Gateway克隆技术,简单易操作
gateway重组技术

Gateway 基因克隆Gateway 基因克隆就是由Invitrogen公司在二十世纪九十年代末发明并应用于分子生物学基因克隆的一项专利技术。
该技术利用专有的重组序列使得DNA片段能够更有效地被转入质粒当中,可应用于大片段的基因克隆,并且在保持正确阅读框的前提下让不同表达载体间的DNA转移成为可能。
这一技术在插入的目的DNA片段两端整合att L1与att L2两个侧端重组序列,来构建一个类似通道的结构并称之为“入门克隆”(Gateway Entry Clone)。
据Invitrogen宣称Gateway 技术使用99%有效且可逆的一小组重组反应,如此使得基因克隆不同于传统的限制性内切酶方法,避免了目的片段内存在切点的问题而使得大片段DNA保持其完整性,大大提高了克隆效率,常应用于大规模的DNA片段整合进同一种表达载体,因此又称之为高通量基因克隆技术(Gateway Cloning Technology)。
一、Gateway 基因克隆的原理及机制Gateway被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后,就不用依赖限制性内切酶,而靠载体上存在的特定重组位点与重组酶,高效、快速地将目的基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway的原理就是建立在噬菌体DNA定点整合到细菌宿主基因组上。
在噬菌体与细菌的整合因子(INF、Int)的作用下,lambda的attP位点与大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中,两侧产生两个新位点:attL与attR。
这就是一个可逆的过程,如果在一个噬菌体编码蛋白Xis与IHF、Int的共同介导下这两个新位点可以再次重组回复为attB与attP位点,噬菌体从细菌基因组上裂解下来(见图1)。
这一过程的方向就是受控于两个重要因素:存在的介导蛋白与重组位点。
gateway技术
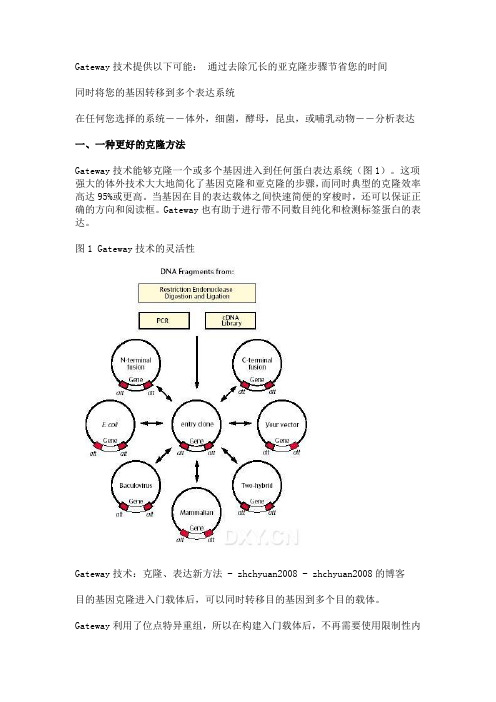
Gateway技术提供以下可能:通过去除冗长的亚克隆步骤节省您的时间同时将您的基因转移到多个表达系统在任何您选择的系统――体外,细菌,酵母,昆虫,或哺乳动物――分析表达一、一种更好的克隆方法Gateway技术能够克隆一个或多个基因进入到任何蛋白表达系统(图1)。
这项强大的体外技术大大地简化了基因克隆和亚克隆的步骤,而同时典型的克隆效率高达95%或更高。
当基因在目的表达载体之间快速简便的穿梭时,还可以保证正确的方向和阅读框。
Gateway也有助于进行带不同数目纯化和检测标签蛋白的表达。
图1 Gateway技术的灵活性Gateway技术:克隆、表达新方法 - zhchyuan2008 - zhchyuan2008的博客目的基因克隆进入门载体后,可以同时转移目的基因到多个目的载体。
Gateway利用了位点特异重组,所以在构建入门载体后,不再需要使用限制性内切酶和连接酶。
一旦您拥有了一个入门克隆,就可以多次使用它,转移您的目的基因到Gateway改造过的各种表达载体(目的载体)。
此外,由于在重组时DNA片段的阅读框和方向保持不变,因而您不必再为新的表达克隆测序担心。
在使用每一种新的表达系统时,将会节省您更多的时间。
二、一项强大而可靠的技术Gateway技术是克隆和亚克隆DNA序列的一项新颖的通用系统,便于功能基因的分析和蛋白质的表达。
一旦进入这个多功能的操作系统,DNA片段可以通过位点特异的重组在载体之间转移。
Gateway技术是基于已研究的非常清楚的λ噬菌体位点特异重组系统(attB x attP →attL x attR)。
BP和LR两个反应就构成了Gateway技术(表1和图2)。
BP反应是利用一个attB DNA片段或表达克隆和一个attP供体载体之间的重组反应,创建一个入门克隆。
LR反应是一个attL入门克隆和一个attR目的载体之间的重组反应。
LR反应用来在平行的反应中转移目的序列到一个或更多个目的载体。
Gateway

Gateway也可以被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后,就不用依赖限制性内切酶,而靠载体上存在的特定重组位点和重组酶,高效、快速地将目的基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway的原理也是建立在噬菌体DNA定点整合到细菌宿主基因组上。
在噬菌体和细菌的整合因子(INF、Int)的作用下,lambda的attP位点和大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中,两侧产生两个新位点:attL和attR。
这是一个可逆的过程,如果在一个噬菌体编码蛋白Xis和IHF、Int的共同介导下这两个新位点可以再次重组回复为attB和attP位点,噬菌体从细菌基因组上裂解下来。
这一过程的方向是受控于两个重要因素:存在的介导蛋白和重组位点。
在Gateway系统中,入门载体包含两个重组位点序列attL1和attL2,大小均为100bp,中间夹着一个自杀基因——ccdB基因。
由于ccdB基因的表达产物能抑制普通的E.coli生长,在克隆时没有切开或者自身环化的载体在转化时不能生长。
在构建含目的基因的入门载体时必须切掉这个基因,接入目的基因。
ccdB基因两端可以选择的酶切位点有限(2个),同时还必须考虑读码框架、启动子、终止密码等问题,因此Gateway系统提供了5种不同的入门载体以供选择。
需要特别注意的是转化用的菌株必须是不含F附加体的,因为它表达的一种产物能阻断ccdB基因,影响筛选结果。
同样,目的载体(Destination Vector)也必须和Gateway系统配套,即目的载体的表达调控元件下游有两个重组位点attR1和attR2,大小均为125bp,同样也夹着一个ccdB自杀基因。
当需要将目的基因从入门载体(Entry Vector)转移到目的载体(Destination Vector)时,只要将两种质粒混合(线性化能有效提高重组率),加入含有Int、IHF、Xis等重组因子的LR重组酶混合物,attR2序列和attL2序列发生重组,生成一个融合质粒。
gateway重组技术

河南农业大学牧医工程学院Gateway 基因克隆Gateway 基因克隆是由Invitrogen公司在二十世纪九十年代末发明并应用于分子生物学基因克隆的一项专利技术。
该技术利用专有的重组序列使得DNA片段能够更有效地被转入质粒当中,可应用于大片段的基因克隆,并且在保持正确阅读框的前提下让不同表达载体间的DNA转移成为可能。
这一技术在插入的目的DNA片段两端整合att L1和att L2两个侧端重组序列,来构建一个类似通道的结构并称之为“入门克隆”(Gateway Entry Clone)。
据Invitrogen宣称Gateway技术使用99%有效且可逆的一小组重组反应,如此使得基因克隆不同于传统的限制性内切酶方法,避免了目的片段内存在切点的问题而使得大片段DNA保持其完整性,大大提高了克隆效率,常应用于大规模的DNA片段整合进同一种表达载体,因此又称之为高通量基因克隆技术(Gateway Cloning Technology)。
一、Gateway 基因克隆的原理及机制Gateway被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后,就不用依赖限制性内切酶,而靠载体上存在的特定重组位点和重组酶,高效、快速地将目的基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway的原理是建立在噬菌体DNA定点整合到细菌宿主基因组上。
在噬菌体和细菌的整合因子(INF、Int)的作用下,lambda的attP位点和大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中,两侧产生两个新位点:attL和attR。
这是一个可逆的过程,如果在一个噬菌体编码蛋白Xis和IHF、Int的共同介导下这两个新位点可以再次重组回复为attB和attP位点,噬菌体从细菌基因组上裂解下来(见图1)。
这一过程的方向是受控于两个重要因素:存在的介导蛋白和重组位点。
Gateway技术能够克隆一个或多个基因进入到任何蛋白表达系统

Gateway技术能够克隆一个或多个基因进入到任何蛋白表达系统,大大简化基因克隆和亚克隆步骤,同时克隆效率高达95%,当基因在目的表达载体之间快速简便的穿梭时,还可保证正确的方向和阅读框。
Gateway技术基于λ噬菌体位点特异重组系统(attB ×attP →attL ×attR)。
BP和LR两个反应就构成Gateway技术,BP反应利用一个attB DNA片段或表达克隆和一个attP供体载体之间的重组反应,创建一个入门克隆;LR反应是一个attL入门克隆和一个attR目的载体之间的重组反应,用于在平行的反应中把目的序列转移到一个或更多目的载体。
Gateway技术也利用了ccdB选择方法,确保高效率的分离重组克隆。
完成构建Gateway表达克隆仅需两步:⑴创建入门克隆,通过PCR将目的基因克隆进入门载体;⑵混合包含目的基因的入门克隆和合适的目的载体及Gateway LR Clonase,产生表达克隆(用来在合适的宿主中进行蛋白的表达和分析)。
构建入门载体PCR重组克隆重组是从PCR产物创建Gateway入门克隆的一种方法。
通过合并attB位点到上游和下游引物上,然后共同孵育扩增PCR产物和pDONR载体(包含attP位点)及GatewayBP ClonaseTM酶混合物。
接着转化进大肠杆菌中,您将会获得包含目的基因的入门克隆,同时目的基因两侧具有attL重组位点。
这个入门克隆可以与任何Gateway目的载体进行重组。
表达系统一旦您构建好Gateway入门克隆,就可以使用Gateway技术进入到几乎是无数种的表达系统。
Gateway技术为进行基因功能分析和蛋白表达提供了广泛深入的方法,通过把目的基因转移到优化构建的体外表达载体,pEXP1-DEST 或pEXP2-DEST,然后开始体外蛋白合成。
目前有很多具有不同启动子的大肠杆菌目的载体,提供一系列表达选择,Gateway目的载体也提供N端或/和C端融合标签的选择,简化表达蛋白的纯化和检测。
实验方案gateway

实验方案Gateway® 反应的基本原理∙BP反应—构建Gatew ay® 入门克隆∙LR 反应—构建Gatew ay® 表达克隆∙一管模式—从PCR 产物构建Gatew ay® 表达克隆∙Gatew ay® 载体转化—将您喜爱的克隆载体转化成Gatew ay® 载体TOPO® TA 克隆- 创建Gateway® 入门克隆∙∙步骤一–制备PCR 产物使用Taq 聚合酶和自己的方案生成PCR 产物。
通过最后7 到30 分钟的延伸步骤,结束PCR 反应。
步骤二- 进行TOPO® 克隆反应1. 按所示顺序使用试剂,以建立以下一种TOPO® 克隆反应。
对于电穿孔,将盐溶液稀释 4 倍以制备稀释盐溶液。
试剂化学转染电穿孔法新鲜的PCR 产物0.5 至 4 µl 0.5 至 4 µl盐溶液 1 µl --稀释盐溶液-- 1 µl无菌水至终体积为 5 µl 至终体积为 5 µlTOPO® 载体 1 µl 1 µl总体积 6 µl 6 µl∙2. 轻轻混合并在室温下孵育 5 分钟。
3. 置于冰上,然后开始转化One Shot® 化学感受态 E. coli,步骤如下步骤三- 转化One Shot® 化学感受态E. coli1. 每次转化时,解冻置于冰上的一小瓶One Shot® E. coli 细胞。
2. 在一小瓶One Shot® 化学感受态E. coli 中加入 2 µl TOPO® 克隆反应液,轻轻混匀。
3. 在冰上孵育 5 至30 分钟。
4. 将细胞置于42°C 下热休克30 秒,不振荡。
立即将试管转移至冰上。
5. 加入250 µl 室温S.O.C. 培养基。
gateway重组技术

Gateway也可以被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后,就不用依赖限制性内切酶,而靠载体上存在的特定重组位点和重组酶,高效、快速地将目的基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway的原理也是建立在噬菌体DNA定点整合到细菌宿主基因组上。
在噬菌体和细菌的整合因子(INF、Int)的作用下,lambda的attP位点和大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中,两侧产生两个新位点:attL和attR。
这是一个可逆的过程,如果在一个噬菌体编码蛋白Xis和IHF、Int的共同介导下这两个新位点可以再次重组回复为attB和attP位点,噬菌体从细菌基因组上裂解下来。
这一过程的方向是受控于两个重要因素:存在的介导蛋白和重组位点。
在Gateway系统中,入门载体包含两个重组位点序列attL1和attL2,大小均为100bp,中间夹着一个自杀基因——ccdB基因。
由于ccdB基因的表达产物能抑制普通的E.coli生长,在克隆时没有切开或者自身环化的载体在转化时不能生长。
在构建含目的基因的入门载体时必须切掉这个基因,接入目的基因。
ccdB基因两端可以选择的酶切位点有限(2个),同时还必须考虑读码框架、启动子、终止密码等问题,因此Gateway系统提供了5种不同的入门载体以供选择。
需要特别注意的是转化用的菌株必须是不含F附加体的,因为它表达的一种产物能阻断ccdB基因,影响筛选结果。
同样,目的载体(Destination Vector)也必须和Gateway系统配套,即目的载体的表达调控元件下游有两个重组位点attR1和attR2,大小均为125bp,同样也夹着一个ccdB自杀基因。
当需要将目的基因从入门载体(Entry Vector)转移到目的载体(Destination Vector)时,只要将两种质粒混合(线性化能有效提高重组率),加入含有Int、IHF、Xis等重组因子的LR重组酶混合物,attR2序列和attL2序列发生重组,生成一个融合质粒。
Gateway技术

Gateway™技术提供以下可能:•通过去除冗长的亚克隆步骤节省您的时间•将您的基因转入到多个表达系统•在任何您选择的系统――体外,细菌,酵母,昆虫,或哺乳动物――分析表达一种更好的克隆方法Gateway™技术能够克隆一个或多个基因进入到任何蛋白表达系统(图1)。
这项强大的体外技术大大地简化了基因克隆和亚克隆的步骤,而同时典型的克隆效率高达95%或更高。
当基因在目的表达载体之间快速简便的穿梭时,还可以保证正确的方向和阅读框。
Gateway™也有助于进行带不同数目纯化和检测标签的表达。
Gateway™利用了位点特异重组,所以在构建入门载体后,不再需要使用限制性内切酶和连接酶。
一旦您拥有了一个入门克隆,就可以多次使用它,转移您感兴趣的基因到Gateway™改造过的的各种表达载体(目的载体)。
此外,由于在重组时DNA片段的阅读框和方向保持不变,因而您不必再为新的表达克隆的测序担心。
在使用每一种新的表达系统时,将会节省您更多的时间。
图1-Gateway™技术的灵活性*目的基因克隆进入门载体后,可以同时转移目的基因到多个目的载体。
一种强大而可靠的技术Gateway™技术是克隆和亚克隆DNA序列的一项新颖的通用系统,便于功能基因的分析和蛋白质的表达。
一旦进入这个多功能的操作系统,DNA片段可以通过位点特异的重组在载体之间转移。
Gateway™技术是基于已研究的非常清楚的λ嗜菌体位点特异重组系统(attB x attP →attL x attR)。
BP和LR两个反应就构成了Gateway™技术(表1和图2)。
BP反应利用一个attB DNA片段或表达克隆和一个attP供体载体之间的重组反应,创建一个入门克隆。
LR反应是一个attL入门克隆和一个attR目的载体之间的重组反应。
LR反应用来在在平行的反应中转移目的序列到一个或更多个目的载体。
Gateway™技术也利用了ccdB选择方法,确保高效率的分离重组克隆。
Gateway

进了新实验室后一直用Gateway做克隆,发现论坛上很少有讨论gateway的帖子,特开此贴,也欢迎对gateway有经验,有兴趣和有疑问的同行们一起交流经验Gateway官方简介:Gateway®克隆技术的基础是lambda噬菌体的位点特异性重组反应,一种准确保存遗传信息的有效的自然的生化途径。
与传统的需要DNA限制性内切酶、DNA连接酶、凝胶电泳和DN***段纯化等多个步骤的单调乏味的亚克隆方法相比,该方法只需一步生化反应而且操作方便、快捷(室温60分钟),节省了大量的时间和劳力。
另外,根据目的蛋白的生产和纯化的不同要求,Invi t rogen公司提供一系列含有不同启动子和融合标签、以及在几种宿主表达的目标载体。
以下是个人总结优点:1. 效率高,空载体含有毒性基因所以避免了大部分的假阳性。
2. 在不同载体之间转换方便,省去了大量的酶切/胶回收/去磷酸/连接的步骤,尤其适合筛选大量基因和突变体的应用。
3. 速度快,大部分反应1小时完成而且转化效率很高,自制的感受态DH10B(1ug=10^8cfu)加1ul反应产物就可以。
4. 反应缓冲液是TE,可以直接电转化5. 兼容TOPO,可以用CmR/ccdB片断构建载体6. 个人感觉容错率较高,对时间和温度的控制要求也不严。
一开始没经验,用水浴加温,拿出来发现一个EP管里的液量神奇的增加了,应该是进了水。
硬着头皮继续做居然成功了。
缺点:1. 酶很贵,真的很贵2. 很贵的酶还很娇贵,冰上几分钟就有可能失活。
虽然现行版本的酶经过改进只需要零下20度但是为了以防万一还是推荐零下80度保存。
3. 10kb以上的片断需要隔夜培育,和传统的酶切-T4连接相比没有优势4. 载体选择比前几年多但是依然较少,invitrogen的质粒很贵,自己构建和扩增载体较繁琐而且需要抗ccdB 的特殊菌株来转化。
下面说一些手册里没有的小技巧1. 手册里的反应体系是10ul, 其实完全可以减半, 不影响效果。
Gateway的原理

Gateway也可以被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后,就不用依赖限制性内切酶,而靠载体上存在的特定重组位点和重组酶,高效、快速地将目的基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway的原理也是建立在噬菌体DNA定点整合到细菌宿主基因组上。
在噬菌体和细菌的整合因子(INF、Int)的作用下,lambda的attP位点和大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中,两侧产生两个新位点:attL和attR。
这是一个可逆的过程,如果在一个噬菌体编码蛋白Xis和IHF、Int的共同介导下这两个新位点可以再次重组回复为attB和attP位点,噬菌体从细菌基因组上裂解下来。
这一过程的方向是受控于两个重要因素:存在的介导蛋白和重组位点。
在Gateway系统中,入门载体包含两个重组位点序列attL1和attL2,大小均为100bp,中间夹着一个自杀基因——ccdB基因。
由于ccdB基因的表达产物能抑制普通的E.coli生长,在克隆时没有切开或者自身环化的载体在转化时不能生长。
在构建含目的基因的入门载体时必须切掉这个基因,接入目的基因。
ccdB基因两端可以选择的酶切位点有限(2个),同时还必须考虑读码框架、启动子、终止密码等问题,因此Gateway系统提供了5种不同的入门载体以供选择。
需要特别注意的是转化用的菌株必须是不含F附加体的,因为它表达的一种产物能阻断ccdB基因,影响筛选结果。
同样,目的载体(Destination Vector)也必须和Gateway系统配套,即目的载体的表达调控元件下游有两个重组位点attR1和attR2,大小均为125bp,同样也夹着一个ccdB自杀基因。
当需要将目的基因从入门载体(Entry Vector)转移到目的载体(Destination Vector)时,只要将两种质粒混合(线性化能有效提高重组率),加入含有Int、IHF、Xis等重组因子的LR 重组酶混合物,attR2序列和attL2序列发生重组,生成一个融合质粒。
利用一步克隆方法快速高效构建Gateway入门载体

利用一步克隆方法快速高效构建Gateway入门载体张艳军; 赵利; 李雨佳; 赵江哲; 张可伟【期刊名称】《《浙江师范大学学报(自然科学版)》》【年(卷),期】2020(043)001【总页数】5页(P72-76)【关键词】入门克隆; SLIC技术; Gateway; LR重组反应【作者】张艳军; 赵利; 李雨佳; 赵江哲; 张可伟【作者单位】浙江师范大学化学与生命科学学院植物遗传发育研究所浙江金华321004【正文语种】中文【中图分类】Q785在研究植物基因功能时,常常需要探究某个基因的时空表达模式、亚细胞定位和互补表型,这就需要研究人员构建多个不同的表达载体,包括GUS融合表达载体、GFP融合表达载体和35S过表达载体等[1].目前大多采用传统的酶切连接法构建这些载体,不同的表达载体可能需要使用不同的酶切位点,整个构建过程需要多个步骤,费时费力.而Invitrogen公司开发的Gateway技术以λ噬菌体的位点特异重组系统为基础[2],首先将扩增的PCR片段构建至Gateway入门载体,然后就可以通过LR反应(一个attL入门克隆和一个attR目的载体之间的重组反应)将目的片段从入门载体转移至不同功能的Gateway目的载体.LR重组过程简单快速,同时保持DNA片段的阅读框和方向不变,从而大大提高了实验效率[3].目前Invitrogen公司开发了多种可供选择的Gateway目的载体,包括用于体外表达的pEXP2-DEST载体、大肠杆菌表达载体pDEST17、酵母表达的pYES2-DEST52等.此外,研究人员也开发了很多与Gateway技术兼容的植物表达载体系统,如Curtis等[4]构建了与Gateway技术兼容的农杆菌双元表达载体,可用于植物基因研究,包括用于蛋白质的亚细胞定位和基因互补试验等相关载体.利用Gateway技术构建目的载体时,首先需要将目的基因构建至入门载体.目前构建入门载体主要有3种方法:1)通过BP反应(一个attB DNA片段或表达克隆与一个attP供体载体之间的重组反应)将PCR片段重组到pDONR[5]和pENTR等入门载体,这些载体可以重复使用,但是BP重组酶价格昂贵,而且插入片段大于2 kb时,效率会大大降低;2)利用TOPO克隆或TA克隆方法将PCR产物连接到pENTR/D-TOPO[6]和pCR8/GW/TOPO等入门载体,但这些载体价格昂贵,且不能够复制和反复使用,大大增加了实验成本;3)利用限制性内切酶将PCR片段连接到入门载体,如GEC和PEC载体[7],该方法成本低廉,但是常受限于载体上的酶切位点位置,并且耗时过长.为了解决上述这些问题,充分利用Gateway技术兼容的植物表达载体系统,本研究开发了一种快速高效低成本构建Gateway入门载体的方法,对于推广Gateway 技术有着重要意义.本研究以实验室保存的pCR8-AtS5H入门克隆载体和水稻异分支酸合成酶基因的启动子(ICS1pro)片段为例,对该方法进行详细阐述.首先利用限制酶EcoR Ⅰ对pCR8-AtS5H入门克隆进行单酶切,并回收pCR8片段,将其作为入门载体;然后扩增并纯化ICS1pro片段,并利用一步SLIC法[8]将纯化后的ICS1pro片段构建至pCR8入门载体,通过菌落PCR和测序方法鉴定得到阳性入门克隆;最后通过LR反应将入门克隆中ICS1pro片段构建至目的载体pMDC163.这种利用不依赖序列和连接的克隆方法快速构建Gateway入门载体的方法操作简便,同时成功率高且成本很低,是一种有广泛应用前景的Gateway入门载体构建方法.1 材料和方法1.1 材料水稻日本晴DNA、大肠杆菌(Escherichia coli)菌株DH5α、质粒pCR8-AtS5H和Gateway目的载体pMDC163均由本实验室保存,其中pCR8-AtS5H质粒是利用TOPO克隆方法将拟南芥S5H基因构建至pCR8/GW/TOPO载体(购自赛默飞世尔科技(中国)有限公司)而获得的入门克隆.KOD FX购自东洋纺(上海)生物科技有限公司;2×Taq Master Mix购自北京康为世纪生物科技有限公司;限制性内切酶EcoR Ⅰ购自宝生物工程(大连)有限公司;T4 DNA聚合酶购自New England Biolabs(北京)有限公司;LR重组试剂购于赛默飞世尔科技(中国)有限公司;质粒提取试剂盒和胶回收试剂盒均购自生工生物工程(上海)股份有限公司.1.2 方法1.2.1 引物设计根据pCR8载体和水稻异分支酸合成酶基因(LOC_Os09g19734)的启动子DNA序列设计扩增引物,分别为ICS1pro-F1:GCAGGCTCCGAATTCTGGCTTGGGTAGGATTATG和ICS1pro-R1:AAGCTGGGTCGAATTCCGGGTGGGAGGAGGAAGAAG,其中划线部分(15或16 bp)为pCR8载体片段两端同源序列.此外,根据水稻异分支酸合成酶基因的启动子(ICS1pro)序列设计菌落PCR引物,分别为ICS1pro-F2:TTGGCTTGGGTAGGATTA和ICS1pro-R2:ACGAAGGAGCGAAGGTTA.上述引物均由杭州擎科梓熙生物技术有限公司合成.1.2.2 ICS1pro片段的扩增与纯化以CTAB方法提取的日本晴DNA为模板进行基因扩增.PCR反应体系为(总体积50 μL):2×KOD FX PCR缓冲液25 μL,2 mmol/L dNTP 10 μL,10 μmol/L引物ICS1pro-F1 1.5 μL,10 μmol/L引物ICS1pro-R1 1.5 μL,KOD FX 1 μL,DNA 1 μL,ddH2O 10 μL.反应条件为:94 ℃预变性3 min;98 ℃变性10 s,55 ℃退火30 s,68 ℃延伸3 min,共30个循环;68 ℃延伸10 min.基因扩增完成后进行琼脂糖凝胶电泳,切胶利用胶回收试剂盒纯化回收PCR产物.最后使用20 μL 10 mmol/L Tris·HC l(pH 8.0)洗脱.1.2.3 pCR8片段的获得利用限制性内切酶EcoR Ⅰ对质粒pCR8-AtS5H进行单酶切,反应体系为:10×H缓冲液5 μL,EcoR Ⅰ 2.5 μL,pCR8-AtS5H质粒(100 ng/μL)20 μL,ddH2O 22.5 μL;37 ℃下酶切1~3 h.酶切时间长短取决于EcoR Ⅰ酶活性,在保证酶切效果(完全酶切)的同时,应尽可能缩短酶切时间,避免造成星活性.酶切产物进行琼脂糖凝胶电泳,切胶并利用胶回收试剂盒纯化回收 pCR8片段(大小为2.8 kb).最后使用20 μL 10 mmol/L Tris-HCl(pH 8.0)洗脱,测定浓度,将浓度调整到大约30 ng/μL.1.2.4 pCR8-ICS1pro质粒的构建利用一步SLIC法将回收的ICS1pro片段和pCR8片段进行连接,其反应体系为:10×NEB缓冲液2.01 μL,100×BSA (NEB)0.1 μL,ICS1pro片段(80 ng/μL)3μL,pCR8片段(30 ng/μL)4 μL,ddH2O 1.9 μL,T4 DNA聚合酶(NEB)0.2μL(最后加入),其中ICS1pro片段和pCR8片段物质的量的比为2∶1.上述混合液在加入T4 DNA聚合酶后立即置于PCR仪中26 ℃下孵育2.5 min,然后立即将其置于冰浴中10 min,然后取2 μL反应产物转化大肠杆菌DH5α感受态(热激法),最后涂板至含有壮观霉素(80 mg/L)的LB培养基平板.此外,首次使用回收的pCR8片段时,应取相应量的pCR8质粒片段单独转化大肠杆菌DH5α作为负对照,用于检验1.2.3 方法中pCR8-AtS5H质粒的酶切效果.如果负对照转化不长斑或仅长几个斑,则获得的pCR8载体片段可用于质粒构建.此外,该步骤的重组反应也可以使用其他商品化的无缝克隆试剂盒替代.1.2.5 pCR8-ICS1pro质粒的菌落PCR验证和测序随机挑取6个单菌落进行菌落PCR验证,其PCR反应体系为:2×Taq Master Mix 5 μL,10 μmol/L引物ICS1pro-F2 1 μL,10 μmol/L引物ICS1pro-R2 1 μL,ddH2O 3 μL.反应条件为:94 ℃预变性3 min;94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min,共32个循环;72 ℃延伸10 min.根据菌落PCR结果,提取阳性克隆质粒,并送至杭州擎科梓熙生物技术有限公司进行测序.测序引物为通用引物M13F(TGTAAAACGACGGCCAGT)和M13R(CAGGAAACAGCTATGACC).1.2.6 LR重组反应将pCR8-ICS1pro质粒和pMDC163质粒(含有ccdB自杀基因)浓度分别调至150 ng/μL,LR重组反应体系为:pCR8-ICS1pro 1 μL,pMDC163 1 μL,LR Clonase II enzyme 0.5 μL.25 ℃孵育1 h后,加入0.25 μL蛋白酶K溶液,混匀后置于37 ℃培养箱反应30 min,然后取1 μL反应产物转化大肠杆菌DH5α感受态(热激法),最后涂板至含有卡那霉素(50 mg/L)的LB培养基平板.1.2.7 pMDC163-ICS1pro质粒的菌落PCR验证和测序随机挑取4个单菌落,按照1.2.5方法进行菌落PCR验证,获得pMDC163-ICS1pro阳性克隆,并提取质粒送至杭州擎科梓熙生物技术有限公司进行测序.测序引物为公共引物M13F(TGTAAAACGACGGCCAGT).2 结果与讨论2.1 ICS1pro片段扩增以日本晴DNA为模板,按照1.2.2方法成功扩增得到ICS1pro的片段,如图1所示.PCR产物大小约3.00 kb左右,与目标片段大小(2.89 kb)相符.对扩增产物进行纯化回收,洗脱时使用20 μL的10 mmol/L Tris-HCl(pH 8.0).1:ICS1pro PCR扩增片段;M:DNA分子标记图1 ICS1pro DNA片段扩增结果2.2 pCR8片段的获得按照1.2.3方法对实验室保存的pCR8-AtS5H质粒进行单酶切,其电泳结果如图2所示.pCR8-AtS5H酶切后产物大小分别为3.00 kb和1.10 kb左右,与理论预测的片段大小相符,其中3.00 kb左右的条带为pCR8片段,利用胶回收试剂盒对其割胶产物进行回收,洗脱时使用20 μL的10 mmol/L Tris·HCl(pH 8.0).1:pCR8-AtS5H 的EcoR Ⅰ酶切后产物;M:DNA分子标记图2 质粒pCR8-AtS5H的EcoR Ⅰ单酶切结果2.3 pCR8-ICS1pro入门克隆的构建按照1.2.4方法对回收的ICS1pro片段和pCR8片段进行连接和转化,并按照1.2.5方法对pCR8-ICS1pro质粒进行菌落PCR验证,其结果如图3所示.6个菌落PCR产物均与阳性对照大小一致,提取1号菌株质粒并送样测序,测序结果显示其序列与水稻中ICS1pro序列完全一致,因此该克隆即为pCR8-ICS1pro入门克隆.M:DNA分子标记;1—6:菌落PCR产物;7:阳性对照;8:阴性对照图3 pCR8-ICS1pro质粒的菌落PCR鉴定结果2.4 pMDC163-ICS1pro目标载体的构建按照1.2.6方法对pCR8-ICS1pro质粒和pMDC163质粒进行LR重组反应,并按照1.2.7方法进行菌落PCR鉴定,其结果如图4所示.4个菌落PCR产物均与阳性对照大小一致,提取1号菌株质粒并送样测序,测序结果显示其序列与水稻中ICS1pro序列完全一致,因此,该克隆即为pDMC163-ICS1pro入门克隆.该结果表明,利用一步SLIC方法构建的Gateway入门载体完全可用于目标载体的构建.M:DNA分子标记;1—4:菌落PCR产物;5:阳性对照;6:阴性对照图4pMDC163-ICS1pro质粒的菌落PCR鉴定结果3 讨论Gateway技术使载体构建更加的方便和快捷,一旦获得了目的基因的入门克隆,就可以非常高效地将目的基因转移到各种Gateway目的载体中[9].目前该技术被广泛用于蛋白表达、蛋白质亚细胞定位、基因文库构建、基因沉默、启动子分析及基因功能研究等[10-12].例如,范芳芳等[13]通过重叠PCR 技术扩增出AttB-TEV-FLAG-AIK 序列,利用 BP 重组反应将目的序列TEV-FLAG-AIK 克隆至供体载体pDONR223中,再通过 LR 重组反应将目的序列转移到目的载体pDEST15中,从而得到GST-AIK 融合蛋白原核表达质粒,实现了GST-AIK 融合蛋白的高效可溶性表达,经亲和层析获得了有生物活性的重组 AIK 多肽,为后续深入研究和大规模制备奠定了基础.虽然Invitrogen公司提供了多种入门载体系统和构建入门克隆的方法,但均存在一定的局限性,且成本相对较高[14],从而影响了Gateway技术的大范围应用.因此,科研人员尝试建立一种简便快速的构建入门克隆的方法.例如,通过对入门载体 pDONR207 进行改造,使其变成一个含有2个Xcm Ⅰ酶切位点和ccdB自杀基因的环状入门载体,经Xcm Ⅰ酶切后即可产生3′端具有单个T末端的线性化入门载体,再通过简单的TA连接方法即构建入门克隆,从而替代了原先较昂贵的BP反应[15].此外,孙全喜等[16]提供了另一种环状入门载体pRMG-C,该质粒含有Sal Ⅰ,Sac Ⅰ,Sph Ⅰ,BamH Ⅰ,Hind Ⅲ,Xho Ⅰ,Pst Ⅰ,Kpn I,EcoR Ⅰ等多个常用限制性酶切位点,可以方便基因的酶切-连接克隆,同时改造后的入门载体为环状质粒,可以自行保存和制备,大大降低了实验的成本.虽然这些改造后的入门载体降低了构建入门克隆的成本,但仍然存在一些缺点,例如TA连接存在非定向克隆的缺点,需要利用PCR方法鉴定片段的插入方向[17].本研究建立了一种利用一步SLIC方法快速构建Gateway入门载体的方法,该方法具有诸多优点:1)非常省时,2.5 min即可完成连接,而BP反应则需要1.5 h;2)利用实验室保存的入门克隆来获得入门载体片段,而且该入门克隆可以保存和反复使用,因此,实验过程中不需要购买昂贵的入门载体(20个反应试剂盒价格高达5 712.0元),同时该方法所用的其他试剂成本也非常低,单个反应所用T4 DNA聚合酶成本仅约2.8元,而单个BP反应成本高达134.8元.因此,本方法可以大大降低构建入门克隆的实验成本(以pCR8-AtS5H入门克隆为例,但其他任何含有合适酶切位点的入门克隆都适用于该方法);3)所利用的一步SLIC技术是一种不依赖序列和连接的克隆方法,不受酶切位点限制,同时连接效率非常高,也不存在TA连接的非定向克隆问题.目前实验室利用该方法成功构建了几十个pMDC系列的Gateway目的载体.LR重组效率取决于目的片段大小、LR酶活性和感受态转化效率等,大多在10~30个菌落/ng的目的载体,与原始pCR8/GW/TOPO载体重组效率相当.总之,本研究建立的利用一步SLIC技术构建入门克隆的方法操作非常简单、效率高且成本低,是一种非常值得推广的构建Gateway入门克隆的新方法.参考文献:【相关文献】[1]黄淦,王潇,金学锋,等.拟南芥谷氧还蛋白GRXC9负调控叶片大小[J].植物学报,2017,52(5):550-559.[2]AKBARI O S,OLIVER D,EYER K,et al.An Entry/Gateway® cloning system for general expression of genes with molecular tags in Drosophila melanogaster[J].BMC CellBiol,2009,10:8.[3]KARIMI M,DEPICKER A,HILSON P.Recombinational cloning with plant gatewayvectors[J].Plant Physiol,2007,145(4):1144-1154.[4]CURTIS M D,GROSSNIKLAUS U.A gateway cloning vector set for high-throughput functional analysis of genes in planta[J].Plant Physiol,2003,133(2):462-469.[5]REHMANY A P,GORDON A,ROSE L E,et al.Differential recognition of highly divergent downy mildew avirulence gene alleles by RPP1 resistance genes from two Arabidopsis lines[J].Plant Cell,2005,17(6):1839-1850.[6]RUARK C D,CHAPLEAU R R,MAHLE D A,et anophosphorus inhibition and characterization of recombinant guinea pig acetylcholinesterase[J].Protein PeptLett,2015,22(10):862-868.[7]WANG X,FAN C,ZHANG X,et al.BioVector,a flexible system for gene specific-expression in plants[J].BMC Plant Biol,2013,13:198.[8]JEONG J Y,YIM H S,RYU J Y,et al.One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies[J].Appl Environ Microbiol,2012,78(15):5440-5443.[9]陈其军,安瑞,周海梦,等.使用与Gateway技术兼容的T载体获得入门克隆[J].生物化学与生物物理进展,2004,31(10):951-954.[10]ALBERTI S,GITLER A D,LINDQUIST S.A suite of Gateway® cloning vectors for high- throughput genetic analysis in Saccharomyces cerevisiae[J].Yeast,2007,24(10):913-919. [11]肖素勤,孙振,轩秀霞,等.用于通路(Gateway)克隆技术的植物表达载体研究进展[J].植物科学学报,2012,30(5):528-544.[12]XIE W,NIELSEN M E,PEDERSEN C,et al.A Split-GFP Gateway cloning system for topology analyses of membrane proteins in plants[J].PLoS One,2017,12(1):e0170118. [13]范芳芳,孙慧莹,徐晖,等.重组阳离子抗肿瘤肽AIK的原核表达、纯化及活性测定[J].生物工程学报,2015,31(12):1753-1763.[14]周洁,王栩鸣,陈斌,等.基于Gateway技术的低成本植物双分子荧光互补分析系统[J].浙江农业学报,2013,25(5):1024-1030.[15]殷宪伦,王春涛,孔祥翔,等.利用TA克隆的方法简便构建入门克隆[J].植物分类与资源学报,2012,34(4):397-402.[16]孙全喜,苑翠玲,王传堂,等.一种改造的入门载体pRMG-C及其应用:CN201610620476.4[P].2016-12-14.[17]廖世奇,张春梅,张雪力,等.一种PCR快速鉴定重组体DNA阳性克隆及插入方向的方法[J].科学技术与工程,2004,4(2):89-90.。
gateway重组技术

Gateway 基因克隆Gateway 基因克隆是由Invitrogen公司在二十世纪九十年代末发明并应用于分子生物学基因克隆的一项专利技术。
该技术利用专有的重组序列使得DNA片段能够更有效地被转入质粒当中,可应用于大片段的基因克隆,并且在保持正确阅读框的前提下让不同表达载体间的DNA转移成为可能。
这一技术在插入的目的DNA片段两端整合att L1和att L2两个侧端重组序列,来构建一个类似通道的结构并称之为“入门克隆”(Gateway Entry Clone)。
据Invitrogen宣称Gateway技术使用99%有效且可逆的一小组重组反应,如此使得基因克隆不同于传统的限制性内切酶方法,避免了目的片段内存在切点的问题而使得大片段DNA保持其完整性,大大提高了克隆效率,常应用于大规模的DNA片段整合进同一种表达载体,因此又称之为高通量基因克隆技术(Gateway Cloning Technology)。
一、Gateway 基因克隆的原理及机制Gateway被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后,就不用依赖限制性内切酶,而靠载体上存在的特定重组位点和重组酶,高效、快速地将目的基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway的原理是建立在噬菌体DNA定点整合到细菌宿主基因组上。
在噬菌体和细菌的整合因子(INF、Int)的作用下,lambda的attP位点和大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中,两侧产生两个新位点:attL和attR。
这是一个可逆的过程,如果在一个噬菌体编码蛋白Xis和IHF、Int的共同介导下这两个新位点可以再次重组回复为attB和attP位点,噬菌体从细菌基因组上裂解下来(见图1)。
这一过程的方向是受控于两个重要因素:存在的介导蛋白和重组位点。
简单易懂的gateway克隆技术

简单易懂的gateway克隆技术Gateway是计算机网络中的重要组件,其主要作用是连接不同的网络,实现数据的转发与路由功能。
随着网络技术的迅猛发展,Gateway的功能和形态也得到不断的升级和改进,其中最重要的就是克隆技术。
Gateway克隆技术是指利用一台正常工作的Gateway硬件设备,通过复制其配置文件和软件程序,生成多台相同的Gateway设备的技术。
它的主要作用是提供快速、简单、可靠且低成本的网络连接方案。
使用Gateway克隆技术可以使得多台设备的配置一致,避免手工配置时产生的差错。
此外,克隆技术还可以提高整个网络的安全性,因为所有的Gateway设备都采用相同的防火墙规则和安全策略。
Gateway克隆技术的实现主要分为以下几个步骤:第一步,备份正常工作的Gateway设备的配置文件和软件程序。
在这个过程中,需要特别注意备份的文件的完整性和正确性,以保证之后的克隆操作能够成功。
第二步,将备份好的文件拷贝到新的Gateway设备中。
这个过程中需要注意新设备的硬件环境和原有设备的差异,如网卡数量、IP地址等,需要根据实际情况进行相应的修改配置。
第三步,启动新的Gateway设备,并进行测试。
在测试过程中需要确认新设备的网卡是否能够正常与外部网络连接,数据是否可以稳定地通过新设备进行传输。
第四步,对新设备进行优化和调整。
根据实际情况对新设备进行一些优化和调整,如修改裸机的内核参数、完善防火墙规则等,以提高设备的性能和安全性。
总的来说,Gateway克隆技术具有快速、简单、可靠、低成本等特点,适用于大规模的企业网络。
但也需要注意,克隆过程中如果文件备份出现差错或者新设备硬件环境设置不当,有可能导致后续操作不能进行或者数据传输不稳定。
因此,在实际应用中需要慎重考虑,以确保数据的安全和稳定。
goldengate克隆技术原理

goldengate克隆技术原理GoldenGate克隆技术原理介绍GoldenGate克隆技术是一种先进的数据复制和实时数据集成解决方案,被广泛应用于数据库和数据仓库的克隆、迁移、高可用和实时数据集成等领域。
本文将从浅入深,逐步解释GoldenGate克隆技术的原理。
原理GoldenGate克隆技术的原理可以简单概括为以下几个步骤:1.源端捕获:GoldenGate的捕获进程会监视源端数据库的日志文件,实时抓取并解析变更的数据。
2.数据转换:捕获到的数据会被GoldenGate进行格式转换,以确保适配目标端数据库的格式和结构。
3.数据传输:转换后的数据会通过网络传输到目标端,并由传输进程负责确保可靠且高效的传输。
4.数据应用:目标端的进程将接收到的数据应用到目标端数据库,确保数据在两端的一致性。
详细解释下面将对每个步骤进行详细解释:源端捕获GoldenGate通过实时监视源端的日志文件,可以捕获到源端数据库中的各种变更操作,包括插入、更新和删除等。
这个捕获的过程是非侵入性的,不会对源端数据库的性能造成影响。
数据转换捕获到的数据并不是直接复制到目标端数据库,而是经过GoldenGate的数据转换进程进行格式转换。
这个过程包括对数据的解析、映射和转换等操作,以确保源端和目标端数据库的数据格式和结构一致。
数据传输经过数据转换的数据会通过网络传输到目标端。
GoldenGate的传输进程负责确保数据的可靠传输,包括数据压缩、流量控制和错误处理等。
传输过程采用高效的网络协议,可以在保证数据安全的同时,提供较低的传输延迟。
数据应用目标端的进程会接收到传输过来的数据,并将这些数据应用到目标端的数据库中。
应用过程包括对数据进行事务管理、冲突检测和应用等操作,以确保目标端数据库与源端数据库的一致性。
结论GoldenGate克隆技术通过源端捕获、数据转换、数据传输和数据应用等步骤,实现了高效、可靠的数据库复制和实时数据集成。
Gateway技术

• 1.3 进行LR反应。 混合包含目的基因的入门克隆和合适的目的 载体以及Gateway LR Clonase酶,构建表达克 隆。(表达克隆用来在合适的宿主中进行蛋 白的表达和分析。)
PCR定向(PCR-Directional)TOPO克隆
• 定向TOPO克隆使得克隆PCR产物和其它的DNA分子更加快 速和有效。简单连接可以产生大于90%的重组子。 • 定向TOPO克隆可以定向克隆平端PCR产物到入门载体,简 化了酶切,连接,筛选等PCR后续步骤。 • 目前有两种定向TOPO克隆载体:pENTR/D-TOPO 和 pENTR/SD/ D-TOPO,有以下特点:
Gate way 技术 的灵 活性
Gateway技术原理
• Gateway技术是基于已研究的清楚的λ噬菌体位 点特异重组系统(attB x attP →attL x attR), 由 BP和LR两个反应过程构成。 • BP反应是利用一个attB DNA片段或表达克隆和 一个attP供体载体之间的重组反应,创建一个 入门克隆。 • LR反应是一个attL入门克隆和一个attR目的载体 之间的重组反应。 LR反应用来在平行的反应中 转移目的序列到一个或更多个目的载体。
Байду номын сангаас Topo原理
• Topo TA克隆原理与TA克隆一样,唯一不同的是TA克隆用的是T4 连接酶把PCR片断连接到T载体上,而Topo TA Cloning用的是DNA Topoisomerase(DNA拓扑异构酶I),该酶同时具有限制酶和连 接酶特性。 其生物学功能是在复制DNA前把超螺旋DNA切割使 之解旋后,再连接成线性DNA。 • Topoisomerase有高效连接的特性把含3`A端的PCR扩增片断快速 连接到3`T端载体上,整个反应只需五分钟!一般T4连接酶需过 夜才能高效连接 • 实验常用牛痘病毒拓扑异构酶I可特异性地识别五核苷酸序列5’(C/T)CCTT-3’,并且可与连接到3´胸腺嘧啶脱氧核苷的磷酸基团 形成共价键。 它切割一条 DNA 链,而使 DNA 解旋。 随后,这 种酶重新连接被切割的链,并使自身从 DNA 上释放出来。 为了 利用拓扑异构酶的再连接活性,我们提供在每个3´磷酸基团连 接有拓扑异构酶 I 的线性化TOPO®载体。 这样,载体便能够轻 易地将 DNA 序列与配伍末端连接在一起
gateway重组技术.pptx

Gateway 被视为一种克隆操作平台:把目的基因克隆到入门载体(Entry Vector)后, 就不用依赖限制性内切酶,而靠载体上存在的特定重组位点和重组酶,高效、快速地将目的 基因克隆到其它的受体载体(Destination Vector,目的载体)上。
Gateway 的原理是建立在噬菌体 DNA 定点整合到细菌宿主基因组上。在噬菌体和细菌 的整合因子(INF、Int)的作用下,lambda 的 attP 位点和大肠杆菌基因组的 attB 位点可以 发生定点重组,lambda 噬菌体DNA 整合到大肠杆菌的基因组DNA 中,两侧产生两个新位 点:attL 和 attR。这是一个可逆的过程,如果在一个噬菌体编码蛋白 Xis 和 IHF、Int 的共 同介导下这两个新位点可以再次重组回复为 attB 和 attP 位点,噬菌体从细菌基因组上裂解 下来(见图 1)。这一过程的方向是受控于两个重要因素:存在的介导蛋白和重组位点。
图 2、gateway 机制 由于在这个反应中 attL1 序列只和 attR1 序列重组,attL2 序列只能与 attR2 序列重组, 这个方向的反应称为LR 反应。LR 反应生成新的位点称为attP1/2(200bp)和 attB1/2(25bp) 序列。 在一定的条件下,attP 和 attB 序列也能发生重组,生成 attL 和 attR 序列,这个反 向反应称为 BP 反应(见图 3)[1]。
Gateway 基因克隆
Gateway 基因克隆是由 Invitrogen 公司在二十世纪九十年代末发明并应用于分子生物 学基因克隆的一项专利技术。该技术利用专有的重组序列使得 DNA 片段能够更有效地被转 入质粒当中,可应用于大片段的基因克隆,并且在保持正确阅读框的前提下让不同表达载体 间的DNA 转移成为可能。
M5 HiPer Gateway BP Clone Kit 使用说明书

101422-1 86M5 HiPer Gateway BP Clone Kit 使用说明书产品名称 单位 货号M5 HiPer Gateway BP Clone Kit 20T MF961-01M5 HiPer Gateway BP Clone Kit 4x20T MF961-04【储存条件】在-80°C 环境中保存。
使用时请置于冰上融解并一直保持低温状态。
【产品简介】Gateway 克隆系列产品是一种崭新的高通量基因克隆技术,可以简单高效地把入门载体 (The EntryClone) 克隆到多种目的载体 (Destination vector) 中。
Gateway 克隆技术无需依赖限制性内切酶和连接酶的传统克隆技术,而是利用天然的 λ 噬菌体与大肠杆菌 (E. coli) 的染色体之间产生的位点 (att)特异性的重组整合,使 DNA 片段在不同的克隆载体之间实现转移。
Gateway 克隆重组技术具有可靠的稳定性,当基因在重组目的表达载体之间快速简便地穿梭时,可以保证基因以正确的方向插入并保持阅读框架不发生改变。
使用 Gateway BP Clone Enzyme 可以使两端带有 attB 位点的基因片段和供体载体 (pDONR) 接合形成入门载体 (The Entry Clone)。
Gateway 克隆体系在细菌转化这个环节使用正向筛选(抗生素)和负向筛选(ccdB 致死基因)两种选择,以确保得到高比例的阳性重组。
【产品特色】灵活便捷:易于把基因克隆到带有不同启动子和标记的多种载体。
方便快速:最大化缩短实验前计划时间,无需限制性内切酶和连接酶。
准确无误:基因以正确的方向插入并保持阅读框架不发生改变。
无需测序:一旦入门载体 (The Entry Clone) 被建立,就可以使用 Gateway 克隆系统克隆至任意Gateway 目的载体 (Destination Vector) ,无需担心引入突变。
Gateway 克隆技术背景原理及其应用

If the normal attB site is deleted from the chromosome, integrase can bind with lower affinity to secondary sites on thechromosome, resulting in integration of lambda at a different site. The frequency of integration into secondary sites is 100-1000 times rarer than integration into attB. The actual crossover occurs between homologous 15 bp core regions on the two sites, but surrounding sequences are required as they contain the binding sites for the recombination proteins (Landy, 1989).
Modifications to the att Sites
Mutations have been made to the core regions of the att sites to eliminate stop codons and to ensure specificity of the recombination reactions to maintain orientation and reading frame. Mutations have been introduced into the short (5 bp) regions flanking the 15-bp core regions of the attB sites to minimize secondary structure formation in single-stranded forms of attB plasmids (e.g. phagemid ssDNA or mRNA). A 43 bp portion of the attR site has been removed to make the in vitro attL x attR reaction irreversible and more efficient (Bushman et al., 1985).
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Gateway克隆技术,简单易操作
在过去数十年,用限制性内切酶产生黏性末端,在DNA连接酶的作用下,连接两个甚至更多片段的克隆方法,是载体构建的经典方案。
但是现如今我们再提到克隆的时候,远远不再只有传统的限制酶克隆一种选择。
其中Gateway克隆技术作为一种可以快速、高效将DNA 序列转移到载体上的克隆方法已经流行开来。
不要觉得Gateway克隆技术高深莫测,其实了解一下你就能懂。
Gateway克隆方法是λ噬菌体感染细菌时发生的整合和切割重组反应的体外形式。
在体内,噬菌体(attP)和细菌(attB)的附着位点发生重组反应,噬菌体整合到细菌基因组中,两侧为两个新的重组位点(attL-left-和attR-right-)。
在某些条件下,attL和attR位点可以重组,导致噬菌体从细菌染色体上切除并重新生成attP和attB位点。
即Gateway技术依赖于下面描述的两个反应:BP反应和LR反应,通过BP反应获得入门克隆(有时候我们也说中间克隆),再通过LR反应将目的DNA以正确的方向连接到各种目的载体上,形成不同的表达载体。
GeneCopoeia的EZShuttle™重组克隆体系应用E.coli 和λ噬菌体特异位点的重组酶促体系使DNA片段在载体间相互转换,其原理与Gateway 克隆技术相同。
BP反应-构建入门载体:
通过PCR将attB位点添加到目的DAN序列两侧,形成attB-PCR产物。
用attB-PCR产物或者含attB位点的供体质粒和含attP位点的质粒发生重组生成入门克隆。
EZRecombinase BP混合物,货号:RCBM-1002-020
LR反应-构建表达载体:
制作表达载体时,选择适合的目标载体非常重要。
这种选择取决于许多因素,如宿主类型,所需的表达水平和实验目的。
选定的attR表达载体与attL-入门载体重组以产生表达载体。
EZRecombinase LR混合液,货号:RCBM-1001-020。