《中国药典》2020—注射用重组人粒细胞巨噬细胞刺激因子原液相关蛋白检测国家药品标准公示稿
《中国药典》2020年版(三部)生物技术产品增修订概况

中国药品标准DrugStandardsofChina2021,22(1) ·5 · 第一作者简介:赵雄,副研究员,研究方向:生物制品研发及质量控制。
Tel:010 67079598;E mail:zhaoxiong@chp org cn 通讯作者简介:郭中平,研究员,研究方向:生物制品。
Tel:010 67079561;E mail:guozhongping@chp org cn《中国药典》2020年版(三部)生物技术产品增修订概况赵雄,王晓娟,曹琰,郭中平(国家药典委员会,北京100061)摘要:生物技术产业的快速发展推动了生物技术产品国家标准的不断提升。
本文介绍了《中国药典》2020年版(三部)生物技术产品及相关通用技术要求的增修订概况,阐释了增修订的考量和特点,旨在为正确理解和执行生物技术产品国家标准提供参考。
关键词:生物技术产品;国家标准;增修订中图分类号:R921 2 文献标识码:A 文章编号:1009-3656(2021)01-0005-05doi:10 19778/j chp 2021 01 001UpdatesandAmendmentsofBiotechnologyProductsintheChinesePharmacopoeia2020(VolumeⅢ)ZHAOXiong,WANGXiaojuan,CAOYan,GUOZhongping(ChinesePharmacopoeiaCommission,Beijing100061,China)Abstract:Therapiddevelopmentofthebiotechnologyindustryhaspromotedthecontinuousimprovementofnation alstandardsforbiotechnologyproducts TheupdatesandamendmentsofbiotechnologyproductsandgeneralrequirementsintheChinesePharmacopoeia2020(VolumeⅢ)areintroduced,andtheconsiderationsandcharac teristicsoftherevisionsareexplained Thataimstoprovideareferencefortheaccurateunderstandingandimple mentationofnationalstandardsforbiotechnologyproducts.Keywords:biotechnologyproducts;nationalstandards;updatesandamendments 生物技术产品是指采用生物技术制备的、临床上用于疾病治疗的大分子生物制品[1]。
重组人粒细胞刺激因子在《中国药典》和《欧洲药典》中质量标准不同点的对比分析

重组人粒细胞刺激因子在《中国药典》和《欧洲药典》中质量标准不同点的对比分析赵先亮;王春雨【摘要】目的对比重组人粒细胞刺激因子(rhG-CSF,Filgrastim)在<中国药典>(2005年版)和<欧洲药典>(6.3版)中标准的不同点,为国内研发机构对基因重组蛋白产品的研发及国内企业对该产品的进出口提供参考.方法按两部药典要求,对国产产品实样进行部分关键指标的检测分析.结果两部药典对该产品的标准描述上存在具体内容的区别.结论 :在现代药物的研发和生产中,对药品的国际及国内标准要进行综合分析而拟定.【期刊名称】《药学研究》【年(卷),期】2010(029)001【总页数】3页(P25-27)【关键词】重组人粒细胞刺激因子;重组人粒细胞刺激因子;欧洲药典;中国药典【作者】赵先亮;王春雨【作者单位】山东科兴生物制品有限公司,山东济南250200;山东科兴生物制品有限公司,山东济南250200【正文语种】中文【中图分类】R921生物技术药品的质量标准和检测方法的研究水平不仅影响着生物技术新药开发的进程,而且决定着生物技术药品产业的质量水平,是药品进入国际市场的一道大关。
从广义上讲,基因重组蛋白药物是生物技术药物的一种。
随着生物技术的飞速发展,尤其是国际市场部分基因重组蛋白药物专利逐个到期,其流通市场的数倍放大,如何制定一个既遵守国内标准又符合国际市场需求的基因重组蛋白药物质量标准,值得大家探讨。
我们以《欧洲药典》(第6.3版)(European Pharmacopoeia 6.3)[1]和《中国药典》2005年版(三部)[2]中共有的重组人粒细胞刺激因子(Recombinant Human Granulocyte Colony-stimulating Factor,rhG-CSF,Filgrastim)为例,对该产品的部分质量标准不同点进行了对比分析,提出了自己的观点,以便对国内企业该产品或类似产品的研发、进出口有所裨益。
《中国药典》2020版—重组人胰岛素注射液国家药品标准公示稿

重组人胰岛素注射液Chongzu Ren Yidaosu ZhusheyeRecombinant Human Insulin Injection本品系将重组人胰岛素原料与适量的抑菌剂、渗透压调节剂等配制而成的无菌水溶液。
1基本要求生产和检定用设施、原辅料、水、器具等应符合“凡例”的相关要求。
2制造2.1原料应符合“重组人胰岛素项下”的规定。
2.2半成品2.2.1配制与除菌按照经批准的配方进行稀释、配制,除菌过滤后即为半成品,保存于适宜的温度。
2.2.2半成品检定按3.1 项进行。
2.3成品2.3.1分批应符合“生物制品分批规程”规定。
2.3.2分装应符合“生物制品分装和冻干规程”及通则 0102 有关规定。
2.3.3规格3ml:300 单位10ml:400 单位2.3.4包装应符合“生物制品包装规程”及通则 0102 有关规定。
3检定3.1半成品检定如需对原液进行稀释或加入其他辅料配制半成品,应确定半成品的质量控制要求,除另有规定外,应按 3.1.1 和 3.1.2 项进行。
3.1.1无菌按薄膜过滤法处理,依法检查(通则 1101),应符合规定。
3.1.2细菌内毒素依法检查(通则 1143),每 100 单位重组人胰岛素中含细菌内毒素的量应小于 80 EU。
3.2成品检定3.2.1性状本品为无色澄明液体。
3.2.2鉴别3.2.2.1取本品,照重组人胰岛素项下的鉴别(1)项试验,显相同的结果。
3.2.2.2在苯酚或间甲酚检查项下记录的色谱图中,供试品溶液中苯酚峰或间甲酚峰的保留时间应与对照溶液中苯酚峰或间甲酚峰的保留时间一致。
3.2.3检查3.2.3.1 pH 值应为 6.9~7.8(通则 0631)。
3.2.3.2有关物质取本品,每 1mL 中加9.6mol/L 盐酸溶液3µl ,作为供试品溶液;取供试品溶液适量(约相当于人胰岛素70µg),照重组人胰岛素项下的色谱条件试验,除去苯酚峰或间甲酚峰,按峰面积归一化法计算,A21 脱氨人胰岛素不得过2.0%,其它有关物质总量不得过 6.0%。
重组人粒细胞-巨噬细胞集落刺激因子的临床新用途解析

重组人粒细胞-巨噬细胞集落刺激因子的临床新用途摘要粒细胞-巨噬细胞集落刺激因子(granulocyte macrophage colony stimulating factor,GM-CSF)是一个具有多项潜能的造血生长因子,因为它不仅能够促进造血前体细胞的增殖、分化和成熟,而且对其他的细胞,如抗原递呈细胞(APC)、成纤维细胞、角质细胞、皮肤粘膜细胞等,均有着不同程度的刺激作用。
由于GM-CSF对骨髓抑制的修复作用,目前在临床上的用途主要是用于治疗恶性肿瘤因放、化疗所致的白细胞减少症,有关这方面的报道已有很多,GM-CSF在升高白细胞方面的作用已经比较肯定。
近年来有许多研究报道表明,GM-CSF对恶性肿瘤放、化疗后所致的口腔粘膜溃疡有着很好的治疗作用,以及在抗病毒、抗真菌感染等的治疗中也有着良好的疗效。
关键词重组人粒细胞- 巨噬细胞集落刺激因子;临床新用途《中国书资料分类法》分类号R730.5; R456自1985年Sieff[1]等发表第1篇有关重组人粒细胞-巨噬细胞集落刺激因子(recombinant human granulocyte macrophage colony stimulatingfactor,rhGM-CSF)的文章以来,rhGM-CSF的临床用途逐渐引起了各位学者的兴趣,这些年来有关rhGM-CSF研究报道的文献每年均在千篇左右。
作为一个多潜能的造血生长因子,它的主要临床应用是促进骨髓造血,目前在临床上rhGM-CSF已成功地用于治疗恶性肿瘤放、化疗后所致的白细胞减少症,以及用于骨髓移植、再生障碍性贫血和某些存在白细胞低下的免疫缺陷性疾病的治疗等。
近些年的研究发现,GM-CSF不仅能够作用于造血干、祖细胞促进骨髓造血,而且还作用于目前已知体内功能最强的抗原提呈细胞——树突状细胞,以促进免疫应答,调节免疫反应[2,3]。
另外,GM-CSF还作用于角质细胞、成纤维细胞、粘膜细胞等。
注射用重组人粒细胞巨噬细胞刺激因子

注射用重组人粒细胞巨噬细胞刺激因子【药品名称】通用名称:注射用重组人粒细胞巨噬细胞刺激因子英文名称:Propranolol Hydrochloride T ablets【成份】本品主要组成成分:活性成分为重组人粒细胞巨噬细胞集落刺激因子(rhGM-CSF)。
非活性成分包括甘露醇,磷酸钠,柠檬酸,聚乙二醇4000和人血白蛋白。
【适应症】适用于治疗和预防骨髓抑制性治疗引起的白细胞减少症。
可预防白细胞减少症伴发的潜在的感染的发生,使患者能更好地耐受化学药物的治疗。
【用法用量】剂量视具体病情而定,应调节剂量使白细胞维持在正常水平。
癌症化疗或放疗后24小时,按3-10μg/kg,皮下注射,每日一次,持续5-10天(注意:本药不应与抗癌药物同时使用,化疗停止24小时后方可使用)。
用1毫升注射用水溶解(切勿剧烈振荡),可在腹部、大腿外侧或上臂三角肌皮肤处进行皮下注射(要求:注射后局部皮肤隆起约1cm2以便药物缓慢吸收)。
【不良反应】大部分不良反应多属轻度中度。
严重的或危及生命的反应罕见,并且一般是当剂量大于推荐剂量范围时才发生。
最常见的不良反应为发热、皮疹,发热可用扑热息痛控制,胸痛、骨痛和腹泻等不良反应较少见。
据国外报道,低血压和低氧综合症在首次给药时可能出现,但以后给药则无此现象,本品在临床研究中,尚未发现,但在使用本品过程中建议医生参考。
另外,据国外研究表明,对肺癌病人使用本药治疗时应特别仔细加以观察。
【禁忌】本药禁用于对rhGM-CSF或该制剂中其它成份有过敏史的病人,同样禁用于自身免疫性血小板减少性紫癜的病人。
【注意事项】1 本药应在专科医生指导下使用,剂量视病人情况而定,病人对rhGM-CSF的治疗反应和耐受性个体差异较大,应调节剂量使病人白细胞数量维持在所期望的水平。
病人在治疗前及开始治疗后定期观察外周血白细胞或中性粒细胞、血小板数据的变化。
血象恢复正常后立即停药或采用维持剂量。
2 接受本药的病人偶而会产生急性过敏反应,应立即停药,并给予适当治疗。
2020年版《中国药典》目录四部目录

附件42020年版《中国药典》目录四部目录通用技术要求目次通则制剂通则1 片剂2 注射剂3 胶囊剂4 颗粒剂5 眼用制剂6 鼻用制剂7 栓剂8 丸剂9 软膏剂乳膏剂10 糊剂11 吸入制剂12 喷雾剂13 气雾剂14 凝胶剂15 散剂16 糖浆剂17 搽剂18 涂剂19 涂膜剂20 酊剂21 贴剂22 贴膏剂23口服溶液剂口服混悬剂口服乳剂24 植入剂25 膜剂26 耳用制剂27 洗剂28 冲洗剂29 灌肠剂30 合剂31 锭剂32 煎膏剂(膏滋)33 胶剂34 酒剂35 膏药36 露剂37 茶剂—1 —38 流浸膏剂与浸膏剂其他通则39 药材和饮片取样法40 药材和饮片检定通则41 炮制通则42 药用辅料43 制药用水44 国家药品标准物质通则45 一般鉴别试验光谱法46 紫外-可见分光光度法47 红外分光光度法48 荧光分光光度法49 原子吸收分光光度法50 火焰光度法51 电感耦合等离子体原子发射光谱法52 电感耦合等离子体质谱法53 拉曼光谱法54 质谱法55 核磁共振波谱法56 X射线衍射法57 X射线荧光光谱法色谱法58 纸色谱法59 薄层色谱法60 柱色谱法61 高效液相色谱法62 离子色谱法63 分子排阻色谱法64 气相色谱法65 超临界流体色谱法66 临界点色谱法67 电泳法68 毛细管电泳法物理常数测定法69 相对密度测定法70 馏程测定法71 熔点测定法72 凝点测定法73 旋光度测定法74 折光率测定法75 pH值测定法76 渗透压摩尔浓度测定法77 黏度测定法78 热分析法79 制药用水电导率测定法80 制药用水中总有机碳测定法其他测定法81 电位滴定法与永停滴定法82 非水溶液滴定法83 氧瓶燃烧法84 氮测定法85 乙醇量测定法86甲氧基、乙氧基与羟丙氧基测定法87 脂肪与脂肪油测定法88 维生素A测定法89 维生素D测定法90 蛋白质含量测定法限量测定法91 氯化物检查法92 硫酸盐检查法93 硫化物检查法94 硒检查法95 氟检查法96 氰化物检查法97 铁盐检查法98 铵盐检查法99 重金属检查法100 砷盐检查法101 干燥失重测定法102 水分测定法103 炽灼残渣检查法104 易炭化物检查法105 残留溶剂测定法106 甲醇量检查法107 合成多肽中的醋酸测定法108 2-乙基已酸测定法特性检查法109 溶液颜色检查法110 澄清度检查法111 不溶性微粒检查法112 可见异物检查法113 崩解时限检查法114 融变时限检查法115 片剂脆碎度检查法116 溶出度与释放度测定法117 含量均匀度检查法118 最低装量检查法119吸入制剂微细粒子空气动力学特性测定法120 黏附力测定法121 结晶性检查法122 粒度和粒度分布测定法123 锥入度测定法124 比表面积测定法125 固体密度测定法126 堆密度和振实密度测定法分子生物学检查法127 聚合酶链式反应法128 细菌DNA特征序列鉴定法生物检查法129 无菌检查法130非无菌产品微生物限度检查:微生物计数法131非无菌产品微生物限度检查:控制菌检查法132 非无菌药品微生物限度标准133 中药饮片微生物限度检查法134 抑菌效力检查法135 异常毒性检查法136 热原检查法137 细菌内毒素检查法138 升压物质检查法139 降压物质检查法140 组胺类物质检查法141 过敏反应检查法142 溶血与凝聚检查法—3 —生物活性测定法143 抗生素微生物检定法144 青霉素酶及其活力测定法145 升压素生物测定法146 细胞色素C活力测定法147 玻璃酸酶测定法148 肝素生物测定法149 绒促性素生物测定法150 缩宫素生物测定法151 胰岛素生物测定法152 精蛋白锌胰岛素注射液延缓作用测定法153 硫酸鱼精蛋白效价测定法154 洋地黄生物测定法155 葡萄糖酸锑钠毒力检查法156 卵泡刺激素生物测定法157 黄体生成素生物测定法158 降钙素生物测定法159 生长激素生物测定法160 放射性药品检定法161 灭菌法162 生物检定统计法中药其他方法163 显微鉴别法164 膨胀度测定法165 膏药软化点测定法166 浸出物测定法167 鞣质含量测定法168 桉油精含量测定法169 挥发油测定法170 杂质检查法171 灰分测定法172 酸败度测定法173 铅、镉、砷、汞、铜测定法174 汞、砷元素形态及价态测定法175 二氧化硫残留量测定法176 农药残留量测定法177 真菌毒素测定法178 注射剂有关物质检查法生物制品相关检查方法含量测定法179 固体总量测定法180 唾液酸测定法181 磷测定法182 硫酸铵测定法183 亚硫酸氢钠测定法184 氢氧化铝(或磷酸铝)测定法185 氯化钠测定法186 枸橼酸离子测定法187 钾离子测定法188 钠离子测定法189 辛酸钠测定法190 乙酰色氨酸测定法191 苯酚测定法192 间甲酚测定法193 硫柳汞测定法194对羟基苯甲酸甲酯、对羟基苯甲酸丙酯含量测定法195 O-乙酰基测定法196 已二酰肼含量测定法197 高分子结合物含量测定法198 人血液制品中糖及糖醇测定法199 人血白蛋白多聚体测定法200 人免疫球蛋白类制品lgG单体加二聚体测定法201 人免疫球蛋白中甘氨酸含量测定法202 人粒细胞刺激因子蛋白质含量测定法203 组胺人免疫球蛋白中游离磷酸组胺测定法204 lgG含量测定法205 单抗分子大小变异体测定法206 抗毒素/抗血清制品分子大小分布测定法207 单抗电荷变异体测定法208 单抗N糖谱测定法化学残留物测定法209 乙醇残留量测定法210 聚乙二醇残留量测定法211 聚山梨酯80残留量测定法212 戊二醛残留量测定法213 磷酸三丁酯残留量测定法214 碳二亚胺残留量测定法215 游离甲醛测定法216 人血白蛋白铝残留量测定法217 羟胺残留量测定法微生物检查法218 支原体检查法219 外源病毒因子检查法220 鼠源性病毒检查法221 SV40核酸序列检查法222 猴体神经毒力试验223血液制品生产用人血浆病毒核酸检测技术要求224 黄热减毒活疫苗猴体试验225禽源性病毒荧光定量PCR(Q-PCR)检查法生物测定法226 免疫印迹法227 免疫斑点法228 免疫双扩散法229 免疫电泳法230 肽图检查法231 质粒丢失率检查法232 外源性DNA残留量测定法233 抗生素残留量检查法234 激肽释放酶原激活剂测定法235 抗补体活性测定法236 牛血清白蛋白残留量测定法237大肠埃希菌菌体蛋白质残留量测定法238假单胞菌菌体蛋白质残留量测定法239酵母工程菌菌体蛋白质残留量测定法240 类A血型物质测定法241 鼠lgG残留量测定法242 无细胞百日咳疫苗鉴别试验243 抗毒素、抗血清制品鉴别试验244A群脑膜炎球菌多糖分子大小测定法245 伤寒Vi多糖分子大小测定法246b型流感嗜血杆菌结合疫苗多糖含量测定法247 人凝血酶活性检查法—5 —248 活化的凝血因子活性检查法249 肝素含量测定法250 抗A、抗B血凝素测定法251 人红细胞抗体测定法252 人血小板抗体测定法253 人免疫球蛋白类制品lgA残留量测定法254 免疫化学法生物活性/效价测定法255 重组乙型肝炎疫苗(酵母)体外相对效力检查法256 甲型肝炎灭活疫苗体外相对效力检查法257 人用狂犬病疫苗效价测定法258 吸附破伤风疫苗效价测定法259 吸附白喉疫苗效价测定法260 类毒素絮状单位测定法261 白喉抗毒素效价测定法262 破伤风抗毒素效价测定法263 气性坏疽抗毒素效价测定法264 肉素抗毒素效价测定法265 抗蛇毒血清效价测定法266 狂犬病免疫球蛋白效价测定法267 人免疫球蛋白中白喉抗体效价测定法268 人免疫球蛋白Fc段生物学活性测定法269 抗人T细胞免疫球蛋白效价测定法(E玫瑰花环形成抑制试验)270 抗人T细胞免疫球蛋白效价测定法(淋巴细胞毒试验)271 人凝血因子Ⅱ效价测定法272 人凝血因子Ⅶ效价测定法273 人凝血因子Ⅸ效价测定法274 人凝血因子Ⅹ效价测定法275 人凝血因子Ⅷ效价测定法276人促红素体内生物学活性测定法277 干扰素生物学活性测定法278 人白介素-2生物学活性测定法279人粒细胞刺激因子生物学活性测定法280人粒细胞巨噬细胞刺激因子生物学活性测定法281牛碱性成纤维细胞生长因子生物学活性测定法282人表皮生长因子生物学活性测定法283鼠神经生长因子生物学活性测定法284 尼妥珠单抗生物学活性测定法285 人白介素-11生物学活性测定法286 A型肉毒毒素效价测定法287Sabin株脊髓灰质炎灭活疫苗效力试验288 康柏西普生物学活性测定法特定生物原材料/动物及辅料289生物制品生产及检定用实验动物质量控制290 重组胰蛋白酶291 新生牛血清292 细菌生化反应培养基293 氢氧化铝佐剂294 生物制品国家标准物质目录药包材检测方法295 121℃玻璃颗粒耐水性测定法296 包装材料红外光谱测定法297 玻璃内应力测定法298 剥离强度测定法299 拉伸性能测定法300 内表面耐水性测定法301 气体透过量测定法302 热合强度测定法303 三氧化二硼测定法304 水蒸气透过量测定法305 药包材急性全身毒性检查法306 药包材密度测定法307 药包材溶血检查法308 药包材细胞毒性检查法309 注射剂用胶塞、垫片穿刺力测定法310 注射剂用胶塞、垫片穿刺落屑测定法试剂与标准物质311 试药312 试液313 试纸314 缓冲液315 指示剂与指示液316 滴定液317 对照品对照药材对照提取物318 标准品与对照品指导原则指导原则319 原料药物与制剂稳定性试验指导原则320 药物制剂人体生物利用度和生物等效性试验指导原则321生物样品定量分析方法验证指导原则322缓释、控释和迟释制剂指导原则323 微粒制剂指导原则324药品晶型研究及晶型质量控制指导原则325 分析方法确认指导原则326 分析方法转移指导原则327 分析方法验证指导原则328 药品杂质分析指导原则329 药物引湿性试验指导原则330 近红外分光光度法指导原则331 中药生物活性测定指导原则332基于基因芯片的药物评价技术与方法指导原则333中药材DNA条形码分子鉴定法指导原则334 DNA测序技术指导原则335 标准核酸序列建立指导原则336药品微生物检验替代方法验证指导原则337非无菌产品微生物限度检查指导原则338药品微生物实验室质量管理指导原则339 微生物鉴定指导原则340药品洁净实验室微生物监测和控制指导原则341无菌检查用隔离系统验证和应用指导原则342 灭菌用生物指示剂指导原则343生物指示剂耐受性检查法指导原则—7 —344 细菌内毒素检查法应用指导原则345 注射剂安全性检查法应用指导原则346 中药有害残留物限量制定指导原则347 色素测定法指导原则348 中药中铝、铬、铁、钡元素测定指导原则349 中药中真菌毒素测定指导原则350 遗传毒性杂质控制指导原则351 生物制品生物活性/效价测定方法验证指导原则352 生物制品稳定性试验指导原则353正电子类放射性药品质量控制指导原则354锝[99m Tc]放射性药品质量控制指导原则355药用辅料功能性相关指标指导原则356 动物来源药用辅料指导原则357预混与共处理药用辅料质量控制指导原则358 药包材通用要求指导原则359 药用玻璃材料和容器指导原则360国家药品标准物质制备指导原则药用辅料品名目次1 乙二胺2 乙基纤维素3 乙基纤维素水分散体4 乙基纤维素水分散体(B型)5 乙酸乙酯6 乙醇7 二丁基羟基甲苯8 二甲基亚砜9 二甲硅油10 二甲醚11 二氧化钛12 二氧化硅13 二氧化碳14 十二烷基硫酸钠15 十八醇16 十六十八醇17 十六醇18 丁香茎叶油19 丁香油20 丁香酚21 丁烷22 丁基羟基苯甲醚23 七氟丙烷(供外用气雾剂用)24 三乙醇胺25 三油酸山梨坦26 三硅酸镁27 三氯叔丁醇28 三氯蔗糖29 大豆油30 大豆油(供注射用)31 大豆磷脂32 大豆磷脂(供注射用)33 小麦淀粉34 山梨酸35 山梨酸钾36 山梨醇37 山梨醇山梨坦溶液38 山梨醇溶液39 山嵛酸甘油脂40 门冬氨酸41 门冬酰胺42 己二酸43 马来酸44 马铃薯淀粉45 无水乙醇46 无水亚硫酸钠47 无水乳糖48 无水枸橼酸49 无水脱氢醋酸钠50 无水碳酸钠51 无水磷酸二氢钠52 无水磷酸氢二钠53 无水磷酸氢钙54 木薯淀粉55 D-木糖56 木糖醇57 中链甘油三酸酯58 牛磺酸59 月桂山梨坦60 月桂氮䓬酮61 月桂酰聚氧乙烯(6)甘油酯62 月桂酰聚氧乙烯(8)甘油酯63 月桂酰聚氧乙烯(12)甘油酯64 月桂酰聚氧乙烯(32)甘油酯65 巴西棕榈蜡66 玉米朊67 玉米油68 玉米淀粉69 正丁醇70 甘油71 甘油(供注射用)72 甘油三乙酯73 甘油磷酸钙74 甘氨酸75 可可脂76 可压性蔗糖77 可溶性淀粉78 丙二醇79 丙二醇(供注射用)80丙交酯乙交酯共聚物(5050)(供注射用)81丙交酯乙交酯共聚物(7525)(供注射用)82丙交酯乙交酯共聚物(8515)(供注射用)83 丙氨酸84丙烯酸乙酯-甲基丙烯酸甲酯共聚物水分散体85 丙酸86 石蜡87 卡波姆共聚物88 卡波姆均聚物—9 —89 卡波姆间聚物90 甲基纤维素91 四氟乙烷(供外用气雾剂用)92 白凡士林93 白陶土94 白蜂蜡95 瓜尔胶96 对氯苯酚97 共聚维酮98 亚硫酸氢钠99 西曲溴铵100 西黄蓍胶101 肉豆蔻酸102 肉豆蔻酸异丙酯103 色氨酸104 交联羧甲纤维素钠105 交联聚维酮106 冰醋酸107 羊毛脂108 异丙醇109 红氧化铁110 纤维醋法酯111 麦芽酚112 麦芽糊精113 麦芽糖114 麦芽糖醇115 壳聚糖116 花生油117 低取代羟丙纤维素118 伽马环糊精119 谷氨酸钠120 肠溶明胶空心胶囊121 辛酸122 辛酸钠123 间甲酚124 没食子酸125 没食子酸丙酯126 尿素127 阿尔法环糊精128 阿司帕坦129 阿拉伯半乳聚糖130 阿拉伯胶131 阿拉伯胶喷干粉132 纯化水133 环甲基硅酮134 环拉酸钠135 苯扎氯铵136 苯扎溴铵137 苯甲酸138 苯甲酸钠139 苯甲醇140 苯氧乙醇141 DL-苹果酸142 L-苹果酸143 松香144 果胶145 果糖146 明胶空心胶囊147 依地酸二钠148 依地酸钙钠149 乳糖150 单双硬脂酸甘油酯151 单亚油酸甘油酯152 单油酸甘油酯153 单硬脂酸乙二醇酯154 单糖浆155 油酰聚氧乙烯甘油酯156 油酸乙酯157 油酸山梨坦158 油酸钠159 泊洛沙姆188160 泊洛沙姆407161 组氨酸162 枸橼酸163 枸橼酸三乙酯164 枸橼酸三正丁酯165 枸橼酸钠166 轻质氧化镁167 轻质液状石蜡168 氢化大豆油169 氢化蓖麻油170 氢氧化钠171 氢氧化钾172 氢氧化铝173 氢氧化镁174 香草醛175 胆固醇176 亮氨酸177 活性炭(供注射用)178 浓氨溶液179 盐酸180 桉油精181 氧化钙182 氧化锌183 氧化镁184 氨丁三醇185 倍他环糊精186 胶态二氧化硅187 胶囊用明胶188 粉状纤维素189 烟酰胺190 烟酸191 DL-酒石酸192 L(+)-酒石酸钠193 酒石酸钠194 海藻酸195 海藻酸钠196 海藻糖197 预胶化羟丙基淀粉198 预胶化淀粉199 黄凡士林200 黄原胶201 黄氧化铁202 硅酸钙203 硅酸镁铝204 硅藻土205 甜菊糖苷206 脱氢醋酸207 脱氧胆酸钠208 羟乙纤维素—11 —209 羟丙甲纤维素210 羟丙甲纤维素邻苯二甲酸酯211 羟丙甲纤维素空心胶囊212 羟丙纤维素213 羟丙基倍他环糊精214 羟丙基淀粉空心胶囊215 羟苯乙酯216 羟苯丁酯217 羟苯丙酯218 羟苯丙酯钠219 羟苯甲酯220 羟苯甲酯钠221 羟苯苄酯222 混合脂肪酸甘油酯(硬脂)223 液状石蜡224 淀粉水解寡糖225 蛋黄卵磷脂226 蛋黄卵磷脂(供注射用)227 维生素E琥珀酸聚乙二醇酯228 琥珀酸229 琼脂230 葡甲胺231 葡糖糖二酸钙232 椰子油233 棕氧化铁234 棕榈山梨坦235 棕榈酸236 棕榈酸异丙酯237 硬脂山梨坦238 硬脂富马酸钠239 硬脂酸240 硬脂酸钙241 硬脂酸锌242 硬脂酸聚烃氧(40)酯243 硬脂酸镁244 硝酸钾245 硫酸246 硫酸钙247 硫酸钠248 硫酸钠十水合物249 硫酸铝250 硫酸铵251 硫酸羟喹啉252 紫氧化铁253 黑氧化铁254 氯化钙255 氯化钠(供注射用)256 氯化钾257 氯化镁258 氯甲酚259 稀盐酸260 稀醋酸261 稀磷酸262 焦亚硫酸钠263 焦糖264 普鲁兰多糖空心胶囊265 滑石粉266 富马酸267 酪氨酸268 硼砂269 硼酸270 微晶纤维素271 微晶纤维素胶态二氧化硅共处理物272 微晶蜡273 腺嘌呤274 羧甲纤维素钙275 羧甲纤维素钠276 羧甲淀粉钠277 聚乙二醇300(供注射用)278 聚乙二醇400279 聚乙二醇400(供注射用)280 聚乙二醇600281 聚乙二醇1000282 聚乙二醇1500283 聚乙二醇4000284 聚乙二醇6000285 聚乙烯醇286 聚山梨酯20287 聚山梨酯40288 聚山梨酯60289 聚山梨酯80290 聚山梨酯80(Ⅱ)291 聚丙烯酸树脂Ⅱ292 聚丙烯酸树脂Ⅲ293 聚丙烯酸树脂Ⅳ294 聚卡波菲295 聚甲丙烯酸铵酯Ⅰ296 聚甲丙烯酸铵酯Ⅱ297 聚氧乙烯298 聚氧乙烯油酸酯299 聚氧乙烯(40)氢化蓖麻油300 聚氧乙烯(60)氢化蓖麻油301 聚氧乙烯(50)硬脂酸酯302 聚氧乙烯(35)蓖麻油303 聚维酮K30304 聚葡萄糖305 蔗糖306 蔗糖八醋酸酯307 蔗糖丸芯308 蔗糖硬脂酸酯309 碱石灰310 碳酸丙烯酯311 碳酸氢钠312 碳酸氢钾313 精氨酸314 橄榄油315 豌豆淀粉316 醋酸317 醋酸纤维素318 醋酸钠319 醋酸羟丙甲纤维素琥珀酸酯320 糊精321 缬氨酸322 薄荷脑323 磷酸324 磷酸二氢钠一水合物325 磷酸二氢钠二水合物326 磷酸二氢钾327 磷酸钙—13 —328 磷酸钠十二水合物329 磷酸氢二钠十二水合物330 磷酸氢二钾331 磷酸氢二钾三水合物332 磷酸氢二铵333 磷酸氢钙二水合物334 磷酸淀粉钠335 麝香草酚。
注射用重组人粒细胞巨噬细胞集落刺激因子说明书

注射用重组人粒细胞巨噬细胞集落刺激因子说明书注射用重组人粒细胞巨噬细胞集落刺激因子用于防治肿瘤病人因化疗、放疗引起的白细胞减少。
下面是店铺整理的注射用重组人粒细胞巨噬细胞集落刺激因子说明书,欢迎阅读。
注射用重组人粒细胞巨噬细胞集落刺激因子商品介绍通用名:注射用重组人粒细胞巨噬细胞集落刺激因子生产厂家: 哈尔滨里亚哈尔生物制品有限公司批准文号:国药准字S1*******药品规格:150μg,冻干粉药品价格:¥159元注射用重组人粒细胞巨噬细胞集落刺激因子说明书【通用名】注射用重组人粒细胞/巨噬细胞集落刺激因子(γhGM-CSF)【商品名】里亚尔【英文名】LEUBENE【汉语拼音】Liyaer【主要成分】活性成份:重组人粒细胞巨噬细胞集落刺激因子(γhGM-CSF)。
非活性成份:甘露醇、氨化钠、磷酸氯二钠、磷酸二氯钾、聚乙二醇(4000)、人血白蛋白。
【性状】白色无菌冻干粉针,易溶于水、pH6.9-7.1。
【适应症】用于防治肿瘤病人因化疗、放疗引起的白细胞减少。
【用法用量】癌症化疗或放疗后。
3-10µg/kg/d,皮下注射持续5-7天(注意:本药不应与抗癌化疗药同时使用,应在化疗停止24小时后可使用),根据白细胞回升速度和水平,确定维持量。
用1ml的无菌生理盐水溶解(切勿剧烈振荡),可在腹部、大腿外侧或上臀三角肌处进行皮下注射(要求:注射后局部皮肤隆起约1cm2,以便药物慢慢吸收)。
【药理作用】里亚尔作用于造血祖细胞,促进其增殖和分化,其很重要作用是刺激粒、单核巨噬细胞成熟,促进成熟细胞向外周血释放,并能促进巨噬细胞及嗜酸性细胞的多种功能。
吸收、分布、消除,自愿者皮下注射3、10或20µg/kg和静注3至30µg/kg可观察到血浓峰值和曲线下面积(AUC)随剂量的增大而增高。
皮下注射里亚尔,在3-4小时血浓达到峰值,静注里亚尔的消除半衰期为1-2小时,皮下注射则为2-3小时。
动态显色法检测注射用重组人粒细胞巨噬细胞刺激因子中内毒素的含量

动态显色法检测注射用重组人粒细胞巨噬细胞刺激因子中内毒素的含量目的建立定量检测注射用重组人粒细胞巨噬细胞集落刺激因子(rhGM-CSF)中内毒素的质控方法。
方法采用动态显色法,通过标准曲线的可靠性验证和干扰试验,建立检测rhGM-CSF中内毒素的定量方法。
结果标准曲线可靠性验证中,曲线的R2均>0.98,标准曲线成立;干扰试验显示,供试品复溶后稀释5倍后加入0.5 EU/ml的标准内毒素作为供试品回收液,经检测其内毒素回收率均接近100%,该稀释倍数下,供试品浓度对内毒素实验的干扰作用较小;另外,日常检测2批供试品的内毒素含量均小于限值,与凝胶法检测结果一致。
结论该动态显色法方法可用于rhGM-CSF中内毒素的定量检测。
标签:动态显色法;注射用重组人粒细胞巨噬细胞集落刺激因子;内毒素;定量细菌内毒素是目前医药工业中最普遍和最主要的外源性致热原[1],它是G-菌细胞壁的脂多糖成分,能够在有机体内作用于单核巨噬细胞,产生肿瘤坏死因子、白细胞介素、前列腺素、凝血素及干扰素等多种细胞因子,这些因子过量时可引起机体发热、心动过速,甚至休克[2]。
因此,为了保证注射剂药品的安全使用,各国药典均将热原检测列为重要检查项目,热原控制也成为药品生产质控的重要对象之一[3-4]。
Levin和Bang于1968年发现并建立了鲎试剂法进行体外检测细菌内毒素[5],该法简便快速、灵敏度高及重现性好的优点使其迅速成为热原检测中家兔法的替代方法[6]。
目前所使用的鲎试剂法多为凝胶限值法,《中国药典》对注射用重组人粒细胞巨噬细胞集落刺激因子(rhGM-CSF)的细菌内毒素检查也采用该法。
凝胶法是目前最常用的细菌内毒素检查法,但其是一种半定量测定方法,无法给出准确的含量。
因此,为了更好满足药品检查和生产过程质控的严格要求,内毒素检测已逐渐向定量方向发展,其中,动态显色法是目前灵敏度最高和检测范围最宽的定量测定法,该法是通过检测反应产物中对硝基苯氨到达某一预设吸光度所需要的反应时间来检测样品中的内毒素含量[7]。
《中国药典》2020版—重组人干扰素α2 类相关产品原液检定增修订内容
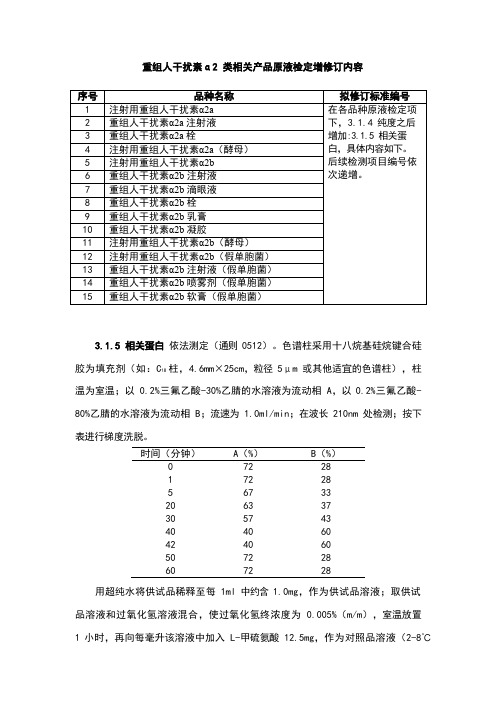
重组人干扰素α2 类相关产品原液检定增修订内容
3.1.5 相关蛋白 依法测定(通则 0512)。
色谱柱采用十八烷基硅烷键合硅胶为填充剂(如:C 18 柱,
4.6mm×25cm,粒径 5μm 或其他适宜的色谱柱),柱温为室温;以 0.2%三氟乙酸-30%乙腈的水溶液为流动相 A ,以 0.2%三氟乙酸- 80%乙腈的水溶液为流动相 B ;流速为 1.0ml/min ;在波长 210nm 处检测;按下表进行梯度洗脱。
用超纯水将供试品稀释至每 1ml 中约含 1.0mg ,作为供试品溶液;取供试品溶液和过氧化氢溶液混合,使过氧化氢终浓度为 0.005%(m/m ),室温放置 1 小时,再向每毫升该溶液中加入 L-甲硫氨酸 12.5mg ,作为对照品溶液(2-8℃
时间(分钟)
A (%)
B (%) 0 72 28 1 72 28 5 67 33 20 63 37 30 57 43 40 40 60 42 40 60 50 72 28 60
72
28。
重组人粒细胞刺激因子在《中国药典》、《美国药典》和《欧洲药典》中的质量标准对比分析

重组人粒细胞刺激因子在《中国药典》、《美国药典》和《欧洲药典》中的质量标准对比分析徐小蓉;郭旺明;黄磊;蔡谨;徐志南;岑沛霖【期刊名称】《药学研究》【年(卷),期】2011(030)002【摘要】目的对比重组人粒细胞刺激因子(rhG- CSF,Filgrastim) 在<中国药典>(2010 年版)、<美国药典>33版初稿和<欧洲药典>(7.0 版)中标准的差异,为国内研发机构在rhG-CSF质量标准的提高研究提供参考.方法按三部药典要求,对rhG-CSF产品进行主要关键指标的检测分析.结果 <中国药典>在对该产品的标准描述上不如<欧洲药典>(7.0 版)和<美国药典>33版初稿,而<美国药典>33版初稿的rhG-CSF标准要求最高.三部药典存在具体内容的区别.结论提高rhG-CSF现行产品质量标准可参照<美国药典>33版初稿.【总页数】4页(P76-79)【作者】徐小蓉;郭旺明;黄磊;蔡谨;徐志南;岑沛霖【作者单位】浙江华阳药业有限公司,浙江,杭州,310053;浙江大学化工系生物工程研究所,浙江,杭州,310027;杭州九源基因工程有限公司,浙江,杭州,310018;浙江大学化工系生物工程研究所,浙江,杭州,310027;浙江大学化工系生物工程研究所,浙江,杭州,310027;浙江大学化工系生物工程研究所,浙江,杭州,310027;浙江大学化工系生物工程研究所,浙江,杭州,310027【正文语种】中文【中图分类】R921【相关文献】1.重组人粒细胞刺激因子在《中国药典》和《欧洲药典》中质量标准不同点的对比分析 [J], 赵先亮;王春雨2.聚乙二醇化重组人粒细胞集落刺激因子的质量标准和质控方法研究 [J], 张翊;韩春梅;高凯;丁有学;饶春明;王军志3.淋巴瘤化疗后粒细胞减少预防中聚乙二醇化重组人粒细胞集落刺激因子的应用观察 [J], 姚达娜;吴福群4.重组人粒细胞刺激因子在儿童感染相关性中性粒细胞减少症中的应用 [J], 陈学高5.聚乙二醇化重组人粒细胞刺激因子在儿童肝母细胞瘤术后化疗中对中性粒细胞减少的预防效果 [J], 支天;张伟令;黄东生;张谊;胡慧敏因版权原因,仅展示原文概要,查看原文内容请购买。
重组人粒细胞刺激因子在《中国药典》和《欧洲药典》中质量标准不同点的对比分析

・ 25 ・
重 组人粒 细 胞 刺激 因子在 《 中国药典 》 《 和 欧洲药 典 》 中 质 量标 准不 同点 的对 比分 析
赵先亮, 王春 雨
( 东科 兴 生 物 制 品 有 限公 司 , 东 济 南 20 0 ) 山 山 5 2 0
摘要 : 目的
对 比 重组 人 粒 细胞 刺 激 因 子 (h r G—C F Flr s m) 《 国 药 典 》 2 0 S , i at 在 中 g i ( 0 5年 版 ) 《 洲 药 典 》 6 3版 ) 和 欧 (. 中标
准的 不 同点 , 国 内研 发 机 构对 基 因 重组 蛋 白产 品 的研 发 及 国 内企 业 对 该 产 品 的 进 出 口提 供 参 考 。 方 法 按 两部 药 典 要 求 , 为
对 国产 产 品 实样 进 行 部 分 关 键 指 标 的 检 测 分 析 。 结 果 两 部 药 典 对 该 产 品 的标 准 描 述 上 存 在 具 体 内容 的 区别 。结 论 : 现 在 代 药物 的 研 发 和 生 产 中 , 药 品 的 国 际及 国 内标 准要 进 行 综 合 分 析 而 拟 定 。 对 关 键 词 : 组 人 粒 细胞 刺激 因 子 重 组人 粒 细胞 刺 激 因子 欧 洲 药 典 中 国药 典 重 中图 分 类 号 : 9 1 文 献标 识 码 : 文章 编 号 :6 2 7 8 2 1 ) 1 0 5 3 R 2 A 1 7 —7 3 【 0 0 0 —0 2 —0
ABS RACT: T 0BJ ECT VE B o a i g t e q a i tn a d fr c mbn n u n g a u o y e c l n - t I y c mp rn h u l y sa d r s o e o i a th ma r n lc t o o y si lt g f c t mu a i a — n t r r G— C F, l r s i ) b t n E r p a a ma o o i ( h . d to ) a d C n s a ma o o i ( 0 5 e ii n , 0 (h S Fi a t g m o h i u o e n Ph r c p ea t e 6 3 e iin n hi e e Ph r c p e a 2 0 d t ) o s memo i ig s g e t n b u h t n a d f h — S o t e s a d r sma e s we e p o o e . n a d t n, o l r — o df n u g s i s a o tt e s a d r so G— C F t h t n a d k r r r p s d I d i o i c u d p o y o r i t
2020年版《中国药典》目录三部

附件32020年版《中国药典》目录三部目录生物制品通则目次1生物制品通用名称命名原则2生物制品生产用原材料及辅料质量控制3生物制品生产检定用菌毒种管理及质量控制4生物制品生产检定用动物细胞基质制备及质量控制5血液制品生产用人血浆6生物制品国家标准物质制备和标定7生物制品病毒安全性控制8生物制品分包装及贮运管理总论目次9人用疫苗总论10人用重组DNA蛋白制品总论11人用重组单克隆抗体制品总论12人用聚乙二醇化重组蛋白及多肽制品总论13人用基因治疗制品总论14螨变应原制品总论15人用马免疫血清制品总论16微生态活菌制品总论各论目次Ⅰ预防类17伤寒疫苗18伤寒甲型副伤寒联合疫苗19伤寒甲型乙型副伤寒联合疫苗20伤寒Vi多糖疫苗21重组B亚单位/菌体霍乱疫苗(肠溶胶囊)22A群脑膜炎球菌多糖疫苗23A群C群脑膜炎球菌多糖疫苗24A群C群脑膜炎球菌多糖结合疫苗25ACYW135群脑膜炎球菌多糖疫苗2623价肺炎球菌多糖疫苗27b型流感嗜血杆菌结合疫苗28吸附白喉疫苗29吸附白喉疫苗(成人及青少年用)30吸附破伤风疫苗31吸附白喉破伤风联合疫苗32吸附白喉破伤风联合疫苗(成人及青少年用)33吸附百日咳白喉联合疫苗34吸附百白破联合疫苗35吸附无细胞百白破联合疫苗36无细胞百白破b型流感嗜血杆菌联合疫苗37皮上划痕用鼠疫活疫苗38皮上划痕人用炭疽活疫苗39皮上划痕人用布氏菌活疫苗40皮内注射用卡介苗41钩端螺旋体疫苗42乙型脑炎减毒活疫苗43冻干乙型脑炎灭活疫苗(Vero细胞)44森林脑炎灭活疫苗45双价肾综合征出血热灭活疫苗(Vero细胞)46双价肾综合征出血热灭活疫苗(地鼠肾细胞)47双价肾综合征出血热灭活疫苗(沙鼠肾细胞)48黄热减毒活疫苗49冻干人用狂犬病疫苗(Vero细胞)50冻干人用狂犬病疫苗(人二倍体细胞)51冻干甲型肝炎减毒活疫苗52甲型肝炎灭活疫苗(人二倍体细胞)53重组乙型肝炎疫苗(酿酒酵母)54重组乙型肝炎疫苗(汉逊酵母)55重组乙型肝炎疫苗(CHO细胞)56甲型乙型肝炎联合疫苗57麻疹减毒活疫苗58腮腺炎减毒活疫苗59风疹减毒活疫苗(人二倍体细胞)60水痘减毒活疫苗61麻疹腮腺炎联合减毒活疫苗62麻疹风疹联合减毒活疫苗63麻腮风联合减毒活疫苗64流感全病毒灭活疫苗65流感病毒裂解疫苗66口服脊髓灰质炎减毒活疫苗(猴肾细胞)67脊髓灰质炎减毒活疫苗糖丸(人二倍体细胞)68脊髓灰质炎减毒活疫苗糖丸(猴肾细胞)69Sabin株脊髓灰质炎灭活疫苗(Vero细胞)70口服Ⅰ型Ⅲ型脊髓灰质炎减毒活疫苗(人二倍体细胞)Ⅱ治疗类71白喉抗毒素72冻干白喉抗毒素73破伤风抗毒素74冻干破伤风抗毒素75马破伤风免疫球蛋白F(ab')2 76多价气性坏疽抗毒素77冻干多价气性坏疽抗毒素78肉毒抗毒素79冻干肉毒抗毒素80抗蝮蛇毒血清81冻干抗蝮蛇毒血清82抗五步蛇毒血清83冻干抗五步蛇毒血清84抗银环蛇毒血清85冻干抗银环蛇毒血清86抗眼镜蛇毒血清87冻干抗眼镜蛇毒血清88抗炭疽血清89抗狂犬病血清90人血白蛋白91冻干人血白蛋白92人免疫球蛋白93冻干人免疫球蛋白94乙型肝炎人免疫球蛋白95冻干乙型肝炎人免疫球蛋白96静注乙型肝炎人免疫球蛋白(pH4)97冻干静注乙型肝炎人免疫球蛋白(pH4)98狂犬病人免疫球蛋白99冻干狂犬病人免疫球蛋白100破伤风人免疫球蛋白101冻干破伤风人免疫球蛋白102静注人免疫球蛋白(pH4)103冻干静注人免疫球蛋白(pH4)104人凝血因子Ⅷ105人纤维蛋白原106人纤维蛋白粘合剂107人凝血酶108人凝血酶原复合物109抗人T细胞猪免疫球蛋白110抗人T细胞兔免疫球蛋白111注射用人促红素112人促红素注射液113注射用人干扰素α1b114人干扰素α1b注射液115人干扰素α1b滴眼液116注射用人干扰素α2a117人干扰素α2a注射液118人干扰素α2a栓119注射用人干扰素α2b120人干扰素α2b注射液121人干扰素α2b滴眼液122人干扰素α2b栓123人干扰素α2b乳膏124人干扰素α2b凝胶125人干扰素α2b喷雾剂126人干扰素α2b软膏127人干扰素α2b阴道泡腾片128注射用人干扰素γ129注射用人白介素-2130人白介素-2注射液131注射用人白介素-2(Ⅰ)132注射用人白介素-11133人粒细胞刺激因子注射液134注射用人粒细胞巨噬细胞刺激因子135外用人粒细胞巨噬细胞刺激因子凝胶136牛碱性成纤维细胞生长因子外用溶液137外用牛碱性成纤维细胞生长因子138牛碱性成纤维细胞生长因子凝胶139牛碱性成纤维细胞生长因子滴眼液140外用人表皮生长因子141人表皮生长因子外用溶液(Ⅰ)142人表皮生长因子凝胶143人表皮生长因子滴眼液144尼妥珠单抗注射液145康柏西普眼用注射液146人胰岛素147人胰岛素注射液148精蛋白人胰岛素注射液149精蛋白人胰岛素混合注射液(30R)150精蛋白人胰岛素混合注射液(50R)151甘精胰岛素152甘精胰岛素注射液153赖脯胰岛素154赖脯胰岛素注射液155注射用人生长激素156注射用鼠神经生长因子157注射用A型肉毒毒素158治疗用卡介苗Ⅲ体内诊断类159结核菌素纯蛋白衍生物160卡介菌纯蛋白衍生物161布氏菌纯蛋白衍生物162锡克试验毒素Ⅳ体外诊断类163乙型肝炎病毒表面抗原诊断试剂盒(酶联免疫法)164丙型肝炎病毒抗体诊断试剂盒(酶联免疫法)165人类免疫缺陷病毒抗体诊断试剂盒(酶联免疫法)166人类免疫缺陷病毒抗原抗体诊断试剂盒(酶联免疫法)167乙型肝炎病毒、丙型肝炎病毒、人类免疫缺陷病毒Ⅰ型核酸检测试剂盒168梅毒螺旋体抗体诊断试剂盒(酶联免疫法)169抗A抗B血型定型试剂(单克隆抗体)通则和指导原则制剂通则1片剂2注射剂3胶囊剂4颗粒剂5眼用制剂6鼻用制剂7栓剂8软膏剂乳膏剂9喷雾剂10凝胶剂11散剂12涂剂分光光度法13紫外-可见分光光度法14荧光分光光度法15原子吸收分光光度法16火焰光度法色谱法17纸色谱法18高效液相色谱法19离子色谱法20分子排阻色谱法21气相色谱法电泳法22电泳法23毛细管电泳法物理检查法24pH值测定法25渗透压摩尔浓度测定法26溶液颜色检查法27澄清度检查法28不溶性微粒检查法29可见异物检查法30崩解时限检查法31融变时限检查法32片剂脆碎度检查法33最低装量检查法34粒度和粒度分布测定法含量测定法35氮测定法36蛋白质含量测定法37干燥失重测定法38固体总量测定法39唾液酸测定法40磷测定法41硫酸铵测定法42亚硫酸氢钠测定法43氢氧化铝(或磷酸铝)测定法44氯化钠测定法45枸橼酸离子测定法46钾离子测定法47钠离子测定法48辛酸钠测定法49乙酰色氨酸测定法50苯酚测定法51间甲酚测定法52硫柳汞测定法53对羟基苯甲酸甲酯、对羟基苯甲酸丙酯含量测定法54O-乙酰基测定法55已二酰肼含量测定法56高分子结合物含量测定法57人血液制品中糖及糖醇测定法58人血白蛋白多聚体测定法59人免疫球蛋白类制品lgG单体加二聚体测定法60人免疫球蛋白中甘氨酸含量测定法61人粒细胞刺激因子蛋白质含量测定法62组胺人免疫球蛋白中游离磷酸组胺测定法63lgG含量测定法64单抗分子大小变异体测定法65抗毒素/抗血清制品分子大小分布测定法66单抗电荷变异体测定法67单抗N糖谱测定法化学残留物测定法68氰化物检查法69水分测定法70炽灼残渣检查法71残留溶剂测定法72乙醇残留量测定法73聚乙二醇残留量测定法74聚山梨酯80残留量测定法75戊二醛残留量测定法76磷酸三丁酯残留量测定法77碳二亚胺残留量测定法78游离甲醛测定法79人血白蛋白铝残留量测定法80羟胺残留量测定法微生物检查法81无菌检查法82非无菌产品微生物限度检查:微生物计数法83非无菌产品微生物限度检查:控制菌检查法84非无菌药品微生物限度标准85抑菌效力检查法86异常毒性检查法87热原检查法88细菌内毒素检查法89支原体检查法90外源病毒因子检查法91鼠源性病毒检查法92SV40核酸序列检查法93猴体神经毒力试验94血液制品生产用人血浆病毒核酸检测技术要求95黄热减毒活疫苗猴体试验96禽源性病毒荧光定量PCR(Q-PCR)检查法生物测定法97免疫印迹法98免疫斑点法99免疫双扩散法100免疫电泳法101肽图检查法102质粒丢失率检查法103外源性DNA残留量测定法104抗生素残留量检查法105激肽释放酶原激活剂测定法106抗补体活性测定法107牛血清白蛋白残留量测定法108大肠埃希菌菌体蛋白质残留量检测法109假单胞菌菌体蛋白质残留量测定法110酵母工程菌菌体蛋白质残留量测定法111类A血型物质测定法112鼠lgG残留量测定法113无细胞百日咳疫苗鉴别试验114抗毒素、抗血清制品鉴别试验115A群脑膜炎球菌多糖分子大小测定法116伤寒Vi多糖分子大小测定法117b型流感嗜血杆菌结合疫苗多糖含量测定法118人凝血酶活性检查法119活化的凝血因子活性检查法120肝素含量测定法121抗A、抗B血凝素测定法122人红细胞抗体测定法123人血小板抗体测定法124人免疫球蛋白类制品lgA残留量测定法125免疫化学法生物活性/效价测定法126胰岛素生物测定法127生长激素生物测定法128重组乙型肝炎疫苗(酵母)体外相对效力检查法129甲型肝炎灭活疫苗体外相对效力检查法130人用狂犬病疫苗效价测定法131吸附破伤风疫苗效价测定法132吸附白喉疫苗效价测定法133类毒素絮状单位测定法134白喉抗毒素效价测定法135破伤风抗毒素效价测定法136气性坏疽抗毒素效价测定法137肉素抗毒素效价测定法138抗蛇毒血清效价测定法139狂犬病免疫球蛋白效价测定法140人免疫球蛋白中白喉抗体效价测定法141人免疫球蛋白Fc段生物学活性测定法142抗人T细胞免疫球蛋白效价测定法(E玫瑰花环形成抑制试验)143抗人T细胞免疫球蛋白效价测定法(淋巴细胞毒试验)144人凝血因子Ⅱ效价测定法145人凝血因子Ⅶ效价测定法146人凝血因子Ⅸ效价测定法147人凝血因子Ⅹ效价测定法148人凝血因子Ⅷ效价测定法149人促红素体内生物学活性测定法150干扰素生物学活性测定法151人白介素-2生物学活性测定法152人粒细胞刺激因子生物学活性测定法153人粒细胞巨噬细胞刺激因子生物学活性测定法154牛碱性成纤维细胞生长因子生物学活性测定法155人表皮生长因子生物学活性测定法156鼠神经生长因子生物学活性测定法157尼妥珠单抗生物学活性测定法158人白介素-11生物学活性测定法159A型肉毒毒素效价测定法160Sabin株脊髓灰质炎灭活疫苗效力试验161康柏西普生物学活性测定法特定生物原材料/动物及辅料162生物制品生产及检定用实验动物质量控制163重组胰蛋白酶164新生牛血清165细菌生化反应培养基166氢氧化铝佐剂试剂试液标准品167试药168试液169试纸170缓冲液171指示剂与指示液172滴定液173生物制品国家标准物质目录其他174灭菌法175生物检定统计法指导原则176分析方法验证指导原则177药品微生物检验替代方法验证指导原则178药品微生物实验室质量管理指导原则179生物制品生物活性/效价测定方法验证指导原则180生物制品稳定性实验指导原则生物制品术语附表181原子量表。
国产重组人粒细胞-巨噬细胞集落刺激因子原液中制品相关蛋白的定性定量分析

药物分析杂志药物分析杂志 标准品溶液b的色谱图 Chromatography of standard solution b药物分析杂志Chinese 质谱测定各制品相关蛋白的相对分子质量Relative molecular weights of different product-related proteins measured by mass spectrometry主峰中蛋白相对分子质量的测定采用液质联用的方式对主峰中的蛋白相对分子4),结果发现:主峰中的蛋白成分并不均一,其中绝大部分为完整的子(理论相对分子质量为14 604.75)N-末端缺少“Met”的蛋白(理论相对分子质量为Journal of Pharmaceutical Analysis 药物分析杂志Chinese药物分析杂志 质谱测定主峰中蛋白相对分子质量Relative molecular mass of proteins in the main peak measured by mass spectrometry。
N-末端缺失1~2个氨基酸残基的GM-CSF 的存在可能跟生产工艺有关,它们可以作为目标产品的一部分而不是制品相关蛋白。
本研究在样品1-2及2-1中发现有相对分子质量不等的肽段出现,而在同一厂家生产的其他批次里未发现相应肽段。
这些肽段的出现可能与生产工艺的细微变化有关,也可能在产品放置过程中形成,具体原因还有待进一步的分析。
研究还发现,rhGM-普遍存在的制品相关蛋白是氧化形式及乙酰化形式,这在其他大肠杆菌表达的蛋白中也比较常。
不同企业由于生产工艺的差异,相应蛋白的14 473.56)以及N-末端缺少“MetAla ”的蛋白(理论相对分子质量为14 402.48)。
除此之外,还有一定数量的相对分子质量为14 719左右的蛋白,经分析为完整蛋白与流动相中的三氟乙酸(TFA )加合形该加合物的理论相对分子质量为14 718.78。
2020版药典变化全汇总

2020版药典变化全汇总一部中药收载2711种,其中新增117种、修订452种。
新增了药材:裸花紫珠。
新增制剂:116个,具体清单如下:二部化学药收载2712种,其中新增117种、修订2387种。
在药品安全性控制方面,要求进一步完善杂质和有关物质的分析方法,推广先进检测技术的应用,重点强化了对有毒有害杂质(特别是基因毒性杂质)的控制,加强了对药品安全性相关控制项目和限度标准的研究制定。
在药品有效性控制方面,要求将药品质量和疗效一致性评价的成果体现在相关制剂的质量标准提高中,进一步完善了常规固体制剂溶出及释放度检测方法,且在整体质量控制方面进一步借鉴国际要求。
2020年版药典二部新增品种(117个),与一部新增品种居然一样!未收载品种(8个),名称变化的品种(5个)。
具体清单如下:未收载品种(8个)本版药典(二部)未收载2015年版药典(二部)中的品种名单:•鱼肝油••重组人生长激素••注射用重组人生长激素••重组人胰岛素••重组人胰岛素注射液••精蛋白重组人胰岛素注射液••盐酸吡硫醇注射液••注射用盐酸吡硫醇•新增品种(117个)本版药典(二部)新增品种名单:1.注射用门冬氨酸鸟氨酸2.马来酸氟伏沙明3.马来酸氟伏沙明片4.扎来普隆5.乌苯美司片6.丙戊酸钠缓释片(I)7.丙泊酚乳状注射液(曾用名:丙泊酚注射液)8.丙酸氟替卡松9.左卡尼汀10.左甲状腺素钠11.左甲状腺素钠片12.右佐匹克隆13.右佐匹克隆片14.卡培他滨15.卡培他滨片16.甲钴胺片17.甲钴胺注射液18.甲磺酸多沙唑嗪19.甲磺酸多沙唑嗪片20.甲磺酸多沙唑嗪胶囊21.甲磺酸瑞波西汀22.甲磺酸瑞波西汀片23.甲磺酸瑞波西汀胶囊24.兰索拉唑肠溶胶囊25.矛头蝮蛇血凝酶(曾用名:蛇毒血凝酶、血凝酶)26.注射用矛头蝮蛇血凝酶(曾用名:注射用蛇毒血凝酶、注射用血凝酶)27.地红霉素肠溶片28.地红霉素肠溶胶囊29.西尼地平胶囊30.西吡氯铵31.西吡氯铵含漱液32.西咪替丁注射液33.西洛他唑片34.托拉塞米35.托拉塞米片36.托拉塞米胶囊37.注射用托拉塞米38.吗替麦考酚酯分散片39.伏格列波糖40.伏格列波糖片41.伏格列波糖胶囊42.米氮平43.米氮平片44.那他霉素45.那他霉素滴眼液46.坎地沙坦酯片47.克霉唑阴道膨胀栓[曾用名:克霉唑栓(指含膨胀棉条的克霉唑栓)]48.更昔洛韦胶囊49.来曲唑50.来曲唑片51.吲哚美辛片52.佐米曲普坦分散片53.阿托伐他汀钙54.阿利沙坦酯55.阿利沙坦酯片56.阿那曲唑片57.苯磺酸左氨氯地平58.苯磺酸左氨氯地平片59.拉西地平60.拉西地平片61.依巴斯汀62.依巴斯汀片63.草酸艾司西酞普兰64.草酸艾司西酞普兰片65.枸橼酸坦度螺酮66.枸橼酸坦度螺酮胶囊67.抗凝血用枸橼酸钠溶液[曾用名:(1)输血用枸橼酸钠注射液(适应症为仅用于单采原料血浆的体外抗凝血)K2)枸橼酸钠抗凝剂]68.枸橡酸钾颗粒69.氟尿苷(曾用名:氟脲苷)70.复方氨基酸(15)双肽(2)注射液71.美司钠72.美司钠注射液73.盐酸乙哌立松74.盐酸乙哌立松片75.盐酸左布比卡因76.盐酸左布比卡因注射液77.盐酸托烷司琼片78.盐酸托烷司琼胶囊79.盐酸曲普利啶80.盐酸伊达比星81.注射用盐酸伊达比星82.盐酸奈福泮胶囊83.盐酸度洛西汀84.盐酸度洛西汀肠溶片85.盐酸度洛西汀肠溶胶囊86.盐酸羟甲唑啉87.盐酸羟甲唑啉喷雾剂88.盐酸羟甲唑啉喷鼻液89.盐酸羟苄唑90.盐酸羟苄唑滴眼液(曾用名:羟苄唑滴眼液)91.盐酸奥布卡因92.盐酸奥布卡因滴眼液93.氨糖美辛肠溶片94.氨糖美辛肠溶胶囊95.脂肪乳注射液(C14~24)(曾用名:脂肪乳注射液)96.酒石酸溴莫尼定97.酒石酸溴莫尼定滴眼液98.酚磺乙胺99.注射用酚磺乙胺100.铝碳酸镁101.铝碳酸镁咀嚼片102.葡萄糖酸钙氯化钠注射液103.硝酸益康唑阴道膨胀栓[曾用名:硝酸益康唑栓(指含膨胀棉条的硝酸益康唑栓)]104.氯沙坦钾105.氯沙坦钾片106.氯沙坦钾胶囊107.酮咯酸氨丁三醇108.酮咯酸氨丁三醇注射液109.腺苷110.腺苷注射液[曾用名:腺苷注射液(供诊断用)]111.去氨加压素片(曾用名:醋酸去氨加压素片)112.注射用去氨加压素(曾用名:注射用醋酸去氨加压素)113.磷酸氟达拉滨114.注射用磷酸氟达拉滨115.磷酸腺嘌呤(曾用名:维生素b4)116.磷酸腺嘌呤片(曾用名:维生B4片)117.碘帕醇注射液三部生物制品收载153种,其中新增20种、修订126种;新增生物制品通则2个、总论4个。
2024年重组人粒细胞剌激因子注射液市场调查报告

2024年重组人粒细胞剌激因子注射液市场调查报告1. 市场概况重组人粒细胞刺激因子注射液(以下简称重组粒刺因注射液)是一种用于治疗骨髓抑制和白细胞减少的药物。
它通过刺激骨髓产生和释放粒细胞,提高患者的免疫功能。
近年来,重组粒刺因注射液在临床应用中得到了广泛关注和使用。
2. 市场需求2.1 患者需求由于人口老龄化和疾病患者数量增加,对于重组粒刺因注射液的需求不断增加。
患者主要需求包括提高免疫功能、预防感染、缓解症状等。
2.2 医疗机构需求医疗机构需要提供高质量、高效率的治疗方案来满足患者的需求。
重组粒刺因注射液正是一种有效的治疗方法之一,因此医疗机构对于该药物的需求较大。
2.3 市场竞争重组粒刺因注射液市场竞争激烈,存在多个品牌和制造商。
不同品牌的产品在质量、价格、服务等方面存在差异,竞争力较强。
3. 市场规模及增长趋势根据市场调查数据显示,重组粒刺因注射液市场规模逐年增长。
2019年市场规模为X亿元,2020年达到X亿元。
预计未来几年市场规模将进一步增加。
4. 市场份额根据市场份额数据显示,目前市场上存在多个重组粒刺因注射液品牌。
其中,A 品牌占据市场份额的XX%,B品牌占据市场份额的XX%。
5. 市场影响因素5.1 政策影响政府对医药行业的政策影响较大,包括药品审批、价格政策等。
这些政策变化将对重组粒刺因注射液市场产生影响。
5.2 可及性和成本重组粒刺因注射液的可及性和成本是影响市场需求的重要因素。
如果产品很难获得或价格过高,市场需求可能受到限制。
5.3 技术进步技术进步对于药物的研发和生产起着重要作用。
新技术的应用将推动重组粒刺因注射液市场的发展。
6. 市场挑战及机遇6.1 挑战市场竞争激烈、政策变化、产品质量问题等是重组粒刺因注射液市场面临的挑战。
制造商需要关注市场变化,不断提高产品质量和服务水平。
6.2 机遇随着医疗技术的不断进步和患者需求的增加,重组粒刺因注射液市场将会面临新的机遇。
创新药物的研发和市场推广有望取得新突破。
HPLC在注射用重组人粒细胞巨噬细胞刺激因子冻干粉针剂主药含量测定中的应用

HPLC在注射用重组人粒细胞巨噬细胞刺激因子冻干粉针剂主药含量测定中的应用周飞燕;孔雯雯;杨丽菲【期刊名称】《中国当代医药》【年(卷),期】2017(024)015【摘要】目的建立注射用重组人粒细胞巨噬细胞集落刺激因子(rhGM-CSF)冻干粉针剂中主要活性成分的HPLC的测定方法.方法 HPLC色谱条件中,色谱柱为TSK gel G3000SW(300.0 mm×7.5 mm,5μm),流动相为0.1 mol/L磷酸盐缓冲液,流速为1 ml/min,自动进样20~40μl,检测波长为280 nm.研究该色谱条件是否能分析rhGM-CSF冻干粉针剂中的各组分、实验重现性以及不同进样体积与各组分峰面积的线性关系.结果检测出rhGM-CSF冻干粉针剂中的主成分为rhGM-CSF蛋白和人血白蛋白,rhGM-CSF蛋白进样量与峰面积呈良好线性关系,R2>0.99,百分含量的RSD值均<3%.结论该法可用于检测rhGM-CSF冻干粉针剂蛋白量在10~20μg范围内的的活性成分含量.【总页数】4页(P104-107)【作者】周飞燕;孔雯雯;杨丽菲【作者单位】广州白云山拜迪生物医药有限公司,广东广州511495;广州白云山拜迪生物医药有限公司,广东广州511495;广州白云山拜迪生物医药有限公司,广东广州511495【正文语种】中文【中图分类】R927.2【相关文献】1.注射用重组人粒细胞-巨噬细胞集落刺激因子致变态反应1例的报告 [J], 王玉卿2.RP-HPLC法测定重组人粒细胞刺激因子注射液主药含量 [J], 杨美花3.动态显色法检测注射用重组人粒细胞巨噬细胞刺激因子中内毒素的含量 [J], 孔雯雯;张明森;林秀菁;魏荣华4.重组人粒细胞-巨噬细胞集落刺激因子凝胶应用于糖尿病患者中厚皮移植术后供皮区的临床效果 [J], 夏卫东;陈光夷;戴文统;赵胜;李粟;林才5.外用重组人粒细胞-巨噬细胞刺激因子凝胶与注射用重组人生长激素联合应用对实验性烫伤动物的疗效 [J], 裴瑾;姜晶;赵丽娟;赵立欣;王班;金磊因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
注射用重组人粒细胞巨噬细胞刺激因子原液相关蛋白检测
依法测定(通则 0512)。
色谱柱采用四烷基硅烷键合硅胶为填充剂(如: C 4 柱,4.6mm×15cm,粒径 5μm 或其他适宜的色谱柱),柱温为室温;以 0.1%
三氟乙酸的水溶液为流动相 A ,以 0.1%三氟乙酸-90%乙腈的水溶液为流动相B ;流速为 1.2ml/min ;在波长 214nm 处检测;按下表进行梯度洗脱。
用稀释液(pH7.0 的磷酸盐缓冲液)将供试品稀释至每 1ml 中约含 0.5mg ,
作为供试品溶液;用稀释液将对照品稀释至每 1ml 中约含 0.5mg ,作为对照品溶液 A ;取对照品溶液 A 与每 1ml 中约含 0.125mg 的人或牛血清白蛋白溶液 (溶剂为稀释液),按体积比 1:4 混匀作为对照品溶液 B 。
取供试品溶液与对照品溶液 A 、B 各 100µl 注入液相色谱仪。
供试品溶液与对照品溶液 A 、B 图谱中,粒细胞巨噬细胞刺激因子主峰的保留时间应一致,约为 22 分钟。
对照品溶液 B 图谱中,主峰与人或牛血清白蛋白峰的分离度应不小于 2,重复进样(不少于 4 次)所得主峰峰面积的 RSD 应不大于 1.5%。
按面积归一化法只计算保留时间为 5~30 分钟的相关蛋白峰面积,单个相关蛋白峰面积应大不于总面积的 1.5%,所有相关蛋白峰面积应不大于总面积的4.0%。
备注:在该品种原液检定项下,3.1.4 纯度之后增加 3.1.5 相关蛋白项(具体内容见上文:注射用重组人粒细胞巨噬细胞刺激因子原液中相关蛋白检测), 后续检测项目编号依次递增。
时间(分钟) A (%) B (%) 0 64 36 30 44 56 35 0 100 45 0 100 50
64 36 60 64 36。