GDC-0980 (RG7422)_I型PI3K PI3Kαβδγ抑制剂_1032754-93-0_Apexbio
miR-802靶向调控PI3K

·论著·miR-802靶向调控PI3K/Akt信号通路促进肝细胞癌血管新生的机制研究刘文豪倪敏沈甫明金涌【摘要】目的探究miR-802在肝细胞癌(HCC)细胞中的表达及其对肿瘤血管新生的影响及作用机制。
方法通过转染miR-802 agomir、miR-802 antagomir上调或抑制人HCC细胞系SMMC-7721中miR-802的表达水平。
将SMMC-7721细胞分为NC组(正常培养细胞)、miR-802 agomir组(转染miR-802 agomir)和miR-802antagomir组(转染miR-802 antagomir)。
采用小管形成实验和Transwell实验检测过表达miR-802的SMMC-7721细胞上清液对人脐静脉内皮细胞(HUVEC)成管能力及迁移能力的影响。
采用蛋白质印迹法检测miR-802 agomir组中磷脂酰肌醇3激酶(PI3K)、磷酸化PI3K(p-PI3K)、蛋白激酶B(Akt)、磷酸化Akt(p-Akt)、血管内皮生长因子(VEGF)的表达水平。
采用免疫荧光实验检测过表达miR-802对VEGF生成的影响。
结果与NC组相比,miR-802 agomir组HUVEC细胞的成管能力及迁移能力均显著增强,miR-802 antagomir组HUVEC细胞的成管能力及迁移能力均显著降低,差异均有统计学意义(P均<0.05)。
与NC组相比,miR-802 agomir组SMMC-7721细胞中VEGF生成量增加,p-PI3K、p-Akt、Akt蛋白表达水平均显著升高,差异均有统计学意义(P均<0.05)。
结论 miR-802可通过调控PI3K/Akt信号通路促进VEGF的释放,参与血管新生,从而促进HCC的发生和进展。
【关键词】肝细胞癌;miR-802;血管新生;PI3K/Akt信号通路DOI: 10. 3969/j. issn. 1673-534X. 2024. 01. 009Mechanism of miR-802 targeting PI3K/Akt signaling pathway in promoting angiogenesisof hepatocellular carcinoma LIU Wenhao, JIN Yong. School of Pharmacy, Anhui Medical University,Hefei 230032, China; NI Min, SHEN Fuming. Department of Pharmacy, Tenth People's Hospital of TongjiUniversity, Shanghai 200072, China【Abstract】 Objective This paper attempts to investigate the expression of miR-802 in hepatocellular carcinoma cells and its effect on tumor angiogenesis and mechanism of action. Methods Theexpression of miR-802 in human hepatocellular carcinoma cell line SMMC-7721 was up-regulated or inhibited by transfection of miR-802 agomir and miR-802 antagomir. SMMC-7721 cells were divided into the NC group (normal cultured cells), the miR-802 agomir group (transfected with miR-802 agomir), and the miR-802 antagomir group (transfected with miR-802 antagomir). The effects of SMMC-7721 cell supernatant overexpressing miR-802 on the tube-forming and migratory abilities of human umbilical vein endothelial cells (HUVEC) were detected by using tubule formation assay and Transwell assay. The expression of phosphatidylinositol 3-kinase (PI3K), phosphorylated PI3K (p-PI3K), protein kinase B (Akt), phosphorylated Akt (p-Akt), and vascular endothelial growth factor (VEGF) was detected by western blotting in the miR-802 agomir group. Immunofluorescence assay was used to detect the effect of overexpression 基金项目:上海市崇明区“可持续发展科技创新行动计划”项目(CKY2022-24)作者单位:230032 安徽合肥,安徽医科大学药学院(刘文豪、金涌);200072 上海,同济大学附属第十人民医院临床药学部(倪敏、沈甫明)通信作者:金涌,of miR-802 on VEGF production. Results Compared with the NC group, the tube-forming and migratory abilities of HUVEC cells in the miR-802 agomir group are enhanced, and the tube-forming and migratory abilities of HUVEC cells in the miR-802 antagomir group are reduced, with statistically significant differences (P<0.05). Compared with the NC group, the production of VEGF of SMMC-7721 cells in the miR-802 agomir group is increased, and the protein expression of p-PI3K, p-Akt, and Akt is elevated, with statistically significant differences (P<0.05). Conclusion miR-802 can promote the release of VEGF through regulating the PI3K/Akt signaling pathway, and then participate in angiogenesis, thus promoting the occurrence and progression of hepatocellular carcinoma. 【Key words】 Hepatocellular carcinoma; miR-802; Angiogenesis; PI3K/Akt signaling pathway原发性肝癌是中国第4位常见恶性肿瘤及第2位肿瘤致死病因,包括肝细胞癌(HCC)、肝内胆管癌(ICC)和混合型肝细胞癌-胆管癌(cHCC-CCA)这3种不同病理类型,其中HCC患者占75%~85%[1-2]。
北医信号转导课件PI3K

PTEN是一种磷酸酶,可将 PI(3,4,5)P3去磷酸化生成
PI(4,5)P2,从而负调控PI3K信号 通路。
SHIP
SHIP也是一种磷酸酶,可将 PI(3,4,5)P3去磷酸化生成PI(3,4)P2 ,同样起到负调控作用。
Ras
Ras蛋白是PI3K信号通路的重要上 游调控因子,可通过激活Raf-1等激 酶间接激活PI3K。
一类特异性催化磷脂酰肌醇( Phosphatidylinositol,PI)3位羟基
ห้องสมุดไป่ตู้磷酸化的激酶。
PI3K由调节亚基和催化亚基组成,其 中调节亚基包含SH2和SH3结构域,负 责与上游信号分子结合;催化亚基具有
激酶活性,负责将PI磷酸化。
根据结构和底物特异性不同,PI3K可 分为I、II、III三种类型,其中I型PI3K 研究最为广泛,与细胞生长、增殖、分
随着对PI3K信号通路认识的深入和药 物研发技术的不断进步,未来将有更 多针对PI3K靶点的创新药物进入临床 试验和临床应用阶段。同时,针对患 者个体差异的精准医疗策略也将成为 未来发展的重要方向。
04
PI3K信号通路与其他通路交互 作用
PI3K与MAPK通路交互作用
PI3K激活后通过产生PIP3招募 并激活PDK1,进而激活AKT等 下游效应蛋白,构成PI3K-AKT
PI3K信号通路与Wnt通路在细 胞极性、迁移和侵袭等过程中 存在交互作用。
PI3K通过激活AKT等下游效应 蛋白,可调控Wnt通路中关键 蛋白的表达和活性。
PI3K与其他信号通路联系
PI3K信号通路与NF-κB通路存在 交互作用,共同参与炎症反应和
免疫应答的调控。
PI3K信号通路与Hippo通路在细 胞增殖、凋亡和器官大小控制等
小分子抑制剂、激动剂、拮抗剂--PI3KAktmTOR信号通路
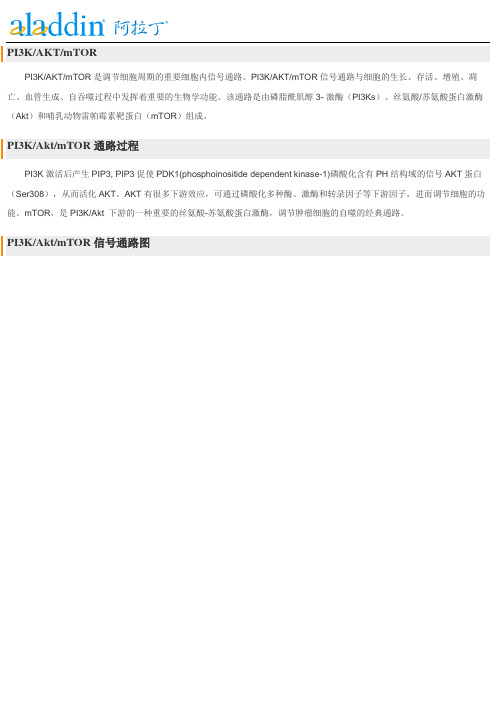
PI3K/AKT/mTORPI3K/AKT/mTOR是调节细胞周期的重要细胞内信号通路。
PI3K/AKT/mTOR信号通路与细胞的生长、存活、增殖、凋亡、血管生成、自吞噬过程中发挥着重要的生物学功能。
该通路是由磷脂酰肌醇3- 激酶(PI3Ks)、丝氨酸/苏氨酸蛋白激酶(Akt)和哺乳动物雷帕霉素靶蛋白(mTOR)组成。
PI3K/Akt/mTOR通路过程PI3K激活后产生PIP3, PIP3促使PDK1(phosphoinositide dependent kinase-1)磷酸化含有PH结构域的信号AKT蛋白(Ser308),从而活化AKT。
AKT有很多下游效应,可通过磷酸化多种酶、激酶和转录因子等下游因子,进而调节细胞的功能。
mTOR,是PI3K/Akt 下游的一种重要的丝氨酸-苏氨酸蛋白激酶,调节肿瘤细胞的自噬的经典通路。
PI3K/Akt/mTOR信号通路图按靶点分类:*PI3KPI3K,是一种胞内磷脂酰肌醇激酶,也具有丝氨酸/苏氨酸(Ser/Thr)激酶的活性。
能够通过PI3K诱发PIP3生成的激活因子,则能够激活Akt 信号途径,包括受体酪氨酸激酶、整合素、B 细胞和T 细胞受体、细胞因子受体、G 蛋白偶联受体等等。
*Akt又称PKB或Rac,是一种丝氨酸/苏氨酸特异性蛋白激酶B,在细胞存活和凋亡中起重要作用,如葡萄糖代谢、凋亡、细胞增殖、转录和细胞迁移。
Akt的Thr308可以被PDK1磷酸化,而被部分激活。
或者473位点上的丝氨酸被mTORC2磷酸化,激发Akt的完全酶活性。
*mTORmTOR是细胞生长和增殖的重要调节因子。
mTOR与其它蛋白质结合,形成两种不同蛋白质复合物,mTORC1和mTORC2,参与调节不同的细胞过程。
*GSK-3。
过氧化物酶体增殖物激活受体

过氧化物酶体增殖物激活受体(PPAR) 是一类由配体激活的核转录因子,属Ⅱ型核受体超家族成员, 存在3种亚型,即PPARα、PPARδ、PPARγ,这三种亚型在结构上有一定的相似性,均含DNA结合区和配体结合区等。
PPAR与配体结合后被激活,与9-顺视黄酸类受体形成异二聚体,然后与靶基因的启动子上游的过氧化物酶体增殖物反应元件(peroxisome proliferator response element,PPRE)结合而发挥转录调控作用。
PPRE 由含相隔一个或两个核苷酸的重复序列AGGTCA组成。
与配体结合后,PPAR在DNA结合区发生变构,进而影响PPAR刺激靶基因转录的能力。
PPARδ几乎在所有组织中表达,浓度低于PPARα及PPARγ,直至最近以前尚未找到此一核受体的选择性配基。
PPARδ是代谢综合征(肥胖、胰岛素抵抗、高血压是与脂质紊乱有关的共同的病态表现)的一个新靶点。
有不少的研究表明:GW501516可作为PPARδ的特异激动剂用于研究。
参考网址:/cjh/2003/shownews.asp?id=156/conference/preview.php?kind_id=03&cat_name=ADA2001&title_id=59219 Regulation of Muscle Fiber Type and Running Endurance by PPARδplos biology,Volume 2 | Issue 10 | October 2004/plosonline/?request=get-document&doi=10.1371%2Fjournal.pbio.0020294NF-KB通路中的抑制剂好像有1.PDTC(pyrrolidine dithiocarbamate),是一种抗氧化剂,主要作用于IκB降解的上游环节(IκBα的磷酸化或IKK的活性水平),2.Gliotoxin 是一种免疫抑制剂,机制可能从多个环节阻断NF-KB的激活,如IκB的降解,NF-KB的核移位和与DNA的结合。
GDC-0084_PI3K和mTOR抑制剂,脑通透性的_1382979-44-3_Apexbio

产品描述:
GDC-0084 is a potent and brain penetrant inhibitor of PI3K and mTOR with Ki values of 2 nM and 70 nM for PI3Kα and mTOR, respectively [1]. Glioblastoma (GBM) is the most common primary brain tumor in adults and aberrant PI3K signaling is associated with more than 80% of cases. The PI3K pathway represents a potential target for the treatment of this disease and the inhibitors would need to freely cross the blood-brain barrier (BBB) [1][2]. GDC-0084 is a potent and brain penetrant inhibitor of PI3K and mTOR. In vitro kinase assay,
参考文献: [1]. Heffron TP1, Ndubakiscovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of PI3K and mTOR. ACS Med Chem Lett. 2016 Feb 16;7(4):351-6. [2]. Salphati L, Alicke B, Heffron TP, et al. Brain Distribution and Efficacy of the Brain Penetrant PI3K Inhibitor GDC-0084 in Orthotopic Mouse Models of Human Glioblastoma. Drug Metab Dispos. 2016 Dec;44(12):1881-1889. Epub 2016 Sep 16.
TGX221PI3K抑制剂-碧云天
使用说明:
1. 收到产品后请立即按照说明书推荐的条件保存。使用前可以在2,000-10,000g离心数秒,以使液体或粉末充分沉淀至管底后 再开盖使用。
2. 对于10mM溶液,可直接稀释使用。对于固体,请根据本产品的溶解性及实验目的选择相应溶剂配制成高浓度的储备液(母 液)后使用。
2.Straub A, et al. Thromb Haemost. 2008, 99(3), 609-615.
3.Lu XY, et al. Appl Microbiol Biotechnol. 2011, 89(5), 1423-1433.
4.Bird JE, et al. Thromb Res. 2011, 127(6), 560-564.
择性作用p110β,作用于PI3K p110β和PI3K p110δ时,IC50分别为8.5和211nM。而且,TGX-221作用于
体外研究 J774.2巨噬细胞,局部降低胰岛素诱导的PKB在Ser473位点磷酸化。TGX-221作用于体外循环(ECC)模
型,抑制血小板-ECC相互作用,血小板凝聚和血小板-粒细胞结合。最新研究显示,用0.2、2和20μM
均肾脏BT。
临床实验 N/A
特征
TGX-221是有效的可渗透细胞的PI3K p110β选择性抑制剂。
相关实验数据(此数据来自于公开文献,碧云天并不保证其有效性):
酶活性检测实验
使用标准脂质激酶活性,及PI作为底物测定IC50值(i)使用100μM冰冻ATP代替10μM,(ii)DMSO浓度为
方法
然后在室温下温育30分钟。板侵泡在PBS溶液中洗三次。烘干板,然后加入100μl 0.1%溶于去离子水的 结晶紫溶液染色,然后在室温下温育20分钟,用去离子水大面积冲洗,移除过量染料,然后烘干板,然 后把染料溶化在100μl 10%乙酸中。使用酶标仪在570nM处直接测量染料抽提物的光密度。
HDAC多靶点抑制剂在癌症治疗中的研究进展

HDAC多靶点抑制剂在癌症治疗中的研究进展朱玉垚;黄坤;王瑜;马俊杰【摘要】在抗癌药物的研发中,组蛋白脱乙酰化酶(HDAC)是极具研究前景的靶标之一.到目前为止,有5种HDAC抑制剂已被批准用于癌症治疗,并有多种HDAC抑制剂也正处于临床试验阶段.现已证实,多靶点抑制剂在预防、治疗和增强协同效应方面较单一靶点药物具有更大优势.本文针对抗癌药物领域,主要对已报道的HDAC 多靶点抑制剂的设计思路和生物活性进行了综述.【期刊名称】《聊城大学学报(自然科学版)》【年(卷),期】2019(032)005【总页数】9页(P71-79)【关键词】抗癌;组蛋白脱乙酰化酶;多靶点抑制剂【作者】朱玉垚;黄坤;王瑜;马俊杰【作者单位】华侨大学医学院,福建泉州362000;华侨大学医学院,福建泉州362000;华侨大学医学院,福建泉州362000;华侨大学医学院,福建泉州362000【正文语种】中文【中图分类】R9660 前言众所周知,癌症的发病机制极其复杂,在发病过程中涉及多种酶、结构蛋白和转录因子.尽管作用于单一靶点的生物活性分子已经被广泛应用于临床,但由于癌细胞可以触发补偿性生存途径,这些治疗方法往往无法提供有效和持久的肿瘤治疗效果.因此,单一靶点的抗癌药通常具有不敏感性和耐药性.现有两种策略可以用来解决该问题:一种是联合用药,但这一策略面临着患者依从性差、药代动力学复杂以及药物与药物相互作用等缺点,严重影响其中一种或多种药物的有效性[1].另一种是将几个生物活性基团结合到一个单一分子中,产生能够同时作用于多个细胞通路的单体化合物,与单一靶向制剂相比具有更高的效率[1, 2],可以有效地克服药代动力学的缺点,并降低开发成本.因此,多靶点药物的开发已经被认为是发现新型抗癌药物的有效途径,具有很大的开发前景[3].表观遗传学在癌症的起源、发展和转移中扮演着重要的作用.组蛋白的乙酰化作为最常见的表观遗传学修饰,在细胞分化、增殖、血管生成和凋亡等正常细胞过程中发挥着至关重要的调节作用,乙酰化失调与癌症的发生和发展息息相关[4].组蛋白和非组蛋白的乙酰化水平由两个拮抗酶家族控制:组蛋白脱乙酰化酶(HDAC)和组蛋白乙酰转移酶(HAT).HDAC是细菌、真菌、植物和动物中普遍存在的酶家族,能够从核心组蛋白和许多非组蛋白中的赖氨酸残基ε-氨基中去除乙酰基[5].已知的HDAC根据其序列同源性分为四类:I类HDAC(1、2、3和8);II类HDAC IIa(4、5、7和9)和IIb(6和10);III类HDAC(sirt1-7);IV类HDAC11[6].沉默或抑制HDAC对细胞周期、细胞生长、染色质异构化、细胞分化、细胞凋亡和血管生成均表现出显著的影响.因此,HDAC已成为治疗癌症的重要研究靶点[7].目前,已有5种HDAC抑制剂:SAHA(1)、Romidepsin(2)、Belinostat(3)、Panobinostat(4)和Chidamide(5)分别被批准用于治疗包括皮肤T淋巴细胞瘤、外周T淋巴细胞瘤(PTCL)和多发性骨髓瘤[8]等癌症.一般来说,这些HDAC抑制剂的药效团可由三部分组成,如图1所示:一个能与HDAC活性口袋边缘相互作用的帽子结构(CAP)、一个锌离子结合基团(ZBG)和一个负责连接CAP与ZBG并可与活性口袋的疏水性通道作用的连接臂(Linker).在这三部分药效团结构中,由于CAP区能够接受较大范围的结构变化.因此,研究人员保持HDAC抑制剂的ZBG 和linker基本不变,通过改变CAP区的结构,开发出了大量的HDAC抑制剂,除了上述5种被批准的HDAC抑制剂外,目前已有多种HDAC抑制剂处于临床试验阶段[9].图1 5种已上市的HDAC抑制剂图2 HDAC多靶点抑制剂的设计策略目前,大部分HDAC抑制剂对血液瘤均能表现出较好的治疗效果,但大部分HDAC抑制剂对实体瘤的治疗效果一般,甚至没有明显的治疗作用[10].为了解决该问题,研究人员提出了开发HDAC多靶点抑制剂的设计思想,在保留HDAC抑制剂的关键药效团ZBG不变的情况下,将对实体瘤有效的靶向抗癌药物(激酶类抑制剂、凋亡类抗癌药物和激素类抗癌药物等)的药效团作为HDAC的CAP区引入到结构中(图2),设计得到多种基于HDAC的多靶点抑制剂,如EGFR/HER2-HDAC抑制剂、VEGFR-HDAC抑制剂、c-Met-HDAC抑制剂、Abl-HDAC抑制剂、PI3K-HDAC抑制剂和P53/MDM2-HDAC抑制剂等,研究表明,根据这种设计策略设计得到的目标化合物不仅可以同时抑制相应的抗癌靶点,而且对实体瘤和血液瘤均能表现出良好的治疗效果[11].一些HDAC多靶点抑制剂已经进入了临床试验阶段,如GUDC-101和GUDC-907.1.1 EGFR/HER2-HDAC多靶点抑制剂埃罗替尼 (erlotinib, 6),表皮生长因子受体/人表皮生长因子受体2 (EGFR/HER2)抑制剂,构效关系研究表明[12],埃罗替尼的喹唑啉和苯胺基团与EGFR受体的ATP结合口袋有着重要的结合作用,是关键的药效基团.但喹唑啉基团上的C-6和C-7处的两个甲氧乙氧基并不参与EGFR受体的结合,是可变基团.因此,Cai等人为了不影响化合物与EGFR 受体的结合,保留了erlotinib的喹唑啉和苯胺基团,在C-6或C-7上引入不同空间体积(醚、酰胺、硫醚和砜)和不同长度的异羟肟酸作为ZBG,替换erlotinib甲氧乙氧基,设计合成得到一系列erlotinib衍生物,如图3.研究表明,这些目标化合物对EGFR、HER2和 HDAC均表现出突出的抑制活性.HDAC抑制剂的活性与linker的长度有关,以6个碳的长度为最佳.同时,linker的结构也影响HDAC的抑制活性.如醚类linker比酰胺类更有效,而砜类linker的抑制活性最低.C-6取代的目标化合物对HDAC的抑制活性优于C-7取代的化合物.其中,CUDC-101(7)活性最强,对HDAC、EGFR和HER2的IC50值分别为4.4、2.4和15.7 nM[13].目前,CUDC-101已进入临床一期研究阶段.体外抗肿瘤活性研究表明,CUDC-101抑制了多种实体肿瘤细胞的增殖,包括肺癌、肝癌、胰腺癌和乳腺癌等,IC50值均低于1 μM,比单独使用erlotinib或SAHA 和erlotinib联合用药要好得多.由于CUDC-101能直接抑制EGFR和HER2信号,间接减弱其他生存信号通路,如Akt、HER3和MET[14],因此除了显著的抗增殖作用外,CUDC-101还能阻止肿瘤细胞迁移和侵袭[15].同时,在体内Hep-G2肝癌模型中,在每日120 mg/kg的剂量下,CUDC-101表现出了显著的抗肿瘤活性,诱导了30%的肿瘤体积消退,比erlotinib和SAHA更加有效[16].图3 埃罗替尼类EGFR/HER2-HDAC多靶点抑制剂1.2 VEGFR-HDAC多靶点抑制剂凡德他尼 (Vandetanib, 8)作为一种有效的血管内皮生长因子-2型(VEGFR2)抑制剂,IC50值为40 nM,同时也抑制VEGFR3和EGFR,IC50分别为110 nM 和500 nM,是首个被批准用于治疗甲状腺髓样癌的药物[17].Shi等人以Vandetanib的药效结构4-取代苯胺喹唑啉为设计模板,在C-6位引入SAHA(1)的ZBG结构-异羟肟酸,设计合成得到一系列VEGFR-HDAC多靶点抑制剂[18],如图4.研究表明,linker的长度仍然是影响HDAC抑制活性的主要因素,当linker的长度(n)为6个碳时,目标化合物的活性最优.此外,在抑制VEGFR-2活性方面,与Vandetanib相比,所有目标化合物均表现出中度到显著的VEGFR-2抑制活性,当苯环的2,4位引入氯原子时,目标化合物9抑制活性最强,IC50值为84 nM,同时对HDAC也表现出突出的抑制活性,IC50值为2.8 nM.体外抗肿瘤活性实验表明,9对肿瘤细胞株MCF-7表现出显著的抑制活性,其IC50值为1.2 μM,优于HDAC抑制剂SAHA(4.5 μM)和Vandetanib(18.5 μM).图4 凡德他尼类VEGFR-HDAC多靶点抑制剂帕唑帕尼 (Pazopanib, 10)作为一种新型的VEGFR抑制剂,于2009年被FDA批准用于治疗肾细胞癌,Pazopanib对VEGFR 1, 2和3的抑制活性分别为10、30和47 nM,可以有效的抑制肿瘤细胞增殖和肿瘤血管生成.Pazopanib和多种HDAC抑制剂的联合疗法已经通过实验验证,并显示出令人欣喜的试验结果[19].因此,设计Pazopanib和HDAC抑制剂的多靶点抑制剂将会是一种非常有效的抗癌方法.与此同时,实体瘤生长和转移所需的关键生理特性是血管的生成,而Pazopanib恰好能有效抑制肿瘤血管的生成.Zhang等人为了克服HDAC抑制剂在实体瘤中治疗效果不佳的缺点,以Pazopanib的结构为基础,将HDAC抑制剂MS-275的ZBG基团(邻苯二胺)引入到Pazopanib中,替换溶剂区的磺酰胺基团,设计合成得到一系列新型的VEGFR-HDAC多靶点抑制剂[20],如图5.研究表明,以邻苯二胺为ZBG的化合物12对HDAC和VEGFR-1, 2, 3的抑制活性最佳,IC50值分别为4.6 μM、37 nM、22 nM和46 nM.同时,对HDAC1, 2和3也表现出显著的抑制活性,IC50值分别为59 nM、91 nM和43 nM.与作者的预期一样,12不仅对血液瘤表现出明显的抑制作用,而且对实体肿瘤细胞株也显示出较强的抗增殖活性,其中对HT29的活性最强,IC50值为1.07 μM.在体外HUVEC血管形成实验中,12在100 nM的浓度下抑制血管形成的效果与Pazopanib相当.此外,在体内,裸鼠HT-29肿瘤异种移植模型中,12在剂量为每天50 mg/kg时能有效地抑制肿瘤生长.图5 帕唑帕尼类VEGFR-HDAC多靶点抑制剂1.3 c-Met-HDAC多靶点抑制剂Xing等人报道了一系列高选择性的c-Met激酶抑制剂,其中化合物13活性最强,IC50值为4.2 nM.构效关系研究表明,喹啉部分的C-7处取代基延伸到c-Met激酶的溶剂区,与c-Met激酶无结合作用,对抑制活性没有明显的影响[21].因此,基于HDAC多靶点抑制剂的设计思想,Lu等人以选择性c-Met抑制剂(13)作为结构骨架,在13的溶剂区引入HDAC抑制剂的ZBG基团(异羟肟酸和邻苯二胺),设计得到了一系列c-Met-HDAC多靶点抑制剂[22],如图6.研究表明,其中化合物14活性最强,抑制c-Met激酶和HDAC1的IC50值分别为0.71 nM和38 nM.同时,对肿瘤细胞系EBC-1和HCT-116也表现出了良好的抗肿瘤活性,IC50值分别为0.058 μM和1.3 μM,优于HDAC抑制剂Chidamide(2.9 μM和7.8 μM)以及c-Met抑制剂13(0.06 μM和>10 μM).图6 c-Met-HDAC混合抑制剂1.4 Abl-HDAC多靶点抑制剂伊马替尼 (imatinib, 15)作为一种Abl抑制剂,IC50为0.6 μM,临床用于治疗慢性髓性白血病和恶性胃肠道间质肿瘤,同时对PDGFR和Kit也表现出突出的抑制作用,IC50均为0.1 μM[23].文献已报道[24],imatinib与HDAC抑制剂联合使用后,显示出协同效应.因此,Mahboobi等人在保留imatinib药效团的前提下,将HDAC抑制剂的ZBG基团(邻苯二胺)引入到imatinib结构中,设计合成得到一系列Abl-HDAC多靶点抑制剂[25].研究表明,大多数目标化合物对HDAC的抑制活性基本保持不变,与SAHA相当,同时对Abl激酶也表现出显著的抑制活性.其中化合物16对HDAC1(IC50=0.208 μM)的选择性高于HDAC6(IC50≥32 μM),同时对Abl激酶的抑制活性也最强,IC50为2 μM,与imatinib(IC50=1 μM)的抑制活性相当.进一步研究表明,16还可以抑制AblT315l突变型,能够克服imatinib带来的耐药性,IC50值为1.1 μM.图7 伊马替尼类的Abl-HDAC多靶点抑制剂1.5 PI3K-HDAC多靶点抑制剂Apitolisib (GDC-0980, RG7422, 17)是一种有效的I型PI3K抑制剂,作用于PI3K-α, β, δ和γ,IC50分别为5、27、7和14 nM,也是mTOR抑制剂,Ki值为17 nM,比作用于其他PI3K家族激酶的选择性要高,目前处于临床二期研究阶段[26].除此之外,还有多种PI3K抑制剂已经被报道,如Pictilisib、PI-103和BKM120等.目前,单独对PI3K途径进行抑制总是存在问题,因为肿瘤其他生存和生长相关途径会被同时激活.实验证据表明,HDAC抑制剂与多个肿瘤生存途径关,HDAC抑制剂SAHA(1)和PI3K抑制剂的联合用药可以对肿瘤细胞的生长产生协同的抑制作用[27-29],这为开发PI3K-HDAC多靶点抑制剂提供了依据.PI3K抑制剂的构效关系研究表明,结构中的吗啉基团对PI3K的抑制活性至关重要,因为吗啉基团可以与PI3K激酶ATP结合域的铰链区形成重要的氢键作用[30].因此,Qian等人保留了PI3K抑制剂的吗啉-嘧啶并噻吩骨架,引入了HDAC抑制剂的ZBG基团(异羟肟酸),设计得到一系列PI3K-HDAC多靶点抑制剂,如图8.活性研究表明[31],其中化合物18(GUDC-907)活性最强,对PI3Kα、β和δ表现出突出的抑制活性,IC50分别为19、54和39 nM.同时,18对I 和 II 类HDAC也表现出广泛且突出的抑制作用,对HDAC1、2、3、10和11的IC50值分别为1.7、5、1.8、2.8和5.4 nM,优于SAHA (1) [32].进一步研究表明,18可以抑制PI3K-AKT-mTOR通路的激活和补偿性信号分子,如RAF、MEK、MAPK和STAT-3等.在体外,18通过激活caspase-3和7的方式诱导HCT-116肿瘤细胞凋亡并阻滞细胞周期G2-M期.在体内,18在口服剂量为每天100mg/kg时观察到了肿瘤生长停滞,且无明显毒性.值得注意的是,18肿瘤治疗效果比SAHA(1)和Pictilisib单独使用或SAHA(1)和Pictilisib联合使用的效果要好.图8 PI3K-HDAC多靶点抑制剂从18的设计中得到启发,考虑到在大部分PI3Ks抑制剂中[33, 34],嘌呤是核心骨架,吗啉基团是活性必需基团,Chen等人采用生物电子等排原理,用吗啉-嘌呤骨架替代了18中的吗啉-嘧啶并噻吩骨架,设计合成得到一类新型PI3K-HDAC 多靶点抑制剂[35].研究结果表明,大部分目标化合物对HDAC1都表现出明显的抑制活性,同时吗啉-嘌呤基团的C-2位的取代基对抑制活性具有显著影响,当C-2位为2-氨基嘧啶基时,化合物19对PI3Kα和HDAC1均表现出突出的抑制活性,IC50值分别为1.33和1.04 nM,优于阳性对照药18 (19和1.7 nM).同时,在体内,19在剂量为10 mg/kg的MV4-11异种移植小鼠模型中也表现出良好的体内抗肿瘤活性,抑瘤率为45.1%.而阳性对照药SAHA(1)在50 mg/kg剂量下没有明显的抑制活性.1.6 JAK-HDAC多靶点抑制剂Pacritinib (SB1518, 20)是一种有效的选择性Janus Kinase 2 (JAK2)抑制剂,IC50为23 nM,临床用于骨髓纤维化和急性骨髓性白血病[36].构效关系研究表明,Pacritinib侧链上的直链状吡咯烷位于JAK2激酶的溶剂区,与JAK2激酶结合口袋没有任何相互作用,不是活性必需基团,而许多潜在的HDAC抑制剂都含有大环结构,例如Romidepsin (2).因此,受到多靶点HDAC抑制剂设计的启发,Dymock等人将JAK抑制剂pacritinib(20)的大环结构作为设计HDAC抑制剂的理想帽子结构,结构中的吡咯烷替换为HDAC抑制剂SAHA (1)的ZBG基团,设计合成得到一系列JAK-HDAC多靶点抑制剂[37],如图9.研究结果表明,所有合成的目标化合物对JAK2均表现出明显的抑制作用,化合物21活性最强, IC50值达到了1.4 nM.同时,对HADC6也表现出较高的选择性和抑制活性,IC50值为2.1 nM.在继续的研究中,Dymock等人基于HDAC抑制剂SAHA (1)和JAK1/2抑制剂ruxolitinib(22)的药效团,保留了能介导ruxolitinib和JAK1/2激酶铰链区之间关键氢键供体-受体相互作用的吡咯-嘧啶结构骨架,将具有不同长度的柔性烷基链的ZBG基团引入到ruxolitinib的吡唑结构中,替换结构中取代的丁腈基团,设计并合成得到另一类新型JAK-HDAC多靶点抑制剂[38].研究表明,含有六个碳长度的ZBG的目标化合物对HDAC和JAK2的抑制活性最佳.其中,化合物23活性最强,对HDAC1、HDAC6和JAK2的IC50值分别为6.9、1.4和75 nM,而ruxolitinib对JAK2的IC50值为56 pM,表明长链的ZBG基团的引入以某种方式阻碍了化合物23和JAK2激酶之间的相互作用.此外,化合物23还可以有效的抑制HDAC2、HDAC3和HDAC10,IC50值分别为5.8、3.9和19 nM.体外抗肿瘤实验表明,23对MDA-MB231、MCF7、HL-60和Jurkat等不同的实体瘤和血液瘤细胞系均表现出显著的抗肿瘤活性,IC50值分别为0.79、0.84、7.36、0.47 μM.图9 JAK-HDAC多靶点抑制剂1.7 P53/MDM2-HDAC多靶点抑制剂Nutlin-3a (24)是Nutlin-3的活性对映体,是一种有效的选择性Mdm2拮抗剂,IC50为90 nM[39].分子对接表明,Nutlin-3a结构中的两个氯苯基和异丙基可以分别进入到Mdm2蛋白中的Leu26、Trp23和Phe 19结合口袋中,形成稳定的疏水作用,而酰基哌嗪酮结构则伸向蛋白的溶剂区,与Mdm2蛋白无相互作用.基于HDAC多靶点抑制剂的设计思想,Sheng等人在酰基哌嗪酮的末端引入HDAC 抑制剂的ZBG基团,设计合成得到一系列p53/Mdm2-HDAC多靶点抑制剂[40],如图10.研究表明,所得到的目标化合物对Mdm2和HDAC1均表现出显著的抑制活性,其中化合物25活性最强,对Mdm2和HDAC1的抑制活性分别为Ki= 0.11 μM和IC50= 0.82 μM,同时,25对HDAC2、3、6和8也表现出突出的抑制活性,IC50值分别为0.42、0.178、0.017和1.224 μM.体外抗肿瘤活性研究表明,25对四株实体瘤细胞系(A549、HCT226、MCF7和NCI-H1299)的抑制活性(IC50)分别为0.91、1.08、4.34和4.16 μM.尤其是在体内,25表现出了显著的体内抗肿瘤活性,抑瘤率为75%,优于HDAC抑制剂SAHA(1)和P53/Mdm2抑制剂Nutlin-3.图10 Mdm2-HDAC多靶点抑制剂2 总结与展望HDAC家族是表观遗传学领域中最重要的促肿瘤酶之一,主要负责组蛋白和非组蛋白底物中乙酰赖氨酸残基(KAc)的去乙酰化,目前已成为开发新型抗肿瘤药物的重要靶点,其中一些HDAC抑制剂已在临床上使用.尽管如此,传统的HDAC抑制剂通常是非选择性的,同时通常伴随着不必要的副作用.此外,HDAC抑制剂对实体瘤的治疗效率有限,严重限制了它们在癌症治疗中的应用.为了克服这些缺陷,构建HDAC多靶点抑制剂已成为克服耐药性,增加敏感性,拓宽临床应用范围的有效途径.到目前为止,已经进入临床试验的HDAC多靶点抑制剂的数量仍然有限,两个HDAC混合抑制剂CUDC-101 (7)和CUDC-907(18)正在临床试验中,这不仅为该设计策略的有效性提供了有力的证据,同时也为开发HDAC多靶点抑制剂奠定了坚实的基础.参考文献【相关文献】[1] Fu R G, Sun Y, Sheng W B, et al. Designing multitargeted agents: An emerging anticancer drug discovery paradigm[J]. Eur J Med Chem, 2017, 136:195-211.[2] Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm[J]. Cheminform, 2006, 37(6): 6523-6543.[3] Brub G. An overview of molecular hybrids in drug discovery[J]. Expert Opin Drug Dis,2016, 11(3): 281-305.[4] Fraga M F, Esteban B, Ana V G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer[J]. Nat Genet, 2005, 37(4): 391-400.[5] Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer[J]. Mol Oncol, 2008, 1(1): 19-25.[6] Zhang L, Han Y T, Jiang Q X, et al. Trend of histone deacetylase inhibitors in cancer therapy: isoform selectivity or multitargeted strategy[J]. Med Res Rev, 2015, 35(1): 63-84.[7] Marielle P, Marina P, Monica B, et al. Histone deacetylase inhibitors: from bench to clinic[J]. J Med Chem, 2008, 51(6): 1505-1529.[8] Stenzel K, H A, Hansen F K, et al. Alkoxyurea-based histone deacetylase inhibitors increase cisplatin potency in chemoresistant cancer cell lines[J]. J Med Chem, 2017, 60(13): 5334-5348.[9] Marson C M. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer[J]. Anti-Cancer Agents Me, 2009, 9(6): 661-692. [10] Zeng H, Qu J, Jin N, et al. Feedback activation of leukemia inhibitory factor receptor limits response to histone deacetylase inhibitors in breast cancer[J]. Cancer Cell, 2016,30(3): 459-473.[11] Mahboobi S, Pilsl B, Sellmer A. Generation and assessment of fusions between hdaci and tki[J]. Methods Mol Biol, 2017, 1510: 405-412.[12] Stamos J, Sliwkowski M X, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor[J]. J Biol Chem, 2002, 277(48): 46265-46272.[13] Cai X, Zhai H X, Wang J, et al. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer[J]. J Med Chem, 2010, 53(5): 2000-2009.[14] Lai C J, Bai R D, Tao X, et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity[J]. Cancer Res, 2010, 70: 3647-3656.[15] Wang J, Pursell N W, Samson M E, et al. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion[J]. Mol Cancer Ther, 2013, 12(6): 925-936.[16] Shimizu T, Lorusso P M, Papadopoulos K P, et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors[J]. Clin Cancer Res, 2014, 20(19): 5032-5040.[17] Wedge S R, Ogilvie D J, Dukes M, et al. ZD6474 Inhibits vascular endothelial growthfactor signaling, angiogenesis, and tumor growth following oral administration[J]. Cancer Res, 2002, 62(16): 4645-4655.[18] Peng F W, Wu T T, Ren Z W, et al. Hybrids from 4-anilinoquinazoline and hydroxamic acid as dual inhibitors of vascular endothelial growth factor receptor-2 and histone deacetylase[J]. Bioorg Med Chem Lett, 2015, 25(22): 5137-5141.[19] Aggarwal R, Thomas S, Pawlowska N, et al. Inhibiting histone deacetylase as a means to reverse resistance to angiogenesis inhibitors: phase i study of abexinostat plus pazopanib in advanced solid tumor malignancies[J]. J Clin Oncol, 2017, 35(11): 1231-1239.[20] Zang J, Liang X, Huang Y, et al. Discovery of novel pazopanib-based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously[J]. J Med Chem, 2018, 61(12): 5304-5322.[21] Xing W, Ai J, Jin S, et al. Enhancing the cellular anti-proliferation activity of pyridazinones as c-met inhibitors using docking analysis[J]. European Journal of Medicinal Chemistry, 2015, 95: 302-312.[22] Lu D, Yan J, Wang L, et al. Design, synthesis, and biological evaluation of the first c-Met/HDAC inhibitors based on pyridazinone derivatives[J]. ACS Medicinal Chemistry Letters, 2017, 8(8): 830-834.[23] Buchdunger E, Zimmermann J, Mett H, et al. Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class[J]. Proc Natl Acad Sci U S A, 1995, 92(7): 2558-2562.[24] Nimmanapalli R, Fuino L, Stobaugh C, et al. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells[J]. Blood, 2003, 101(8): 3236-3239.[25] Mahboobi S, Dove S, Sellmer A, et al. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: a series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases[J]. J Med Chem, 2009, 52(8): 2265-2279.[26] Sutherlin D P, Bao L, Berry M, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-Kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer[J]. J Med Chem, 2011,54(21): 7579-7587.[27] Takashi Y, Shingo Y, Takeshi Y, et al. Combination of a novel HDAC inhibitor OBP-801/YM753 and a PI3K inhibitor LY294002 synergistically induces apoptosis in human endometrial carcinoma cells due to increase of Bim with accumulation of ROS[J]. Gynecol Oncol, 2013, 129(2): 425-432.[28] Leigh E, Yu K S, Swathi R, et al. Combinatorial antitumor effect of HDAC and the PI3K-Akt-mTOR pathway inhibition in a Pten defecient model of prostate cancer[J]. Oncotarget,2013, 4(12): 2225-2236.[29] Takeshi Y, Mano H, Masahide S, et al. A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma[J]. Int J Oncol, 2013, 43(4): 1080-1086.[30] Martin A, Jan K, Daniel J, et al. Phosphatidylinositol 3-Kinase (PI3K) and phosphatidylinositol 3-kinase-related kinase (PIKK) inhibitors: importance of the morpholine ring[J]. J Med Chem, 2015, 58(1): 41-71.[31] Qian C, Lai C J, Bao R, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling[J]. Clin Cancer Res, 2012, 18(15): 4104-4113.[32] Mondello P, Derenzini E, Asgari Z, et al. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma[J]. Oncotarget, 2017, 8(8): 14017-14028.[33] Kolev V N, Wright Q G, Vidal C M, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells[J]. Cancer Res, 2015, 75(2): 446-455.[34] Subramaniam P, Whye D, Efimenko E, et al. Targeting nonclassical oncogenes for therapy in T-ALL[J]. CANCER Cell, 2012, 21(4): 459-472.[35] Chen Y, Wang X, Xiang W, et al. Development of purine-based hydroxamic acid derivatives: potent histone deacetylase inhibitors with marked in vitro and in vivo antitumor activities[J]. J Med Chem, 2016, 59(11): 5488-5504.[36] Hart S, Goh K C, Novotny-Diermayr V, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies[J]. Leukemia, 2011, 25: 1751-1759.[37] Yang E G, Mustafa N, Tan E C, et al. Design and synthesis of janus kinase 2 (JAK2) and histone deacetlyase (HDAC) bispecific inhibitors based on pacritinib and evidence of dual pathway inhibition in hematological cell lines[J]. J Med Chem, 2016, 59(18): 8233-8262. [38] Yao L, Mustafa N, Tan E C, et al. Design and synthesis of ligand efficient dual inhibitors of janus kinase (JAK) and histone deacetylase (HDAC) based on ruxolitinib and vorinostat[J]. J Med Chem, 2017, 60(20): 8336-8357.[39] Vassilev L T, Vu B T, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2[J]. Science, 2004, 303(5659): 844-848.[40] He S, Dong G, Wu S, et al. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): discovery of novel multitargeting antitumor agents[J]. J Med Chem, 2018, 61(16): 7245-7260.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
参考文献: [1] Wallin JJ1, Edgar KA, Guan J, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, Salphati L, Olivero AG, Sutherlin DP, O'Brien C, Spoerke JM, Patel S, Lensun L, Kassees R, Ross L, Lackner MR, Sampath D, Belvin M, Friedman LS. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011 Dec;10(12):2426-36. doi: 10.1158/1535-7163.MCT-11-0446. Epub 2011 Oct 13.
产品说明书
化学性质
产品名: Ca22) 1032754-93-0 498.61 C23H30N8O3S
产品名: GDC-0980 (RG7422) 修订日期: 6/30/2016
化学名:
SMILES: 溶解性: 储存条件: 一般建议:
运输条件:
生物活性
(2S)-1-[4-[[2-(2-aminopyrimidin-5-yl)-7-methyl-4-morpholin-4-ylthie no[3,2-d]pyrimidin-6-yl]methyl]piperazin-1-yl]-2-hydroxypropan-1-o ne
CC1=C(SC2=C1N=C(N=C2N3CCOCC3)C4=CN=C(N=C4)N)CN5CCN(CC5 )C(=O)C(C)O
GDC-0980(RG7422)是一种选择性的、新型的、有效的 1 类 PI3K / mTOR 激酶的口服抑制剂,
作用于 mTOR 激酶的 Ki 值为 17 nmol/ L [1]。 在前列腺癌、乳腺癌和 NSCLC 细胞系中,GDC-0980(RG7422)已经显示出有效的抑制作用, IC50 值小于 200 nmol/ L。此外,研究表明,在 KPL4 细胞中,GDC-0980(RG7422)通过抑制 细胞周期和诱导细胞凋亡来降低细胞活力,抑制细胞周期的 IC50 值为 109 nM,诱导凋亡的 EC50 值为 78 nM。口服 GDC-0980(RG7422)显著抑制异种移植模型的肿瘤应答。值得注意 的是,在体实验研究表明,GDC-0980(RG7422)和抗肿瘤剂 docetaxel 共同使用可以加强抗 肿瘤活性[1]。
Evaluation sample solution : ship with blue ice All other available size: ship with RT , or blue ice upon request
靶点 :
PI3K/Akt/mTOR Signaling
信号通路:
mTOR
产品描述:
>21.35mg/mL in DMSO
Store at -20°C
For obtaining a higher solubility , please warm the tube at 37°C and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20°C for several months.
特别声明
产品仅用于研究,
不针对患者销售,望谅解。
每个产品具体的储存和使用信息显示在产品说明书中。ApexBio 产品在推荐的条件下是稳定 的。产品会根据不同的推荐温度进行运输。许多产品短期运输是稳定的,运输温度不同于长 期储存的温度。我们确保我们的产品是在保持试剂质量的条件下运输的。收到产品后,按照 产品说明书上的要求进行储存。