分子设计与合成的研究则可以提高药物研发的效率,减少对动物实验和临床试验的依赖。
第五章:新型药物分子设计与合成研究的挑战5.1 多因素影响在药物分子设计与合成的过程中,往往需要综合考虑多种因素,如分子结构、活性和毒性等。
这些因素之间的复杂相互关系增加了研究的难度。
5.2 多学科交叉新型药物分子设计与合成的研究需要涵盖化学、生物学、药学等多个学科领域的知识。
不同学科之间的交叉合作和交流是推动研究进展的关键。
第六章:新型药物分子设计与合成研究的新进展6.1 基于人工智能的药物设计人工智能技术的发展为药物分子的设计提供了新的思路。
【2020考研】中国药科大学710 药学基础综合(一) 适用专业汇总
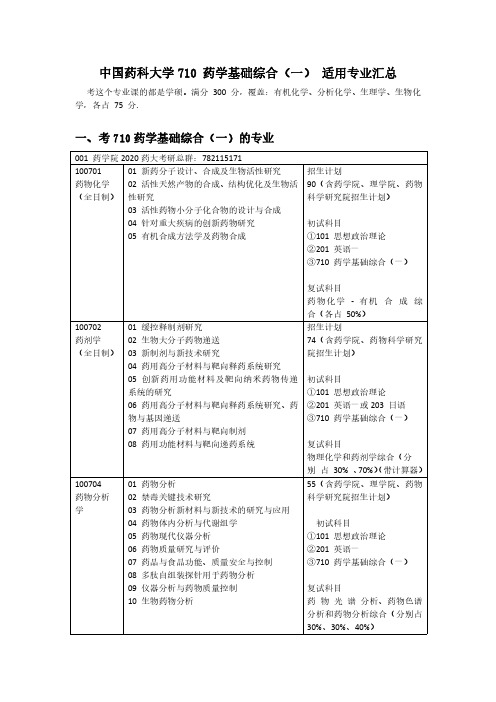
中国药科大学710药学基础综合(一)适用专业汇总考这个专业课的都是学硕。
满分300分,覆盖:有机化学、分析化学、生理学、生物化学,各占75分.一、考710药学基础综合(一)的专业001药学院2020药大考研总群:782115171100701药物化学(全日制)01新药分子设计、合成及生物活性研究02活性天然产物的合成、结构优化及生物活性研究03活性药物小分子化合物的设计与合成04针对重大疾病的创新药物研究05有机合成方法学及药物合成招生计划90(含药学院、理学院、药物科学研究院招生计划)初试科目①101思想政治理论②201英语一③710药学基础综合(一)复试科目药物化学-有机合成综合(各占50%)100702药剂学(全日制)01缓控释制剂研究02生物大分子药物递送03新制剂与新技术研究04药用高分子材料与靶向释药系统研究05创新药用功能材料及靶向纳米药物传递系统的研究06药用高分子材料与靶向释药系统研究、药物与基因递送07药用高分子材料与靶向制剂08药用功能材料与靶向递药系统招生计划74(含药学院、药物科学研究院招生计划)初试科目①101思想政治理论②201英语一或203日语③710药学基础综合(一)复试科目物理化学和药剂学综合(分别占30%、70%)(带计算器)100704药物分析学01药物分析02禁毒关键技术研究03药物分析新材料与新技术的研究与应用04药物体内分析与代谢组学05药物现代仪器分析06药物质量研究与评价07药品与食品功能、质量安全与控制08多肽自组装探针用于药物分析09仪器分析与药物质量控制10生物药物分析55(含药学院、理学院、药物科学研究院招生计划)初试科目①101思想政治理论②201英语一③710药学基础综合(一)复试科目药物光谱分析、药物色谱分析和药物分析综合(分别占30%、30%、40%)100706药理学01结构药理02抗炎免疫药理03神经药理、神经精神药理04生化药理05心血管药理06代谢药理07分子药理毒理、高通量高内涵药物筛选08肝脏药理、毒理09肿瘤药理10医学生物信息学11光功能探针用于疾病标记物检测研究12纳米载物材料用于疾病诊疗81(含药学院、药物科学研究院、基础医学与临床药学学院招生计划)初试科目①101思想政治理论②201英语一③710药学基础综合(一)复试科目药理学-药物毒理学综合(分别占70%、30%)1007Z6药物代谢动力学01创新药物的药物代谢动力学研究02代谢组学、蛋白质组学研究03中药复方活性物质组及药效机制研究04内源性物质代谢调控与免疫05药物代谢转运系统的调控机理及药物相互作用研究06转化药动/药效新模型研究07代谢调控08基于代谢-蛋白组学技术的内源性小分子及药物的靶标发现研究09内源性活性物质代谢调控与免疫10药代动力学-药效动力学结合模型11药物代谢酶/转运体表达调控机制研究12中药生物大分子体内外过程研究13复杂组分代谢处置与药效物质基础研究14生物药物的药代动力学研究15临床前药代动力学、临床药代动力学新理论和新模型研16中药PK-PD17生理药代动力学45(含药学院、药物科学研究院、基础医学与临床药学学院招生计划)初试科目①101思想政治理论②201英语一③710药学基础综合(一)复试科目药物代谢动力学-药理学综合(各占50%)002中药学院2020药大考研总群:782115171100703生药学01生药鉴定、活性成分与质量评价、质量控制及标准研究02药用植物资源与质量评价研究03中药活性成分与生物技术研究04中药活性成分质量控制与体内分析研究05中药药效物质基础与质量控制研究06天然药物分子药理学07天然药物活性组分与创新中药研究08天然药物体内合成及作用机制的蛋白质组学研究09中药活性成分发现与质量评价45(含中药学院、药物科学研究院招生计划)初试科目①101思想政治理论②201英语一③710药学基础综合(一)复试科目生药学1007Z9天然药物化学01天然产物结构修饰、合成和生物活性研究02天然药物与中药的活性成分研究21初试科目①101思想政治理论②201英语一或203日语③710药学基础综合(一)复试科目药物化学-有机合成综合(各占50%)或天然药物化学及波谱解析(各占50%)或药理学1008Z1中药化学01中药化学成分及质量标准研究02中药化学成分研究及新药开发18初试科目①101思想政治理论②201英语一或203日语③710药学基础综合(一)复试科目天然药物化学及波谱解析(各占50%)1008Z2中药生物技术学01中药资源利用与生物技术02中药活性成分体内过程与生物技术03中药新药研发与生物技术7初试科目①101思想政治理论②201英语一③710药学基础综合(一)复试科目中药生物技术1008Z3中药药理学01中药及复方药理学02中药及天然药物分子药理学03中药抗代谢性疾病药理学04中药抗炎免疫药理学05中药抗肿瘤药理学06中药神经精神药理学及毒理学28①101思想政治理论②201英语一③710药学基础综合(一)药理学1008Z4中药制剂学01药物制剂新剂型与新工艺的研究02中药新药创制研究03中药制剂新剂型与新技术11①101思想政治理论②201英语一③710药学基础综合(一)中药药剂学1008Z5中药分析学01现代中药分析02中药活性成分质量控制与体内分析研究03中药谱效关系研究5①101思想政治理论②201英语一③710药学基础综合(一)中药分析学1008Z6中药资源学01中药资源与新药开发02中药资源与质量2①101思想政治理论②201英语一③710药学基础综合(一)中药资源学003生命科学与技术学院2020药大考研总群:782115171100705微生物与生化药学01微生物药物和生化与生物技术药物的开发与应用02抗感染药物的药效及机理研究03生物新药的基因工程和蛋白质工程研究04抗体药物研究与开发05微生物药物资源开发与利用06微基因药物与基因治疗07药物相关基因的表达与调控08功能片段和肽疫苗的设计研究09微生物和生化药物相关的基础研究64①101思想政治理论②201英语一③710药学基础综合(一)微生物学与生物技术1007Z3药物生物信息学01计算机辅助药物设计2①101思想政治理论②201英语一③710药学基础综合(一)生物信息学1007Z4海洋药物学01海洋天然活性产物与海洋药物的研究2①101思想政治理论②201英语一③710药学基础综合(一)微生物学与生物技术008工学院2020药大考研总群:7821151711007Z1制药工程学01制药分离工程02制药污染控制工程03制药装备的设计、优化和在线监测04肿瘤靶向药物研究19①101思想政治理论②201英语一③710药学基础综合(一)制药工程学-化工原理综合(各占50%)(带计算器)009基础医学与临床药学学院1007Z5临床药学01合理用药与临床药物评价02临床药物代谢动力学7①101思想政治理论②201英语一③710药学基础综合(一)临床医学概论和临床药理学综合(各占50%)。
中国药科大学cadd重点概括

第1章概论一、药物发现一般过程新药的研究有三个决定阶段:先导化合物的发现,新药物的优化研究,临床与开发研究。
计算机辅助药物设计的主要任务就是先导化合物的发现与优化。
二、合理药物设计1、合理药物设计(rational drug design)是依据与药物作用的靶点,即广义上的受体,如酶、受体、离子通道、病毒、核酸、多糖等,寻找和设计合理的药物分子。
通过对药物和受体的结构在分子水平甚至电子水平的全面准确了解进行基于结构的药物设计和通过对靶点的结构、功能、与药物作用方式及产生生理活性的机理的认识基于机理的药物设计。
CADD通过内源性物质或外源性小分子作为效应子作用于机体的靶点,考察其形状互补,性质互补(包括氢键、疏水性、静电等),溶剂效应及运动协调性等进行分子设计。
2、方法分类(1)合理药物设计有基于靶点结构的三维结构搜索和全新药物设计等方法。
后者分为模板定位法、原子生长法、分子碎片法(碎片连接法和碎片生长法)。
(2)根据受体是否已知分为直接药物设计和间接药物设计。
前者即通过结构测定已知受体或受体-配体复合物的三维结构,根据受体的三维结构要求设计新药的结构。
受体结构测定方法:同源模建(知道氨基酸序列不知道空间结构时),X射线衍射(可结晶并得到晶体时),多维核磁共振技术(溶液状态)。
后者通过一些配体的结构知识(SAR,计算机图形显示等)推测受体的图像,提出家乡受体,采用建立Pharmacophore模型或3D-QSAR和基于药效团模型的三维结构搜索等方法,间接进行药物设计。
三、计算化学计算化学包括分子模型、计算方法、计算机辅助分子设计(CAMD)、化学数据库及有机合成设计。
计算方法包括很多种,但基本上可以分为两大类:分子力学和量子力学(分为从头计算方法和半经验方法)。
常用的计算应用有:(1)单点能计算:根据模型中原子的空间位置给出相应原子坐标的势能;(2)几何优化:系统的修改原子坐标使原子的三维构象能量最小化;(3)性质计算:预测某些物理化学性质,如电荷、偶极矩、生成热等;(4)构象搜索:寻找能量最低的构象;(5)分子动力学模拟:模拟分子的构象变化。
药物化学中的靶向药物设计与合成

药物化学中的靶向药物设计与合成药物化学是一门综合性较强的学科,旨在研发新型药物及改良现有药物的性能与效果。
其中,靶向药物设计与合成是药物化学领域的一个重要方向。
本文将探讨靶向药物设计的原理以及最新的合成方法与技术。
一、靶向药物设计的原理靶向药物设计的核心目标是将药物直接作用于疾病相关的靶点,从而提高疗效并减少副作用。
靶点可以是具有特定功能的蛋白质、细胞表面受体或其他与疾病有关的生物分子。
药物设计师常常通过研究靶点的结构与功能,利用计算机辅助设计技术来发现新的药物候选化合物并进行优化。
在药物设计的初期阶段,设计师首先利用药物库中的化合物或已知药物分子作为起始点,与靶点结构进行分子对接实验。
通过计算机模拟,可以预测药物分子与靶点之间的结合模式、亲合力等参数,从而筛选出具有较高亲和力的候选化合物。
其次,药物设计师利用有机合成的方法,通过合成一系列结构相关的化合物,并进行生物活性评价。
这一过程中需要考虑化合物的结构与性质,以及与靶点的相互作用。
通过不断优化化合物的结构和性能,筛选出具有较好药效和选择性的化合物。
最后,经过体外和体内的测试验证,合适的候选化合物将进一步进行药代动力学和安全性的评估,常通过严格的临床试验来验证其疗效和安全性。
二、靶向药物合成的方法与技术靶向药物的合成是药物化学中的一项重要任务。
合成出高纯度、高收率的目标化合物是靶向药物设计成功的基础。
目前,靶向药物的合成主要依赖于有机合成化学的一系列方法与技术。
其中,常用的包括保护基策略、反应活性基团的引入与修饰、立体化学控制以及合成路径的设计等。
在药物合成过程中,常常需要引入保护基,以防止非特定反应的发生。
例如,保护羟基、胺基或羧基等,可以通过选择适合的保护基来实现特定位点的修饰。
此外,针对药物分子中的具有反应活性的基团,如亲电基团或亲核基团等,可以通过选择适当的反应条件和试剂来引入或修饰这些基团,实现有选择性的反应。
在合成路径的设计中,灵活的合成顺序和选择适当的试剂、溶剂以及反应条件对于提高产率和纯度至关重要。
药物合成实验

《药物合成反应》实验部分教学大纲(非独立设课的课程实验)药物合成实验是制药工程专业的一门重要的专业方向选修实验课,通过对这门课程的教学,达到培养学生动手能力、自学能力、观察和思维理解能力、分析与解决问题能力以及创新意识,培养学生良好的实验工作方法和工作习惯、实事求是和严谨的科学态度、理论联系实际的能力,进一步巩固课堂理论知识,为今后学习后继课程和科学研究打下良好的基础的目的。
作为未来的制药领域的科技人员,也需要通过本课程的学习获得必要实验技能。
三、实验教学内容与要求1、绪论部分四、作业(报告)要求:实验报告:其内容包括:实验目的、路线设计、仪器药品、具体操作方法、实验结果及讨论。
其中路线设计、具体操作方法是实验报告的重要部分,并对实验现象进行分析和解释。
五、实验考核方式:实验考核方式采用动手操作结合平时成绩。
实验考核安排在学期实验结束后进行。
六、成绩评定:(1)平时成绩:含预习报告、实验操作态度和实验报告占50%;(2)实验操作考核成绩占50%。
七、必要的说明执笔人:聂丽审核人:张强制(修)订时间:2011年7月15日药物合成反应实验实验一苯佐卡因的制备一、实验目的1、通过制备对氨基苯甲酸了解酯化反应的原理和方法。
2、掌握回流、重结晶、萃取等基本操作技能。
二、实验原理本实验以对氨基苯甲酸为原料,在强酸性条件下与乙醇发生酯化反应,生成目标产物对氨基苯甲酸乙酯。
三、主要仪器与试剂仪器:圆底烧瓶、直形冷凝管、分液漏斗、抽滤装置试剂:对氨基苯甲酸、浓硫酸、95%乙醇、10%碳酸钠溶液、乙醚、无水硫酸镁四、实验步骤在50mL圆底烧瓶中加入2g对氨基苯甲酸和25mL 无水乙醇,振摇烧瓶使大部分固体溶解。
将烧瓶置于冰水浴中冷却,加入2mL浓硫酸,立刻产生大量沉淀(在接下来的回流中沉淀将逐渐溶解),将反应混合物在水浴上加热回流1.5h,并不时振摇。
回流结束后将反应混合物转入烧杯中,冷却后分批(缓慢?)加入固体碳酸钠粉末,可观察到有气体逸出,并产生泡沫(发生了什么反应?),直至无明显气泡产生。
化学制药工艺学PDF课件合集(中国药科大学)

一、类型反应法
二、分子对称法
三、逐步综合法 四、追溯求源法 �逆合成分析�
工艺路线
一、类型反应法
反应时间对异辛酸铋盐合成的影响
定义和思维方法
利用常见的典型有机化学反应与合成方 法 �按功能基形成的单元反应�比如卤化、 酯化等�把各单元反应串联起来�形成一 条工艺路线 �进行药物合成设计的思考方 法。其中包括应用各类化学结构的有机合 成物的通用合成法�官能团的形成、转换、 保护等合成反应单元以及重要的人名反应。
三、逐步综合法
抗结核药异烟肼�25�的合成
三、逐步综合法
�二�功能基的生成、保护与转化
多功能基药物的合成路线设计
具有多个功能基的药物的合成设计与基团之间 的相互影响�电子效应和立体效应�、各自的 理化特点、功能基引入的先后次序、保护基的 运用、活化部位利用等都有关系。设计时必须 综合考虑反应路线长短、反应顺序及反应条件 等多种因素。
和收率。
• 2、天然物的全合成及结构改造 • 发展有机化学理论和有机合成方法。 • 3、创新药物和已经上市药物�注意知识产
权�的生产。
• 生产的现实性、经济的合理性和技术的先进 性。
生产的现实性
• (1)原材料品种以少为好�并能保障供应。 • (2)原材料价格是否便宜。 • (3)尽可能避免使用有毒、易燃易爆的原材料。 • (4)尽可能简化合成反应及后处理操作,缩短
对于有明显类型结构特点以及官能团特点的化合物�可以 采用此法进行设计。
二、分子对称法
一、类型反应法
二、分子对称法
生物碱鹰爪豆碱�sparteine�16�的合成
2
二、分子对称法
抗麻风病药物克风敏�Clofazimine, 17�
中国药科大学药物化学复试真题

CPU08复试真题药化部分:一.写出下列药物作用靶点和临床用途。
5题10分二.问答。
50分1.组胺H1-R拮抗剂有镇静副作用,采用哪些方法降低其镇静副作用。
2.血管紧张素2受体拮抗剂抗高血压药物的发展。
3.生物电子等排体定义,分类,举例说明其优点(差不多之类的,记得不太清楚了)4.抗肿瘤药物的分类,近年来的发展。
5.前药的定义,分类,举例说明其目的,用途。
三。
合成题。
15分,四选三(都给出了化学式)1.萘普生2.氟康唑3.氟哌啶醇4.雷尼替丁有机合成部分:四.用反应式,举例说明,10分1.Knoevenagel反应2.场效应(F)3.Vilsmeier试剂4.Mitsunobu反应(不知写的对不对,去年考过了)5.Mannich反应五.完成反应式,有一个没见过,好像是什么Dimoth重排六.选择题5题10分七.合成题.35分1.以1-甲基-1,2,3,4-四氢萘-2-酮和丙酮合成,不好意思,不会命名,应该是菲类,大致描述一下:甾类化合物去掉D环(环戊环),A环是苯环,9位有甲基,8,13之间成双键,12位氧代。
其他试剂任选。
2.以香草醛,邻氨基苯甲酸,还有一个不记得了,合成曲尼司特(给了化学式)3.以醋酸甲酯合成3-甲基2-戊酮,要求产物中六个碳原子均来自醋酸甲酯衍生物。
CPU09复试真题药物化学部分一。
根据药物结构写出临床作用或者靶点(5x2)有喹诺酮类,1,4二氢吡啶类,吗啡类。
剩下两个我觉得是h1受体拮抗剂,和一个局部麻醉。
二。
简答题(意思是那样的)1根据药物代谢,说明其对新药研究和设计的作用2根据作用部位的不同,说明抗高血压药物的分类,并举一例3抗代谢肿瘤药物设计原理,举例说明4新药研发前期和后期的主要内容5喹诺类药物的构效关系三合成题(3x5)(题中均写了药物结构的)1氯霉素的合成主要原料对硝基苯已酮2雷诺嗪的合成3左氧氟沙星主要原料1,2,3,三氟—4-硝基苯药物合成一名词解释用反映举例加以说明(5x2)1氮杂环中的Dimoth重排2meerwein-ponndorf还原3claisen重排4curtius重排5blanc反应二选择题(5x2)三完成反应式(10x2)有个wittig-horner反应,mannich反应,烯丙胺的反应,fisher 吲哚成环的反应,其他的记不住了四合成(35)(题中均写了药物结构的)1用二乙醇胺为主要原料合成环磷酰胺(10)2慢心律的合成主要原料2,6—二甲基苯酚,和环氧丙烷(11)3名字不好说,叙述一下。
吡啶类IRAK4抑制剂的设计、合成及生物活性评价

吡啶类IRAK4抑制剂的设计、合成及生物活性评价朱宝;金双龙;郭毅;李月珍;张奕华;赖宜生【期刊名称】《中国药科大学学报》【年(卷),期】2017(48)6【摘要】在现有IRAK4抑制剂MK-32和AU-5的基础上,利用开环策略和拼合原理,结合分子对接技术,设计并合成了12个以吡啶为基本母核的目标化合物。
采用放射性同位素标记法评价了目标化合物对IRAK4激酶的抑制活性。
结果表明,该类化合物对IRAK4显现出良好的抑制活性。
其中5个化合物的IC50小于1μmol/L,值得进一步研究。
【总页数】5页(P670-674)【关键词】IRAK4抑制剂;设计;合成;生物活性【作者】朱宝;金双龙;郭毅;李月珍;张奕华;赖宜生【作者单位】中国药科大学新药研究中心,南京210009;中国药科大学有机化学教研室,南京210009;中国药科大学江苏省代谢性疾病药物重点实验室,南京210009【正文语种】中文【中图分类】R914.5【相关文献】1.吡咯并吡啶类整合酶抑制剂的设计、合成和生物活性研究 [J], 剧仑;毛志杰;王小利;刘伟;曾程初;胡利明2.组蛋白去乙酰化酶抑制剂N-(6-氧代-6-(苯氨基)己基)-2-氨基苯甲酰胺类衍生物的设计、合成与活性评价 [J], 朱小花;贺殿3.苯磺酰胺类mIDH2抑制剂的设计、合成及生物活性评价 [J], 刘鹏宇;姚坤;刘海鹏;杨杰;魏清筠;曹鹏;赖宜生4.联苯类PD-L1抑制剂的设计合成及生物活性评价 [J], 岳露; 金双龙; 高健; 郭文洁; 徐强; 赖宜生5.萘环类PD-1/PD-L1抑制剂的设计、合成及生物活性评价 [J], 赵磊;余龙波;欧阳宜强;郭文洁;徐强;高健;赖宜生因版权原因,仅展示原文概要,查看原文内容请购买。
药物设计与合成方法研究

药物设计与合成方法研究一、药物设计方法概述药物设计是指利用先进和现代的技术,结合对生物分子结构和功能的深入认识和探索,开发新颖的药物,提高药物的疗效和安全性的一项科学技术。
药物设计方法可以分为基于靶标、基于分子作用机制和基于结构活性关系等几个主要方向。
1.基于靶标的药物设计该方法的核心是对药物与靶标相互作用的认识,靶标又称受体,是指生物体内对物质的特异性结合部位。
当前,利用X-射线晶体学、核磁共振等技术,人们对生物大分子的结构已有了较深入的认识,尤其是人们借助计算机模拟技术,不断深入探索药物-受体相互作用机理,从而大大提高了基于靶标的药物设计的准确性。
2.基于分子作用机制的药物设计该方法是在对药物分子与生物体分子作用机制的深入认识下进行的药物设计。
药物分子在体内与生物分子发生特定的相互作用,从而产生预期的生物效应。
借助计算机模拟、分子为基础的组学等技术,可以对药物的作用机理进行深入研究,并在此基础上进行新药物的设计。
3.基于结构活性关系的药物设计这是一种传统的药物设计方法,早期经常采用。
它认为药物分子与靶标的相互作用是由于分子结构的特异性而导致的。
因此,设计良好的药物分子必须具有一定的分子结构特征和活性规律,从而使得药物分子能准确的与靶标相符合,从而达到预期效果。
二、合成方法的分类药物合成的方法可以分为化学方法和生物合成方法两种。
1.化学合成方法化学合成方法,是通过化学反应来完成新药物的生产制备。
这种方法通常需要选择合适的原料和条件,掌握强大的化学知识和技能,进行一系列复杂的化学反应,将原料转化为需要的药物,并进行后续的纯化和分离工作。
化学合成方法有着广泛的适用范围,同时也有着重要的局限性:产量低、制备时间长、成本高等,因此这种方法在选择适用范围较窄和制备难度较高的药物时更为常见。
2.生物合成方法生物合成方法即通过微生物或细胞进行合成的方法。
和化学合成方法不同,这种方法利用微生物、细胞及其代谢活动,从生物体内产生药物。
中国药科大学710 药学基础综合(一)专业分析--适用专业
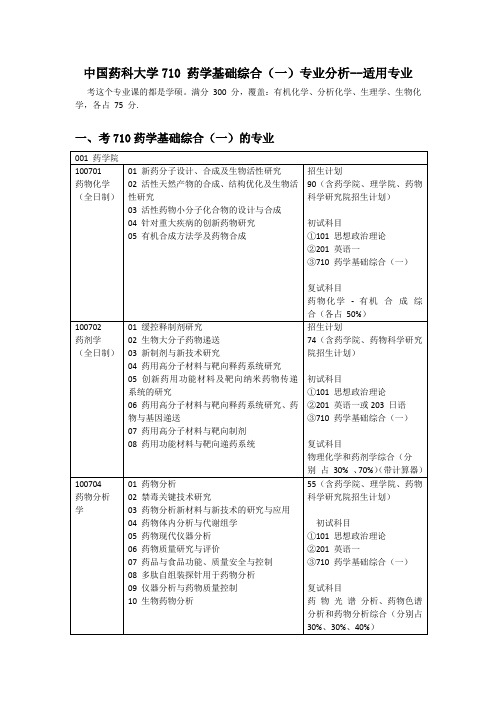
03新制剂与新技术研究
04药用高分子材料与靶向释药系统研究
05创新药用功能材料及靶向纳米药物传递系统的研究
06药用高分子材料与靶向释药系统研究、药
物与基因递送
07药用高分子材料与靶向制剂
08药用功能材料与靶向递药系统
招生计划
74(含药学院、药物科学研究院招生计划)
初试科目
①101思想政治理论
②201英语一
③710药学基础综合(一)
复试科目
药理学-药物毒理学综合(分别占70%、30%)
1007Z6
药物代谢
动力学
01创新药物的药物代谢动力学研究
02代谢组学、蛋白质组学研究
03中药复方活性物质组及药效机制研究
04内源性物质代谢调控与免疫
05药物代谢转运系统的调控机理及药物相互作用研究
06转化药动/药效新模型研究
②201英语一
③710药学基础综合(一)
生物信息学
1007Z4
海洋药物
学
01海洋天然活性产物与海洋药物的研究
2
①101思想政治理论
②201英语一
③710药学基础综合(一)
微生物学与生物技术
008工学院
1007Z1
制药工程
学
01制药分离工程
02制药污染控制工程
03制药装备的设计、优化和在线监测
04肿瘤靶向药物研究
性研究
03活性药物小分子化合物的设计与合成
04针对重大疾病的创新药物研究
05有机合成方法学及药物合成
招生计划
90(含药学院、理学院、药物科学研究院招生计划)
初试科目
①101思想政治理论
②201英语一
药物合成与药物设计实践

药物合成过程中可能产生大量废弃物和有毒有害物质,对环境和人类健康造成潜在威胁。
合成步骤复杂
药物合成往往涉及多步骤反应,每步反应都需要精确控制条件,使得整个合成过程既耗时又易出错。
绿色合成技术
未来药物合成将更加注重环保和可持续发展,采用绿色合成技术减少废弃物和有毒有害物质的产生。
智能化合成与自动化生产
05
06
03
02
药物合成实验步骤一般包括原料准备、反应条件设置、反应过程监控、产物分离纯化等。
以阿司匹林合成为例,演示典型药物合成实验操作
准备原料:水杨酸、乙酸酐、浓硫酸等。
设置反应条件:将原料按一定比例混合,加热至一定温度进行反应。
监控反应过程:通过TLC等方法监控反应进程,确保反应完全。
产物分离纯化:通过萃取、重结晶等方法对产物进行分离纯化,得到纯净的阿司匹林。
CHAPTER
药物设计实践案例分析
靶点选择与确认
药物分子设计与筛选
体外与体内药效评价
临床试验与上市
针对特定癌症类型,确定关键靶点蛋白,如激酶、受体等。
通过细胞实验和动物模型验证药物分子的抗肿瘤活性及安全性。
基于靶点结构,运用计算机辅助药物设计(CADD)方法进行分子对接和虚拟筛选,获得潜在活性分子。
在临床试验中验证抗病毒药物的疗效和安全性后,广泛应用于临床治疗。
深入研究神经系统疾病的发病机制和关键靶点,如神经递质受体、离子通道等。
神经系统疾病机制研究
基于靶点结构和功能特点,设计并评价具有潜在治疗作用的神经系统药物分子。
药物分子设计与评价
通过体外细胞实验和体内动物模型验证药物分子的神经保护作用、抗癫痫作用等药效学特性。
CHAPTER
药物化学中的化合物设计与合成

药物化学中的化合物设计与合成在药物研发的过程中,化合物设计与合成是至关重要的环节。
通过合理的化合物设计和高效的合成方法,研究人员可以开发出具有良好药理活性和药代动力学特性的化合物,为新药的发现和开发提供有力支持。
一、化合物设计的基本原理化合物设计是指根据目标疾病的靶点或生理过程,利用计算化学、结构生物学等方法,设计出具有良好活性的化合物。
在化合物设计的过程中,需要考虑以下几个方面的因素:1. 靶点结构和特点:了解目标蛋白的结构、功能以及与疾病相关的生理过程,从而确定合适的靶点。
2. 结构活性关系:通过分析已有活性物质的结构特点和活性数据,找到结构活性关系的规律,为设计新的化合物提供指导。
3. 药物代谢动力学特性:考虑化合物的溶解度、渗透性、代谢稳定性等药代动力学特性,以确保药物在体内具有良好的吸收、分布和排泄特性。
4. 安全性和毒性考虑:考虑化合物对机体的毒性作用,预测和评估潜在的不良反应,以确保化合物在体内的安全性。
二、合成方法的选择与优化化合物设计确定后,接下来就是合成该化合物的问题。
合成方法的选择与优化对于药物的研发至关重要,主要包括以下方面的内容:1. 合成路线设计:根据化合物的结构特点和合成难易程度,设计合成路线,确定所需原料和反应条件。
2. 反应选择和优化:选择合适的反应类型和反应条件,优化反应条件以提高产率和纯度。
3. 保护基的选择与去除:在合成过程中,可能需要保护一些活性基团以避免其在反应中被破坏,同时在必要时去除保护基。
4. 紧凑性和高效性:合成路线尽可能紧凑,化合物转化步骤尽可能少,以提高合成效率和减少中间体的制备。
5. 检验和纯化:合成后需要对化合物进行结构鉴定和纯化,确保化合物的纯度和结构与设计一致。
三、前沿技术在化合物设计与合成中的应用随着科学技术的不断进步,越来越多的前沿技术被应用于药物化学中的化合物设计与合成。
其中,计算化学和合成生物学是两个较为重要的方向。
1. 计算化学:通过在计算机上进行模拟和计算,预测化合物的生理活性、药效和药代动力学性质,为化合物设计提供理论指导。
新型药物的设计和合成研究

新型药物的设计和合成研究近年来,随着生物技术和化学合成技术的不断进步,新型药物的设计和合成研究已成为药物研究领域的一个重要研究方向。
新型药物的研发对临床医学应用、人类健康和医疗保健产业的发展都具有极为重要的意义。
药物研究中,新型药物的研发流程主要分为以下几个步骤:药物的设计,化合物预筛选,活性物质筛选,化学结构最优化,临床前期研究和临床研究等。
其中,药物设计和化学结构最优化是最关键的两个环节。
药物的设计主要包括以下几个方面:基于生物机制的药物设计,基于结构活性关系的药物设计和基于计算机辅助设计的药物设计。
基于生物机制的药物设计主要是通过对药物与靶点相互作用的机制进行研究,从而设计出更加高效的药物。
基于结构活性关系的药物设计则是通过化学结构的调整,来改变药物的药效,从而设计出更加具有选择性和高效的药物。
基于计算机辅助设计的药物设计则是利用计算机技术对药物进行运算和模拟,从而设计出更加优秀的药物结构。
化学结构最优化是将药物的分子结构进行改造,以达到更好的药效。
常用的化学结构最优化方法包括化学合成和药物修饰。
化学合成是通过化学反应,将化合物进行改造,以达到更好的药效。
药物修饰是针对已有的药物结构进行微调和改造,以提高药效和降低副作用。
对于对新型药物的研究,首先需要对有一定代表性的靶点进行筛选和确认。
通过对靶点的研究和筛选,可以获得有关靶点的化学性质和生物机制等信息,为药物的设计和化学结构最优化提供依据。
在筛选的靶点中,不仅包括疾病的主要靶点,还包括疾病的治疗辅助靶点,以及一些具有重要作用的靶点。
基于已有的药物结构,在药物的设计和合成过程中,需要注意药物的结构和生物活性之间的关系。
药物分子的结构特征、物理化学性质和生物反应活性都是相互关联的。
化学结构最优化过程中,需要考虑生物活性的因素,如药物对靶点的亲和力和选择性等。
同时还需要考虑药物分子的理化性质,如溶解度、生物利用度、药代动力学等。
在药物的研发过程中,除了药物的设计和化学结构最优化,药物的制备也是一个至关重要的环节。
盐酸右哌甲酯的合成及工艺改进

盐酸右哌甲酯的合成及工艺改进
吴增;余永强;朱雍;陆涛
【期刊名称】《海峡药学》
【年(卷),期】2010(022)009
【摘要】目的合成盐酸右哌甲酯,并对合成工艺进行改进.方法以苯甲酰甲酸甲酯为原料,经过十步反应得到盐酸右哌甲酯.结果总收率为26.24%.经核磁共振谱及质谱确证结构.结论该工艺收率与文献报道基本持平,但简化了步骤,缩短了反应时间并降低了成本.
【总页数】3页(P211-213)
【作者】吴增;余永强;朱雍;陆涛
【作者单位】中国药科大学有机化学教研室,南京,210009;中国药科大学有机化学教研室,南京,210009;中国药科大学有机化学教研室,南京,210009;中国药科大学有机化学教研室,南京,210009
【正文语种】中文
【中图分类】TQ460.4
【相关文献】
1.盐酸托莫西汀与盐酸哌甲酯治疗儿童注意缺陷多动障碍的对比研究 [J], 朱舒虹;李思涛;符平
2.盐酸托莫西汀和盐酸哌甲酯治疗注意缺陷多动障碍门诊患儿的随机双盲对照研究[J], 李飞;苏林雁;刘军;朱焱
3.盐酸哌甲酯控释片与盐酸托莫西汀治疗儿童注意缺陷多动障碍对比研究 [J], 李
瑾
4.盐酸托莫西汀(ATX)与盐酸哌甲酯(MPH)治疗儿童注意缺陷多动障碍(ADHD)的临床疗效及安全性比较 [J], 梁淑晶
5.FDA批准30分钟起效的盐酸右哌甲酯缓释胶囊 [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
化学合成新药物研发技术

化学合成新药物研发技术已经成为当今医药领域的主流,因为它可以用更高效的方式来发现新药物和治疗疾病。
下文将从以下几方面介绍新药物化学合成的研发技术。
一、分子设计分子设计是化学研发新药物的核心技术之一。
它是通过分子模拟、药效团识别和结构相似性分析等技术,在药物候选化合物中找到有可能存在药效和药物代谢稳定性好的分子组合。
一般来说,分子设计的方式有双向模型和单向模型两种。
其中,双向模型侧重于药效团的引入和各种构象的优化,而单向模型则注重分子内部化学键交换和表面化学反应的分子变换。
无论是哪种模型,分子设计都是中的重要环节。
二、药物合成药物合成是为获得药物的诸多药代动力学性质、药效、药物毒性等而进行的一项繁琐的工作。
药物合成是化学合成药物需要进行的非常精确的步骤,这些步骤需要考虑药物中间体、合成反应、离子化、最终中产品等多种化学反应。
遗传工程和化学合成一起,可以让研究人员获得各种不同于自然来源的药物治疗剂。
三、活性分子筛选活性分子筛选是HiThroughput技术中比较重要的一部分。
它利用增量分析识别活性分子。
在分析活性分子之前,研究人员在大量的小分子化合物的库中筛选出几个具有活性物质的化合物,然后通过不断的筛选、细化和优化,最终获得一种有效的药物。
四、高通量技术高通量技术又称高通量筛选技术(HTS),是一种用于在最短时间内对高通量结果进行开发和协调的技术。
它将小分子化合物库与蛋白质系统集成在一起,从而通过大量的小分子药物的极高速度筛选出自然源中药效高的组分。
五、新型载体新型载体是利用不同的载体把药物和分子组合物结合起来,并加速其运输到需要治愈的部位。
它可以通过化学变化、招募基因、靶向蛋白质等不同的机制来增强药物的治疗效果。
新型载体可以使药物更容易、更人性化的给到患者,并且可以彻底解决药物在体内的不稳定性和代谢难度问题。
六、仿生合成仿生合成是利用生物合成学的原理,在生物场合下进行化学反应,从而获得更高分子量的化合物和更高的反应效率。
新药设计与合成——中国药科大学
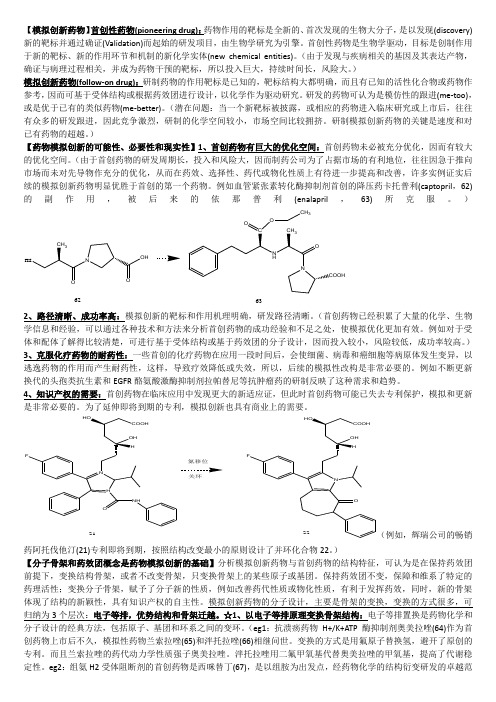
【模拟创新药物】首创性药物(pioneering drug):药物作用的靶标是全新的、首次发现的生物大分子,是以发现(discovery)新的靶标并通过确证(Validation)而起始的研发项目,由生物学研究为引擎。
首创性药物是生物学驱动,目标是创制作用于新的靶标、新的作用环节和机制的新化学实体(new chemical entities)。
(由于发现与疾病相关的基因及其表达产物,确证与病理过程相关,并成为药物干预的靶标,所以投入巨大,持续时间长,风险大。
)模拟创新药物(follow-on drug):研制药物的作用靶标是已知的,靶标结构大都明确,而且有已知的活性化合物或药物作参考,因而可基于受体结构或根据药效团进行设计,以化学作为驱动研究。
研发的药物可认为是模仿性的跟进(me-too),或是优于已有的类似药物(me-better)。
(潜在问题:当一个新靶标被披露,或相应的药物进入临床研究或上市后,往往有众多的研发跟进,因此竞争激烈,研制的化学空间较小,市场空间比较拥挤。
研制模拟创新药物的关键是速度和对已有药物的超越。
)【药物模拟创新的可能性、必要性和现实性】1、首创药物有巨大的优化空间:首创药物未必被充分优化,因而有较大的优化空间。
(由于首创药物的研发周期长,投入和风险大,因而制药公司为了占据市场的有利地位,往往因急于推向市场而未对先导物作充分的优化,从而在药效、选择性、药代或物化性质上有待进一步提高和改善,许多实例证实后续的模拟创新药物明显优胜于首创的第一个药物。
例如血管紧张素转化酶抑制剂首创的降压药卡托普利(captopril ,62)的副作用,被后来的依那普利(enalapril ,63)所克服。
)NOHONCCOOHCH 3O62632、路径清晰、成功率高:模拟创新的靶标和作用机理明确,研发路径清晰。
(首创药物已经积累了大量的化学、生物学信息和经验,可以通过各种技术和方法来分析首创药物的成功经验和不足之处,使模拟优化更加有效。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
【模拟创新药物】首创性药物(pioneering drug):药物作用的靶标是全新的、首次发现的生物大分子,是以发现(discovery)新的靶标并通过确证(Validation)而起始的研发项目,由生物学研究为引擎。
首创性药物是生物学驱动,目标是创制作用于新的靶标、新的作用环节和机制的新化学实体(new chemical entities)。
(由于发现与疾病相关的基因及其表达产物,确证与病理过程相关,并成为药物干预的靶标,所以投入巨大,持续时间长,风险大。
)模拟创新药物(follow-on drug):研制药物的作用靶标是已知的,靶标结构大都明确,而且有已知的活性化合物或药物作参考,因而可基于受体结构或根据药效团进行设计,以化学作为驱动研究。
研发的药物可认为是模仿性的跟进(me-too),或是优于已有的类似药物(me-better)。
(潜在问题:当一个新靶标被披露,或相应的药物进入临床研究或上市后,往往有众多的研发跟进,因此竞争激烈,研制的化学空间较小,市场空间比较拥挤。
研制模拟创新药物的关键是速度和对已有药物的超越。
)【药物模拟创新的可能性、必要性和现实性】1、首创药物有巨大的优化空间:首创药物未必被充分优化,因而有较大的优化空间。
(由于首创药物的研发周期长,投入和风险大,因而制药公司为了占据市场的有利地位,往往因急于推向市场而未对先导物作充分的优化,从而在药效、选择性、药代或物化性质上有待进一步提高和改善,许多实例证实后续的模拟创新药物明显优胜于首创的第一个药物。
例如血管紧张素转化酶抑制剂首创的降压药卡托普利(captopril ,62)的副作用,被后来的依那普利(enalapril ,63)所克服。
)NOHONCCOOHCH 3O62632、路径清晰、成功率高:模拟创新的靶标和作用机理明确,研发路径清晰。
(首创药物已经积累了大量的化学、生物学信息和经验,可以通过各种技术和方法来分析首创药物的成功经验和不足之处,使模拟优化更加有效。
例如对于受体和配体了解得比较清楚,可进行基于受体结构或基于药效团的分子设计,因而投入较小,风险较低,成功率较高。
)3、克服化疗药物的耐药性:一些首创的化疗药物在应用一段时间后,会使细菌、病毒和癌细胞等病原体发生变异,以逃逸药物的作用而产生耐药性,这样,导致疗效降低或失效,所以,后续的模拟性改构是非常必要的。
例如不断更新换代的头孢类抗生素和EGFR 酪氨酸激酶抑制剂拉帕替尼等抗肿瘤药的研制反映了这种需求和趋势。
4、知识产权的需要:首创药物在临床应用中发现更大的新适应证,但此时首创药物可能已失去专利保护,模拟和更新是非常必要的。
为了延伸即将到期的专利,模拟创新也具有商业上的需要。
21氮移位关环药阿托伐他汀(21)专利即将到期,按照结构改变最小的原则设计了并环化合物22。
)【分子骨架和药效团概念是药物模拟创新的基础】分析模拟创新药物与首创药物的结构特征,可认为是在保持药效团前提下,变换结构骨架,或者不改变骨架,只变换骨架上的某些原子或基团。
保持药效团不变,保障和维系了特定的药理活性;变换分子骨架,赋予了分子新的性质,例如改善药代性质或物化性质,有利于发挥药效,同时,新的骨架体现了结构的新颖性,具有知识产权的自主性。
模拟创新药物的分子设计,主要是骨架的变换,变换的方式很多,可归纳为3个层次:电子等排,优势结构和骨架迁越。
☆1、以电子等排原理变换骨架结构:电子等排置换是药物化学和分子设计的经典方法,包括原子、基团和环系之间的变环。
(eg1:抗溃疡药物 H+/K+ATP 酶抑制剂奥美拉唑(64)作为首创药物上市后不久,模拟性药物兰索拉唑(65)和泮托拉唑(66)相继问世。
变换的方式是用氟原子替换氢,避开了原创的专利。
而且兰索拉唑的药代动力学性质强子奥美拉唑。
泮托拉唑用二氟甲氧基代替奥美拉唑的甲氧基,提高了代谢稳定性。
eg2:组氨H2受体阻断剂的首创药物是西咪替丁(67),是以组胺为出发点,经药物化学的结构衍变研发的卓越范例,其后继的模拟创新药物如雷尼替丁(68)和法莫替丁(69)等,分别是用呋喃和噻唑环代替了西咪替丁的咪唑环,同时对侧链的取代基作适当的变换以调整分子的碱性,使得模拟创新的替丁超越了首创分子。
)NHNSH 2CNOCH 3CH 3H 3CH 3COO 64HNSH 2CNOCH 2CF 3H 3CO 65N HNSH 2CNOCH 3H 3COF 2HCOO 66OSN HN H3NCH 3H 3CNO 2HNNSN HN HCH 3NCNCH 3SNSN HNH 2NNH 2H 2NNSO 2NH 26768692、以优势结构为导向的变换骨架结构:治疗男性勃起障碍的磷酸二酯酶5抑制剂西地那非(sildenafil ,70)是首创药物,虽是偶然发现的,却具有划时代的意义。
伐地那非(vardenafil ,71)是将母核骨架异嘌呤的氮原子易位,成为新的骨架,乌地那非(udenafil ,72)是韩国2005上市的模拟新药,其药效学强度和选择性以及药代动力学性质均优于西地那非,且研发的时间与成本也低于西地那非。
NNH 3CSOC 2H 5NHNNNCH 3CH 3OOONNC 2H 5SOC 2H 5NNHNNCH 3CH 3OOON HSONHNNNCH 3CH 3OOON CH3CH 3707172CCCH 3CH 3CH 376773、以结构-活性演化的方式进行骨架迁越:钠葡萄糖共转运蛋白2(sodium glucose cotransporter-2,SGLT-2)是治疗2型糖尿病的药物靶标,最初发现SGLT 抑制剂是天然产物二氢查耳酮根皮苷(phlorizin ,73),经骨架迁越将苯酚环变成苯并呋喃得到T-l095(74),现处于I1期临床研究。
Serglitlozin(75)是将天然产物的两个苯环距离缩短成一个碳原子,为碳酸酯前药,处于II 期临床研究。
Dapagliflozin(76)模拟77的二苯甲基骨架,但将0-葡萄糖苷变换成 C-糖苷,提高了稳定性。
76处于III 期临床研究阶段。
化合物77是通过螺环将糖环固定,对构象加以限制,成为新结构类型的SGLT 抑制剂。
右芬氟拉明(dexfenfluramine ,78)为 5HT2C 受体激动剂,最初作为减肥药批准上市,但一年后(1997)被终止使用,系具有使心脏瓣膜发生变形的严重副作用。
后来证明心脏瓣膜的副作用是由于右芬氟拉明同时对5HT2B 的激动作用,所以,消除右芬氟拉明激动 5HT2B 的作用,提高对5HT2c 亚型的选择性活性,是研发减肥药的重要途径。
为此,对右芬氟拉明加以构象限制,得到苯并氮卓化合物lorcaserin(79),它对5HT2c 的选择性作用强于5HT2B 100倍,每日口服 l0mg 两次连续一年可降低体重3.6 kg ,未见心脏瓣膜的不良变化,目前FDA 已受理其上市申请。
F 3CNHCH 3NHClH 3C7879【双靶标药物】新药研究的两种模式:1、以表型为基础的研究模式。
在此之前,研制新药和评价化合物的活性,主要是以动物模型、离体器官或组织以及细胞的生理学或表型变化为指标,观察生物体的宏观现象的变化作为新药的评价体系。
其后,以研究正常组织与病理状态的蛋白质差异为切入点,开始了以生物靶标为核心的新药研发模式。
☆表型(phenotype)泛指有机体呈现的各种可观测的性质或特征,例如形态,发育,生化,生理性质以及行为等,起因于基因的表达或环境因素的影响或者两者之间的相互作用。
√以表型为基础的药物研究,将机体、器官或组织作为研究对象,药物作为探针,观察与疾病相关的模型出现的生理效应,过去是先导物的发现与优化的主要方法,研制的起始点为化学所驱动。
由于评价方法的限制,化合物的筛选数量有限,时间较长。
在药物靶标被确定之前,大都采用这种模式。
这种模式的优点在于,用整体动物或组织器官作宏观评价,犹如“生物平台”,在评价药效的同时,一定程度上还反映了药物的吸收、分布等药代行为和化合物的安全性,而且呈现的药效也是生理学的总体表现,所以有较高的成药效率。
2、以生物靶标为核心的研究模式:以生物靶标为核心的药物研究,认为大多数的疾病的发生、发展与形成是由于某种蛋白的异常而产生的,纠正或调节异常蛋白功能趋于正常状态,有可能达到控制或治愈的目的。
这种研发模式由于简便或快捷给人们带来了巨大的期望。
随着表达和纯化蛋白技术的成熟化,引发了组合化学、高通量筛选和基于结构的理性设计,已成为新药研发的主要模式。
近30年来,全球投入新药研发的经费剧增,但新分子实体(NME)的数量并没有相应增加,投入产出比失调,分析其原因是:第一,所选定的靶标未必或较少与疾病关联,缺乏临床数据或动物模型数据的支持;第二,离体的蛋白与机体中的蛋白所处的环境差异很大,例如体外实验是分子直接相互作用,而体内蛋白所处的环境有血液、营养物和激素的供应以及反馈机制的调节,会减弱或缓冲药物对蛋白的作用;第三,疾病作为一个系统具有稳定性,一些重要的疾病如肿瘤、代谢性和CNS疾病等一旦形成就非常顽强和皮实,以致抑制单一靶标不能影响疾病的整体状态。
双靶标作用的优势:同时干扰两个或多个环节可提高治疗的效果。
(多数的中枢神经系统和心脑血管的药物大都作用于多种受体靶标。
奥氮平(olanzapine,80)对至少10个受体亚型的拮抗作用达nmol/L水平,最初被贬为“赖药”(dirty drug),却是世界销量领先的抗精神病药物。
非甾体抗炎药阿司匹林、降血糖药二甲双胍以及抗白血病药物伊马替尼(imatinib,81)等都是作用子多靶标的药物。
)多种药物组合治疗效果优于单一药物的治疗,也说明多靶标作用的优势。
在不少情况下,作用于双靶标的药物要比高选择性的单一靶标的药物更优胜。
两个药物同时作用于两个靶标,产生协同作用,表现为活性强度的增加或药效的提高。
这种效果也可以在药物的不同剂量或浓度比例的组合下发生,Borisy等用实验方法设置不同组合的浓度与比例,得到浓度-效应的三维图,提示以不同浓度和比例的组合,都可产生增效的协同效果。
NHNS CH3NNCH3NNNHNH3CHNNNCH38081【双或多靶标药物的实现方法】1、药物组合实现多靶标的作用:为了达到双靶标药物治疗目的,可有两种策略:一是将不同的药物作组合治疗,或组合成单一制剂,实现双靶标治疗,这是临床上普遍应用的方法;另一是作用于两个或多个靶标的单一化合物的策略,这是当今新药分子设计的活跃领域。
2、多种药物的组合策略:1).用药灵活,可以调节药物组分间的比例,并由此揭示靶标的分布、强度以及靶标的生物化学计量;2).组合的药物治疗类型和化学结构非常广泛,速度快,投入低;3).实现个性化给药,根据患者的药理遗传组学,调整不同的组方和剂量;4).在临床证明单一药物有效、作用机理明确的基础上合并用药,因而成功的概率高;5).可以实现序贯性给药,发挥最大的治疗效果。