中科大 Materials Studio 培训教程4
Materials Studio 培训教程

Materials Studio 培训教目录Materials Studio 快速入门教程⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯2 Visualizer 模块快速入门教程⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯11用第一性原理预测AlAs 的晶格参数⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯36 CO 分子在Pd(110)表面的吸附⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯43Pd(110)面上的CO 分子电荷密度变化⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯55模拟CO_Pd(110)体系的STM 图⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯61使用DMol3 中的离域内坐标对固体进行几何优化⋯⋯⋯⋯⋯⋯64 用LST/QST 搜索过渡态⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯69气体在聚合体中扩散的测量⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯76聚合物与金属氧化物表面的相互作用⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯86计算共存相之间的界面张力⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯96运行简单的MesoDyn 模拟⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯99使用粉末衍射图进行分析⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯108指标化粉末衍射图⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯117无机物的Rietveld 精修⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯125使用Reflex Plus 来解析3-氯-反-苯乙烯酸的结构⋯⋯⋯⋯⋯⋯⋯133 无机化合物FIN31 的结构确定⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯142创腾科技有限公司Neotrident Technology Limited 2Materials Studio 快速入门教程该教程将介绍Materials Studio 软件的基本功能,在这一部分,你将学到:1.生成Projects2.打开并且观察3D 文档3.绘制苯甲酰胺分子4.观察并且处理研究表格文档5.处理分子晶体:尿素6.建造Alpha 石英晶体7.建造多甲基异丁烯酸盐8.保存Project 并结束1. 生成Projects(1).运行Material Visualizer从运行菜单中运行或者在桌面点击快捷方式。
中科大MS--第四章Reflex模块的初步应用

7
Like Level 5, but 4 additional impurity peaks allowed at low 2-theta
Create Cell 然后重命名
通过Refine对比结果:
选上零点优化
对C2重做优化
练习指标化及精修过程
无机物的精修
输入ak100的结构和衍射图
选择用于指标化的衍射峰:
尽量低角度衍射峰:峰强,重叠少
避免用可疑峰(杂峰会大大提高运算量),弱峰
原则上选取20-25个衍射峰即可
输入NPHU的衍射 扣除背底(it=300) 放大8.2o处的峰, 估计半高全宽 FWHM~0.2o
平滑
X-Cell指标化
Maximum number of calculated peaks = Number of observed peaks / detection level.
练习操作并输入数据
16.82 21.84 22.89 25.81 28.08 29.04 31.86 32.20 33.04 34.08 35.58 39.26 39.98 42.16 43.84 46.82 48.22 49.52
放大操作:R+鼠标左键 选择放大区域
导入fin31的结构, 并与index得到的结 构对比
设置还原
/~fuzp/course/ ftp://210.45.126.12/temp/material studio
XRD图的指标化(续)
Fin31
输入fin31的衍射数据并扣除背底噪音
建立空表格以手动输入峰位
手动输入峰位 右键拷贝黏贴到谱上
两种方法: 背底扣除方法 相分析方法
精确的相分析需要 预先知道各纯相的 衍射
MaterialsStudio培训教程

None
PPT文档演模板
Dashed line
Line
Stick
MaterialsStudio培训教程
Lighting
在TON.xsd 的 3D Viewer 上单 击右键,选择 Lighting 选项,该选 项将指定加光情况。在此选项卡内 可以设定三个光源,并改变光源的 照射位置(照射位置用箭头显示)。
msi ?
PPT文档演模板
MaterialsStudio培训教程
2.调整显示方式 在 3D Viewer 上按右键,出右键菜单,选 Display Style ,Display Style 对话
框中的各选项的意义如下:
Atom 栏: Display Style: Line:线状模型。 Stick:棍状模型。 Ball and stick:球棍模型。 CPK:球堆砌模型。 Polyhedron:多面体堆积模型(晶体)。
Materials Studio 使用了多种类型的文件,如3D Atomistic and Mesoscale、 text、chart、 HTML、 study table、grid、script、 和 forcefield documents。在 后面进行计算时,这些文件将逐个显示在projects中,反映了计算的过程。 现在 的教学中, 主要出现的是 3D Atomistic 类型的文件。
3. 改变3D结构的视图
可以使用3D Viewer 工具栏上的工具来改变3D 视图。
3D Viewer工具栏
通过选择相应的工具并在3D 结构上拖动来改变结构视图。
Rotate:旋转结构视图。使用三键鼠标,右键是旋转操作。 Zoom:向上或者右侧拖动可以增大所选结构的视图;向下或者向左侧拖动会缩小所 选结构的视图。使用三键鼠标,也可用鼠标上的滚轮进行3D结构的放大、缩小。 Translation:将结构沿着不同的方向平移。 对于三键鼠标来说,左键执行所选操作,右键则是旋转操作,同时按下左健和右键 则会完成缩放操作。此外还可以将键盘和鼠标联用来完成上述操作。
materials studio操作手册

materials studio操作手册(实用版)目录1.Materials Studio 简介2.操作手册的主要内容3.如何使用 Materials Studio 进行基本操作4.高级操作技巧与示例5.材料建模与模拟的实践应用6.常见问题与解决方案正文【1.Materials Studio 简介】Materials Studio 是一款专业的材料科学模拟软件,广泛应用于材料研究、教育等领域。
该软件集成了多种模拟方法,如第一性原理、分子动力学、蒙特卡洛模拟等,能够实现对材料的结构、性能、缺陷等方面的研究。
Materials Studio 具有用户友好的界面,支持可视化操作,使得用户可以轻松地搭建模型、设置参数、运行模拟和分析结果。
【2.操作手册的主要内容】Materials Studio 操作手册主要包括以下几个方面的内容:(1)软件安装与配置:介绍如何安装 Materials Studio 及其依赖库,以及配置环境变量等。
(2)界面与基本操作:介绍 Materials Studio 的操作界面,包括菜单栏、工具栏、状态栏等,以及如何进行文件的保存、导入、导出等基本操作。
(3)模型构建与参数设置:介绍如何添加原子、分子、晶体等模型,以及如何设置模拟参数,如温度、压力、晶格常数等。
(4)模拟运行与结果分析:介绍如何运行模拟,以及如何分析结果,如计算能量、力、电荷密度等。
(5)高级操作技巧与示例:介绍如何进行高级操作,如自定义模拟算法、编写脚本等,并提供典型示例。
(6)材料建模与模拟的应用:介绍如何应用 Materials Studio 进行材料研究,如晶体结构预测、材料性能优化等。
【3.如何使用 Materials Studio 进行基本操作】(1)打开软件:在 Windows 系统下,点击“开始”菜单,找到“Materials Studio”并双击;在 Mac 和 Linux 系统下,进入终端,输入命令并回车。
中科大MaerialsSudio培训教程包你学会精品PPT课件

2.调整显示方式 在 3D Viewer 上按右键,出右键菜单,选 Display Style ,Display Style 对话
框中的各选项的意义如下:
Atom 栏: Display Style: Line:线状模型。 Stick:棍状模型。 Ball and stick:球棍模型。 CPK:球堆砌模型。 Polyhedron:多面体堆积模型(晶体)。
3. 在此可改变模块和 图示工具的设置值。 初学者慎用。
在layer builder中试 试。
二. 打开并且观察3D 文档 目的: 介绍Materials Studio 中文档 documents 的概念 模块: Materials Visualizer 前提: 已生成一个Project
Materials Studio 使用了多种类型的文件,如3D Atomistic and Mesoscale、 text、chart、 HTML、 study table、grid、script、 和 forcefield documents。在 后面进行计算时,这些文件将逐个显示在projects中,反映了计算的过程。 现在 的教学中, 主要出现的是 3D Atomistic 类型的文件。
3D Viewer工具栏
通过选择相应的工具并在3D 结构上拖动来改变结构视图。 Rotate:旋转结构视图。使用三键鼠标,右键是旋转操作。 Zoom:向上或者右侧拖动可以增大所选结构的视图;向下或者向左侧拖动会缩小所 选结构的视图。使用三键鼠标,也可用鼠标上的滚轮进行3D结构的放大、缩小。 Translation:将结构沿着不同的方向平移。 对于三键鼠标来说,左键执行所选操作,右键则是旋转操作,同时按下左健和右键 则会完成缩放操作。此外还可以将键盘和鼠标联用来完成上述操作。
Materials Studio 培训教程4(包你学会!)请将这一系列全看完,一定有收获。
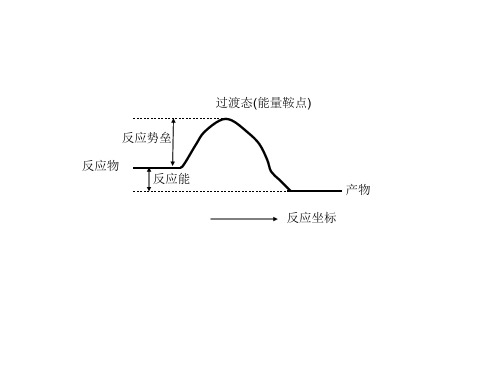
反应势垒 反应物 反应能 反应坐标
产物
用LST/QST 搜索过渡态
目的: 介绍如何使用 DMol3 和 Reaction Preview 工具进行过渡态搜索的计算。 对简 单反应,这种方法是有效的。 模块: Materials Visualizer, DMol3 前提: 用局域内坐标对固体进行结构优化。 背景 对任何反应的势能面的探索都要求知道反应进程中每一步的结构和能量, 或者动力学和热动力学的快照(snapshots)。特别重要的是决定反应速率的那一步, 这通常需要找到那些难以捕获的过渡态结构。有一些方法对找到过渡态的结构是 很有效果的,其中比较知名的就是线性同步度越(linear synchronous transit, LST)和二次同步度越(quadratic synchronous transit,QST)。 本例中,我们将介绍DMol 中的LST /QST 工具的使用,将会看到如何使用 LST/QST 搜索乙烯醇转变为乙醛的H转移反应 的过渡态结构。 CH2CHOH → CH3CHO 本例包括以下内容: ������ 1. 建立一个计算模型 ������ 2. 优化分子结构 ������ 3.定义原子对 ������ 4.用LST/QST 的方法计算过渡态 ������ 5.优化过渡态结构
1.建立一个计算模型 选择 creating a new project,建立名为vinylOH 的project 。
在本单元中,你要在两个不同的3D Atomistic 界面中建立反应物和产物模型。第 一步就是打开一个新的3D Atomistic界面,构建反应物乙烯醇(vinyl alcohol)。 点击工具栏里的New button,选择3D Atomistic。
点击碳-碳键一次,选中。点击Sketch工 具条上的Modify Bond Type 键 ,选 择双键,从而把单键变成双键。点击别处, 取消选择碳-碳键。
MaterialsStudio快速入门教程

材料性质预测
分子动力学模拟:预测材料力学性 质
弹性常数计算:评估材料稳定性
添加标题
添加标题
添加标题
添加标题
密度泛函理论:计算材料电子结构
声子谱分析:研究材料热力学性质
分子结构优化
目的:通过优化 分子结构来提高 材料的性能
方法:使用 MaterialsStudi o软件中的模块 进行分子结构优 化
目的:预测材料的物理、化学和机械性能,为材料设计和优化提供 依据
方法:利用MaterialsStudio的高级功能,如X射线衍射、中子衍 射和电子显微镜等手段进行实验测量和数据处理
应用:广泛应用于材料科学、化学、物理学和工程等领域
Part Five
常见问题与解决方 案
常见问题汇总
材料计算软件 运行缓慢
量子力学计算
MaterialsStudio中的量子力 学计算模块可用于模拟分子的 电子结构和性质
支持多种量子力学方法,如密 度泛函理论、分子力学等
可用于研究分子的电子结构、 能量、振动频率等性质
用户可以通过简单的界面和操 作完成量子力学计算
晶体结构分析
定义:通过MaterialsStudio软件对晶体结构进行分析,了解材料 的性质和行为
应用场景:在 MaterialsStudi o中,蒙特卡罗 模拟可用于模拟 材料的物理性质, 如热导率、电导 率等。
优势:蒙特卡罗 模拟可以快速得 到近似解,对于 大规模复杂系统 具有很高的计算 效率。
操作步骤:在 MaterialsStudi o中,用户可以 通过选择 “Simulate”菜 单下的“Monte Carlo”选项来 进行模拟。
步骤:选择优化 算法、设置优化 参数、执行优化 计算、分析优化 结果
Materials-Studio培训学习教程资料

Materials-Studio培训学习教程资料MaterialsStudio 培训学习教程资料在当今科技迅速发展的时代,材料科学的研究和应用变得越来越重要。
而 MaterialsStudio 作为一款功能强大的材料模拟软件,为科研人员和工程师提供了有力的工具。
对于想要深入学习和掌握这款软件的人来说,一套系统全面的培训学习教程资料是必不可少的。
首先,我们来了解一下 MaterialsStudio 软件的基本情况。
它涵盖了众多的模块,能够对材料的结构、性能、热力学等方面进行精确的模拟和分析。
无论是在化学、物理、材料科学还是工程领域,都有着广泛的应用。
那么,一份好的 MaterialsStudio 培训学习教程资料应该包含哪些内容呢?基础知识部分是重中之重。
这包括软件的安装与配置,让学习者能够顺利地在自己的电脑上搭建起学习和工作的环境。
同时,要详细介绍软件的界面和操作流程,让初学者能够快速熟悉各个功能区域和操作按钮。
对于材料结构的建模,教程资料应当有清晰的步骤和示例。
从简单的晶体结构构建,到复杂的分子体系建模,都要逐步引导学习者掌握。
并且,要讲解如何优化模型结构,以获得更准确的计算结果。
在性能计算方面,要涵盖诸如能带结构、态密度、光学性质等重要内容。
通过实际案例,演示如何设置计算参数,解读计算结果,并分析材料的性能特点。
热力学性质的计算也是不可忽视的一部分。
比如,相图的绘制、热膨胀系数的计算等,这些内容对于研究材料的稳定性和相变过程具有重要意义。
除了理论知识和操作方法,实际案例的分析也是教程资料的关键组成部分。
通过实际的科研项目或工程应用案例,让学习者能够看到MaterialsStudio 在解决实际问题中的强大能力。
同时,也能帮助他们更好地理解和运用所学的知识。
为了让学习者更好地掌握所学内容,教程资料还应当配备相应的练习和作业。
这些练习可以是针对某个具体知识点的小题目,也可以是综合性的项目,让学习者在实践中巩固和提高。
_Materials_Studio_培训教程共78页

1. 输入一个结构
File / Import,打开输入文件对话框
(注意,此对话框也可用工具栏上的输入按钮
打开)。
选择 Examples / Documents / 3D Model / TON.msi,单击 Import 按钮。
在d盘上建文件夹class 1: d:\class 1
一. 生成一个Project 目的: 介绍Materials Studio 中 project 概念 模块: Materials Visualizer
1. 建立一个新文件夹D:\ MS teach \ class1 2. 运行 Materials Studio,生成名称为My quickstart 的Project
在桌面双击快捷方式
或从运行菜单中运行:所有程序\ Accelrys Materials Studio 4.4 \ Materials Studio
选择此文件夹存放数据
这样就产生了新的 Materials Studio project,开始了Materials Studio 运行
写入My quickstart
3. 在此可改变模块和 图示工具的设置值。 初学者慎用。
在layer builder中试 试。
二. 打开并且观察3D 文档 目的: 介绍Materials Studio 中文档 documents 的概念 模块: Materials Visualizer 前提: 已生成一个Project
Materials Studio 使用了多种类型的文件,如3D Atomistic and Mesoscale、 text、chart、 HTML、 study table、grid、script、 和 forcefield documents。在 后面进行计算时,这些文件将逐个显示在projects中,反映了计算的过程。 现在 的教学中, 主要出现的是 3D Atomistic 类型的文件。
Materials-Studio教程

Materials Studio 实用指南目录Preface.前言专题1.COF晶胞的构建专题2.CMP模型的构建Reflex模块介绍Forcite模块介绍Sorption模块教程DFTB+模块介绍VAMP模块介绍DMol3模块介绍CASTEP模块介绍GULP模块介绍专题3.综合应用专题4 Materials Studio的安装与设置专题1. COF晶胞的构建在这一节,我们来了解一下如何用Materials Studio内的建模工具构建COF晶胞。
COF材料在近几年内获得了越来越多的关注,相关研究涉及的层面也越来越广泛。
但是从材料的构成来看,COF与其他高分子材料并无本质的不同,为什么COF就这么受到瞩目?因为COF材料是晶体材料。
如果一种材料是晶体材料,从理论角度说,通过研究其单个晶胞的性质,就可以推知该材料的宏观特性,从应用角度说,只要得到特征的XRD衍射线,就可以断定得到了预期的材料,且其结构与性质必定与具备相同XRD 谱线的同类样品完全一致,这是只有晶体材料才能具备的特性。
也正因为COF具备晶体材料的这些优良特性,才能被学术界如此看重。
COF材料的晶体属性为相关研究带来了相当广阔的发挥空间,但也对研究者提出了更高的要求,如果要在这一领域开展深入探索,不但要掌握合成方面的必要技能,还要对晶体的结构与性质具有一定程度的了解,最好还能够独立操作相关软件设计COF结构,构建COF晶体模型,并进行一些基本性质的计算。
在本专题,我们就如何用MS平台构建COF晶胞开展一些初步的探索,也算是回应COF材料带给我们的挑战的第一步。
COF材料最初由Yaghi小组合成,并发表在2005年的Science上。
在通篇文献中,我认为最引人注目的就是右边这张COF材料的结构模拟图。
在该文献支持信息中我们还会了解到,正文中涉及的COF结构均是由Cerius2软件完成——包括XRD谱图的解析,晶胞的构建以及结构优化——该软件是Materials Studio早期的Unix工作站版本,我们也可以把它看成是目前的Materials Studio的前身,两者主要模块及功能完全一致,所以我们可以肯定,用相对容易接触到的MS也肯定能完成相同的工作。
Materials Studio 培训教程
Materials Studio 培训教目录Materials Studio 快速入门教程⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯2 Visualizer 模块快速入门教程⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯11用第一性原理预测AlAs 的晶格参数⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯36 CO 分子在Pd(110)表面的吸附⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯43Pd(110)面上的CO 分子电荷密度变化⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯55模拟CO_Pd(110)体系的STM 图⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯61使用DMol3 中的离域内坐标对固体进行几何优化⋯⋯⋯⋯⋯⋯64 用LST/QST 搜索过渡态⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯69气体在聚合体中扩散的测量⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯76聚合物与金属氧化物表面的相互作用⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯86计算共存相之间的界面张力⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯96运行简单的MesoDyn 模拟⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯99使用粉末衍射图进行分析⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯108指标化粉末衍射图⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯117无机物的Rietveld 精修⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯125使用Reflex Plus 来解析3-氯-反-苯乙烯酸的结构⋯⋯⋯⋯⋯⋯⋯133 无机化合物FIN31 的结构确定⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯142创腾科技有限公司Neotrident Technology Limited 2Materials Studio 快速入门教程该教程将介绍Materials Studio 软件的基本功能,在这一部分,你将学到:1.生成Projects2.打开并且观察3D 文档3.绘制苯甲酰胺分子4.观察并且处理研究表格文档5.处理分子晶体:尿素6.建造Alpha 石英晶体7.建造多甲基异丁烯酸盐8.保存Project 并结束1. 生成Projects(1).运行Material Visualizer从运行菜单中运行或者在桌面点击快捷方式。
中科大MaterialsStudio培训教程包你学会

选择 File / Save 或单击工具栏上的 Save button 中建立了新的 my_benzamide.xsd 3D文件。
。这样就在my quickstart project
2. 设置球棍模型为默认显示方式 从菜单栏中选择 Modify / Default Atom Style ,打开 Default Atom Style 对话
用 None、Dashed line、Line和 Stick styles显 示zeolite Theta-1 的结构. 注意3D Viewer边框的 变化 .
将显示固定在Line.
None
Dashed line
Line
Stick
Lighting
在TON.xsd 的 3D Viewer 上单 击右键,选择 Lighting 选项,该选项 将指定加光情况.在此选项卡内可以 设定三个光源,并改变光源的照射位 置(照射位置用箭头显示).
选择 Tools \ Settings Organizer ,打开Settings Organizer 对话框.
1. 在这此处的Materials Studio icon,选 中所有的模块和图示工具.
2. 单击Reset,所有的模块和图示工具都恢复 Accelrys默认值.
若干次操作后,已有 一些参数设置.由于 错误等原因,要重复 前面的一个过程.为 保存两次操作一样, 需返回MS的默认设置.
在d盘上建文件夹class 1: d:\class 1
一. 生成一个Project 目的: 介绍Materials Studio 中 project 概念 模块: Materials Visualizer
1. 建立一个新文件夹D:\ MS teach \ class1 2. 运行 Materials Studio,生成名称为My quickstart 的Project
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
点击Build | Bonds ,打开Bond
Calculation对话框,勾选上化学键 计算对话框上的Monitor bonding, 关闭对话框。
设置动画演示方式,按下动 画工具条上的Play 按钮,观看反 应物到产物的变化。 看完后按下Stop 按钮。
4. 使用LST/QST/CG 方法计算过渡态 Note:reactant_product.xtd 包含了DMol3 需要的重要信息,第一桢是反应物的,最 后一桢是产物的。 现在准备设置使用DMol3 计算过渡态。 从菜单条中选择Modules | DMol3 | Calculation,或使用DMol3图标 DMol3onian 和计算的精度。精度决定了使用的基组(basis set) 和轨道的截断cutoff。这里基组为DND。可以在Electronic 栏里检查这些参数的设置。 现在需要应用电子分布热平滑thermal smearing来加快结构优化的收敛。 点击Electronic 标签。检查SCF 是不是设为Medium。按下More…按钮,显示了 DMol3 的Electronic 选项对话框。在SCF 标签栏里,勾选上Using smearing 选项。关 闭DMol3 Electronic选项对话框。
* 在这个新的3D 界面文件中,点击O-H 键。按下键 盘上的DELETE 键。点击Sketch Atom 按钮 ,然 后是孤立的H 原子,以及亚甲基团中的C 原子。如 果画分子有问题,则删除3D Atomistic.xsd上的原子, 再进行CTRL+C、CTRL+V。
点击一次C-O 键,由单键改为双键。
在Project中双击reactant.xtd文件, 动画显示工具按钮Animation 激活。
如果动画(Animation)工具条 是不可见的, 则按右侧的操作,使用观看 (View)菜单让它显示。
设置显示方式,按播放键
。
B-O近似,体系的电子能量是核构型的函数。
在继续工作之前,需要关闭Materials Visualizer 中的所有文件。 关闭DMol3 计算对话框。选择File\Save Project,然后Window\Close All。 双击几何优化子文件夹中的reactant.xsd 和product.xsd。 现在工作区域中只有两个优化了的结构。
按下Match…按钮。 寻找等价原子(Find Equivalent Atoms) 对话框显示出来了, 从中可以看到,一 个原子(O)匹配了, 而仍有六个原子(C、 H)没有匹配。
双击反应物栏(reactant column)中的2xC。 在产物栏里的对应的文件夹同时打开了。反 应物栏包含了1:C 和2:C,它们应该直接和 产物栏里的对应物相匹配,以下的步骤将对 此加以确认。
在Sketch 工具条上,点击Sketch Atom 按钮 。 将鼠标移至 3D Atomistic界面,连续点击三次鼠标,画三个连接的 碳原子。按一下键盘上的ESC 键。
改为球棍显示
在3D Atomistic上 ,点击选择第三个 C 原子。点击Modify Element 按钮上 的选择箭头,选择氧元素。刚才被选 的原子由碳原子变成了氧原子。
仍有3个H原子没有匹配,重复上面 的手动匹配步骤。双击反应物栏 (reactant column)中的3xH。在产物栏里 的对应的文件夹同时打开了。反应物栏 包含了4:H、5:H 和7:H,分别点击反应 物框里的4:H和产物框里的4:H,两个对 话框里的H原子被选上,两个3D 文件里 的碳原子也同时被选中。
从菜单条中选择Tools | Reaction Preview,打开了反 应预览(Reaction Preview)对话 框。
从Reactant 的 下拉树 形图中的几何优化文件 夹中选择reactant.xsd 。
同样从Product 的下拉树 形图中的几何优化文件夹 中选择product.xsd。
当第一个计算结束后,对product.xsd 重复刚才的操作。 把当前工作文件换为product.xsd,点击DMol3 计算对话框上的Run 按钮。 计算过程中,计算的进程用图表和文本文件的形式展现出来。
当两个计算都完成的时候,两个新的文件夹出现在工作浏览器中,分别叫做 reactant DMol3 GeomOpt 和product DMol3 GeomOpt。最后的优化结构包含在 reactant.xsd 和product.xsd 文件中,计算的输出结果在reactant.outmol 和 product.outmol 文件中。 几何优化文件夹包含了.xtd 文件,这是能量最小化过程中的轨迹文件,可以显 示几何优化过程。下面演示反应物的结构优化过程。从reactant Energies.xcd图中可 以看出,反应物经过12步才优化结束,我们可以看到每一步结构的变化。
点击Set Match,还剩下5H、7H未匹配。重复这个过程,继续点击Set Match来匹配 剩下的没有配对的原子。
反应物、产物的原子已配对。现在可以预览一下反应物和产物之间原子的匹配 情况。 点击反应物或者产物栏中列表里的任意一个原子,可以看到匹配的另一原子。 考察匹配情况,直到满意为止。关闭Find Equivalent Atoms 对话框。
运用DMol3的 LST/QST功能来搜索过渡态,需要 在反应物和产物之间创建一条通道,这也是DMol3 计 算时所要求的输入文件。 在反应预览(Reaction Preview)对话框中,把桢数 提高到100;勾选上Superimpose structures;单击 Preview按钮,关闭反应预览(Reaction Preview)对话 框。
在设置(Setup)标签栏里,把Task 由
几何优化改为TS Search。确定计算精度 为Medium,泛函为GGA 和BP。 点击More…按钮显示DMol3 过渡 态搜索(DMol3 Transition State Search) 对话框。确认搜索协议(Search
protocol)设置为Complete LST/QST,
2. 优化分子结构 为了优化LST/QST的计算性能,需要对反应物和产物的结构进行优化。这个工作可 以通过DMol3 的几何优化功能来完成。 点击别处,取消选 择结构。按下工具 条上的DMol3 按 钮 ,然后选 择下拉条中的 Calculation。 DMol3 的计算对话 框显示出来。
将Task 由 Energy 改为 Geometry Optimization。 确认Quality 设为Medium。 将泛函改为 GGA \BP。
点击碳-碳键一次,选中。点击Sketch工 具条上的Modify Bond Type 键 ,选 择双键,从而把单键变成双键。点击别处, 取消选择碳-碳键。
按下Adjust Hydrogen 按钮 ,点 击一次Clean 按钮 ,拖动结构模 型,使得和下图相似,以球棍模型显 示。
在Project浏览器内,右击 3D Atomistic.xsd,选择 Rename,将其重新命名为 reactant.xsd。
在几秒钟内,一个名为 reactant-product.xtd 的新的3D Atomistic Trajectory 文件显示 出来;可以对这个文件进行 DMol3 计算;可以使用动画 (Animation)工具条来播放轨迹 文件。 动画用Bounce模式观看效 果最佳。把化学键监测(bond monitoring)打开,这样每变化一 步,就会对化学键重新计算。
1.建立一个计算模型 选择 creating a new project,建立名为vinylOH 的project 。
在本单元中,你要在两个不同的3D Atomistic 界面中建立反应物和产物模型。第 一步就是打开一个新的3D Atomistic界面,构建反应物乙烯醇(vinyl alcohol)。 点击工具栏里的New button,选择3D Atomistic。
现在准备开始计算了。 让reactant.xsd 成为当前工作文件。点击Job Control 标签。按下More…按钮, 显示了DMol3的工作控制选项对话框。确认Update structure,Update graphs 和 Update textual results 三项被勾选上。关闭Job Control 选项对话框,点击Run 按钮。
连续双击C-C 键,C-C 键就会由双键变为三键,然后又变 成单键。
点击一次Clean 按钮 。 现在结构就和下面的看上去相似了。
现在需要把该结构的文件名改为 product.xsd。右击工作浏览器 Project内的3D Atomistic.xsd, 将其名称改为product.xsd,回 车。
精度为Medium。关闭DMol3 Transition State Search 对话框。
电子Hamiltonian 的设置与几何优化计 算的设置一样。
这次需要计算频率(Frequency)相关的性质。 点击Properties 标签栏,勾选上Frequency。
最后,需要对工作描述(Job Description)加以设 置。 点击Job Control 标签,确认Automatic 没有被勾 选上;在Job Description 一栏里打上TS。 按下Run 按钮。关闭DMol3 Calculation 对话框。 等待计算完毕。
过渡态(能量鞍点)
反应势垒 反应物 反应能 反应坐标
产物
用LST/QST 搜索过渡态
目的: 介绍如何使用 DMol3 和 Reaction Preview 工具进行过渡态搜索的计算。 对简 单反应,这种方法是有效的。 模块: Materials Visualizer, DMol3 前提: 用局域内坐标对固体进行结构优化。 背景 对任何反应的势能面的探索都要求知道反应进程中每一步的结构和能量, 或者动力学和热动力学的快照(snapshots)。特别重要的是决定反应速率的那一步, 这通常需要找到那些难以捕获的过渡态结构。有一些方法对找到过渡态的结构是 很有效果的,其中比较知名的就是线性同步度越(linear synchronous transit, LST)和二次同步度越(quadratic synchronous transit,QST)。 本例中,我们将介绍DMol 中的LST /QST 工具的使用,将会看到如何使用 LST/QST 搜索乙烯醇转变为乙醛的H转移反应 的过渡态结构。 CH2CHOH → CH3CHO 本例包括以下内容: ������ 1. 建立一个计算模型 ������ 2. 优化分子结构 ������ 3.定义原子对 ������ 4.用LST/QST 的方法计算过渡态 ������ 5.优化过渡态结构