Alzheimer_s疾病相关蛋白质相互作用网络构建及其相互作用预测
蛋白质相互作用的预测方法

蛋白质相互作用的预测方法全文共四篇示例,供读者参考第一篇示例:蛋白质相互作用是生物体内细胞信号传递以及代谢调控的核心机制之一。
研究蛋白质相互作用对于理解生命活动的规律以及疾病的发生发展具有重要意义。
在过去的几十年里,科学家们提出了许多方法来预测蛋白质相互作用,其中包括生物物理学方法、生物信息学方法以及机器学习方法等。
在生物物理学方法中,双杂交技术是最常用的方法之一。
这是一种通过将感兴趣的两个蛋白质分子分别与酵母细胞的DNA结合,来判断它们是否有相互作用的技术。
双杂交技术可以大规模地筛选出潜在的蛋白质相互作用,但是其结果需要后续的验证。
生物信息学方法主要利用蛋白质的序列信息以及结构信息来预测蛋白质相互作用。
基于同源结构的方法通过比对蛋白质序列及结构来发现具有相似结构的蛋白质,从而提前推测它们可能具有相似的功能与相互作用关系。
还有一些基于蛋白质结构的模拟方法,如分子对接技术,通过计算两个蛋白质的结构与相互作用方式,来预测它们之间的相互作用模式。
近年来,随着人工智能技术的发展,机器学习方法在蛋白质相互作用预测领域也取得了一定的进展。
机器学习方法通过训练大量的蛋白质相互作用数据,来构建预测模型并对新数据进行预测。
支持向量机、神经网络以及随机森林等方法都被广泛应用于蛋白质相互作用的预测。
除了以上提到的方法外,一些综合方法也被提出来提高蛋白质相互作用预测的准确性。
将生物物理学方法和生物信息学方法相结合,可以综合利用蛋白质序列、结构以及相互作用信息来进行预测。
还有一些基于网络的方法,通过构建蛋白质相互作用网络,来分析蛋白质之间的关联性以及预测潜在的相互作用关系。
预测蛋白质相互作用是一个复杂的问题,需要多种方法的综合应用。
随着科学技术的不断进步,我们相信未来会有更多更准确的方法被提出来帮助我们更好地理解蛋白质相互作用的规律,从而为生命科学研究和药物研发提供更多的帮助。
第二篇示例:蛋白质相互作用是细胞内复杂生物过程中的一部分,它对于细胞的正常功能以及疾病的发生起到非常重要的作用。
阿尔茨海默病的信号通路与药物靶点

阿尔茨海默病的信号通路与药物靶点阿尔茨海默病(Alzheimer's disease)是一种神经退行性疾病,主要影响老年人的记忆、认知和行为功能。
这个疾病在全球范围内已经成为一种严重的公共卫生问题,目前还没有有效的治疗方法。
然而,通过对阿尔茨海默病信号通路及药物靶点的研究,或许能够为治疗该病提供新的途径。
阿尔茨海默病信号通路的研究,主要围绕神经元的损伤和退行性改变展开。
研究发现,淀粉样蛋白以及tau蛋白在阿尔茨海默病的发病过程中起到重要的作用。
淀粉样蛋白主要以β-淀粉样蛋白聚集形式存在于老年斑和神经原纤维缠结中,而tau蛋白异常磷酸化并聚集在神经纤维缠结中。
这种异常的蛋白聚集不仅引起了神经元的死亡和功能损失,还导致了炎症反应的发生。
药物靶点是指药物作用的特定分子或结构,通过干预这些靶点的功能来发挥治疗作用。
对于阿尔茨海默病来说,药物靶点主要包括针对淀粉样蛋白和tau蛋白的药物靶点,以及针对炎症反应的药物靶点。
目前,研究者们正致力于开发能够抑制淀粉样蛋白聚集的药物靶点。
一些研究表明,抑制淀粉样蛋白聚集可减轻阿尔茨海默病的病理过程,并且改善认知功能。
这些药物靶点主要包括β-淀粉样蛋白沉积、酶切或聚集过程中的相关酶类以及淀粉样蛋白的清除途径等。
与此同时,针对tau蛋白的研究也取得了一定的进展。
tau蛋白异常的磷酸化和聚集与阿尔茨海默病的临床症状密切相关。
因此,开发抑制tau蛋白异常磷酸化和聚集的药物靶点是阿尔茨海默病研究的另一个重要方向。
近年来,一些药物靶点通过调节磷酸化酶和磷酸化酶的活性,从而改善tau蛋白的磷酸化状态和聚集程度。
此外,炎症反应在阿尔茨海默病中也扮演着重要的角色。
研究发现,炎症反应的增加与神经元损伤和认知功能下降密切相关。
因此,抑制炎症反应成为防治阿尔茨海默病的重要策略之一。
药物靶点主要包括一些调节炎症细胞因子释放的药物以及调节炎症信号通路的药物。
虽然对阿尔茨海默病信号通路和药物靶点的研究还在持续进行中,但这些研究已经为未来的治疗提供了新的方向。
基于机器学习的蛋白质相互作用预测模型构建

基于机器学习的蛋白质相互作用预测模型构建蛋白质相互作用是生物体内许多重要生命活动的基础。
通过研究和理解蛋白质之间的相互作用,我们可以揭示生物系统的结构和功能,从而为疾病治疗和药物设计等领域提供重要启示。
然而,实验室实验对蛋白质相互作用的研究往往费时费力,并且很难涵盖到所有可能的蛋白质相互作用。
因此,构建基于机器学习的蛋白质相互作用预测模型成为了一项重要的研究工作。
机器学习是一种通过算法和模型从数据中自动学习和提取知识的方法。
在构建蛋白质相互作用预测模型时,我们可以利用机器学习的方法利用已知的蛋白质相互作用数据来训练模型,并用该模型预测新的蛋白质相互作用。
首先,为了构建一个准确可靠的蛋白质相互作用预测模型,我们需要大量的蛋白质相互作用数据作为训练集。
这些数据可以来自于已有的实验结果,也可以通过计算方法进行预测。
在数据预处理阶段,我们需要对数据进行清洗和标准化,去除噪音和异常值,以提高模型的质量和准确度。
接下来,选择合适的特征表示是构建蛋白质相互作用预测模型的关键步骤。
蛋白质相互作用预测中常用的特征包括蛋白质的氨基酸序列、结构信息和生物化学属性等。
这些特征可以通过生物信息学方法、结构分析工具和实验技术等手段获取。
在选择特征时,需要考虑特征的相关性、可解释性和计算效率等因素,并使用特征选择技术进行优化。
然后,选择合适的机器学习算法进行模型训练和预测。
在蛋白质相互作用预测中常用的机器学习方法包括支持向量机(SVM)、随机森林(Random Forest)、神经网络(NeuralNetwork)等。
这些算法根据不同的模型结构和学习策略,可以捕捉蛋白质相互作用的特征和模式,并进行准确的预测。
在训练模型时,需要使用交叉验证等技术来评估模型的性能和泛化能力,以防止过拟合和提高模型的鲁棒性。
同时,还可以使用集成学习、深度学习和迁移学习等方法来进一步提高模型的性能和稳定性。
最后,构建好的蛋白质相互作用预测模型需要进行评估和应用。
基于网络药理学方法探究天麻素抗阿尔兹海默症的作用机制
基于网络药理学方法探究天麻素抗阿尔兹海默症的作用机制作者:***来源:《云南中医中药杂志》2022年第09期摘要:目的利用網络药理学方法探究天麻素抗阿尔兹海默症(alzheimer’s disease,AD)的作用机制。
方法通过PhrmMapper平台预测天麻素的潜在靶点,检索GeneCards 数据库中AD的靶点,取潜在靶点与疾病靶点的交集,作为天麻素抗AD的候选靶点。
将候选靶点导入STRING数据库,绘制蛋白互作网络并进行分析,度值高的靶点作为天麻素抗AD的重要靶点。
将候选靶点导入DAVID数据库进行基因功能注释和KEGG通路富集分析。
结果经PhrmMapper预测和筛选后得到69个的潜在靶点,检索GeneCards 数据库中得到AD相关靶点10972个,获得候选靶点共55个;蛋白互作网络包含ESR1、CASP3、HSPA8、CCNA2、LDHB、PLAU等44个蛋白。
基因功能注释结果得到46个生物过程(BP)条目,8个分子功能(CC)条目,25个细胞组成(MF)条目。
KEGG 通路富集得到8条信号通路(P<0.05),包括氨基糖和核苷酸糖代谢、氮代谢、AMPK信号通路、胰岛素信号通路、淀粉和蔗糖代谢、半胱氨酸和蛋氨酸代谢、甲状腺激素信号通路、细胞周期。
结论天麻素可能通过ESR1、CASP3、HSPA8、CCNA2、LDHB、PLAU等靶点,调控氨基糖和核苷酸糖代谢、氮代谢、AMPK信号通路、胰岛素信号通路、淀粉和蔗糖代谢、半胱氨酸和蛋氨酸代谢、甲状腺激素信号通路、细胞周期,发挥治疗AD的作用。
关键词:天麻素;阿尔兹海默症;网络药理学;作用机制中图分类号:R285.6 文献标志码:A 文章编号:1007-2349(2022)09-0039-08Exploring the Mechanism of Action of Gastrodin against Alzheimer's Disease Based on Network PharmacologyZHANG Ke(General Hospital of Tianjin Medical University, Tianjin 300052, China)【Abstract】Objective: To explore the mechanism of action of gastrodin against Alzheimer's disease (AD) by network pharmacology. Methods: The potential targets of gastrodin were predicted by PhrmMapper platform, the AD targets in the GeneCards database were searched, and the intersection of potential targets and disease targets was taken as the candidate targets of gastrodin against AD. The candidate targets were imported into the STRING database, and the protein interaction network was drawn and analyzed. The candidate targets were imported into the DAVID database for gene function annotation and KEGG pathway enrichment analysis. Results: 69 potential targets were predicted and screened by PhrmMapper, and 10972 AD-related targets were retrieved from the GeneCards database, and a total of 55 candidate targets were obtained. The protein interaction network included ESR1, CASP3, HSPA8, CCNA2, LDHB, PLAU and other 44 proteins. Gene function annotation results obtained 46 biological process (BP) entries, 8 molecular function (CC) entries, and 25 cellular composition (MF) entries. KEGG pathway enrichment resulted in 8 signaling pathways (P<0.05), including amino sugar and nucleotide sugar metabolism, nitrogen metabolism, AMPK signaling pathway, insulin signaling pathway,starch and sucrose metabolism, cysteine and methionine metabolism, thyroid Hormone signaling pathways and cell cycle. Conclusion: Gastrodin may regulate amino sugar and nucleotide sugar metabolism, nitrogen metabolism, AMPK signaling pathway, insulin signaling pathway, starch and sucrose metabolism, cysteine and methionine metabolism, thyroid hormone signaling pathway and cell cycle through ESR1, CASP3, HSPA8, CCNA2, LDHB, PLAU and other targets and play a role in the treatment of AD.【Key words】Gastrodin; Alzheimer's Disease; Network Pharmacology; Mechanism of Action阿尔兹海默症(alzheimer’s disease,AD)是一种发病隐匿的中枢神经系统退行性疾病,65岁以上的人群多发,确诊后很难控制,且目前治疗效果不佳[1-3]。
蛋白质互作网络预测和分析

蛋白质互作网络预测和分析蛋白质是生命体内不可或缺的一种基本物质,其在细胞内起着举足轻重的作用。
蛋白质互作网络指的是蛋白质之间的相互作用关系,是细胞内最为庞大和复杂的调控系统之一。
在生物学研究中,预测和分析蛋白质互作网络是十分重要的工作。
蛋白质互作网络的预测和分析方法有很多种,其中重要的方法之一是基于生物信息学和系统生物学的分析工具开发。
生物信息学是将计算机科学和生物学相结合的学科,通过研究分子生物学的大量数据,揭示生物学中的基本原理。
系统生物学则是建立在生物信息学的基础之上,其目的是探究生命系统的组成、运行原理和调控机制。
通过这两个学科的研究,可以得到蛋白质互作网络的预测和分析方法。
蛋白质相互作用网络的预测方法在研究中得到了广泛的应用。
这些方法主要可以分为两类:基于基因组学和基于结构学。
基于基因组学的方法通过分析基因组信息,提取蛋白质间相互作用关系,比如双杂交和蛋白质复合物沉淀等。
而基于结构学的方法通过分析蛋白质结构信息,预测蛋白质与蛋白质之间的相互作用关系。
这些方法虽然在预测精确性上各有优劣,但是在细菌和酵母等模型生物的蛋白质互作网络中,能够预测出大量的功能关系。
在预测了蛋白质的相互作用关系后,需要对这些相互作用关系进行分析。
蛋白质互作网络的分析方法也有很多种,其中主要包括网络拓扑分析、功能注释和生物通路分析等。
网络拓扑分析主要是通过对网络结构和特征的研究,揭示网络中节点的核心性质和功能。
在运用网络拓扑分析研究蛋白质相互作用网络时,可以揭示网络中自组织功能模块和关键节点,为揭示蛋白质相互作用网络中的物质转运、细胞信号传导和基因调控等方面的生物学意义提供了依据。
除了网络拓扑分析,还有一种重要的蛋白质互作网络分析方法是功能注释。
功能注释是通过对基因和蛋白质注释信息的研究,发现蛋白质相互作用关系的生物学意义。
在蛋白质互作网络研究中,功能注释主要是通过分析蛋白质的分子功能、细胞定位和信号通路位置等信息,深入了解蛋白质之间的关系,为了解生命系统中的许多基本生理过程、疾病的发生和发展等方面提供新的思路和方法。
蛋白质相互作用网络构建和分析方法

蛋白质相互作用网络构建和分析方法蛋白质相互作用是细胞内发生的一种重要的分子级相互作用,它在细胞的信号传导、代谢调节、基因转录和疾病发生等多个生物学过程中扮演着关键角色。
蛋白质相互作用网络是描述蛋白质间相互作用关系的图形化数据模型,它有助于我们理解蛋白质功能、预测未知蛋白质间相互作用以及揭示疾病相关基因网络的特征。
本文将介绍蛋白质相互作用网络的构建和分析方法。
蛋白质相互作用网络的构建通常包括两个步骤:第一步是蛋白质相互作用实验的数据采集,第二步是数据处理和网络构建。
蛋白质相互作用实验有多种技术可供选择,包括酵母双杂交、质谱分析、蛋白质芯片等。
其中,酵母双杂交是最常用的实验方法之一。
它通过将目标蛋白质与酵母菌中的转录因子结合,观察酵母生长情况来判断是否存在蛋白质相互作用。
此外,质谱分析也被广泛应用于蛋白质相互作用研究中。
质谱分析能够通过检测蛋白质之间的物理交互作用来鉴定蛋白质相互作用对,其优势在于可以检测全细胞水平的蛋白质相互作用。
不同的方法有不同的检测原理和适用范围,研究者可以根据具体的研究目的选择合适的实验方法。
数据处理和网络构建是蛋白质相互作用网络构建的关键步骤。
首先,蛋白质相互作用实验得到的原始数据需要进行质控和过滤,去除假阳性和假阴性的结果。
然后,通过建立蛋白质相互作用矩阵来表示蛋白质之间的相互作用关系。
这个矩阵可以使用邻接矩阵来表示,其中每个元素表示不同蛋白质之间的相互作用情况,0代表无相互作用,1代表有相互作用。
最后,通过将相互作用关系抽象成节点和边的形式,构建蛋白质相互作用网络图。
蛋白质相互作用网络的分析可以帮助我们揭示网络的特征和功能。
常用的蛋白质相互作用网络分析方法包括网络节点中心性分析、功能模块发现和生态系统级功能分析。
网络节点中心性分析是衡量网络节点重要性的一种方法。
常用的节点中心性指标包括度中心性、介数中心性和接近中心性。
度中心性是指网络中节点的连接数,即节点与其他节点相互作用的数量。
蛋白质互作网络及其功能预测

蛋白质互作网络及其功能预测细胞是生命的基本单位,而蛋白质则是构成大部分细胞内部和外部结构的重要组成成分。
蛋白质通过相互作用形成复杂的互作网络,这种互作网络对于维持细胞的正常功能至关重要。
了解蛋白质互作网络以及预测蛋白质功能的方法,有助于揭示蛋白质间相互作用的机制,并为疾病诊断和药物开发提供重要的指导。
蛋白质互作网络是由蛋白质之间的相互作用所构成的复杂网络系统。
这种网络关系可以由实验技术如亲和纯化、酵母双杂交等得到,也可以基于已知的蛋白质序列和结构进行推测。
通过分析蛋白质互作网络,科学家们可以研究蛋白质之间的相互作用模式、功能模块等信息,并从中发现新的蛋白质互作关系,挖掘未知的生物学模式。
功能预测是指对尚未被研究过的蛋白质进行功能推断的过程。
在大规模基因组测序技术的推动下,许多新的蛋白质序列被快速确定,但关于它们的功能却仍然知之甚少。
传统方法如同源性比对、基于结构的预测等可以为蛋白质的功能预测提供有用的线索,但这些方法的准确性和可靠性仍然需要进一步改进。
近年来,随着大数据和机器学习技术的发展,基于蛋白质互作网络的功能预测方法逐渐受到关注。
如何预测蛋白质功能是一个具有挑战性的问题。
目前,主要的预测方法可分为两大类:归纳方法和演绎方法。
归纳方法通过学习已知蛋白质的属性和功能,建立模型来推断未知蛋白质的功能。
这种方法能够扩展已知的功能注释到无注释的蛋白质上,但模型的可靠性和泛化能力也是一个问题。
演绎方法则是基于蛋白质的互作网络,通过聚类、模块发现等方法来预测蛋白质的功能。
这种方法具有相对较好的可靠性,但一些不包含在互作网络中的蛋白质仍然难以预测。
蛋白质互作网络的预测与挖掘是一项艰巨且复杂的任务。
首先,需要构建蛋白质互作网络,收集大量的蛋白质相互作用实验数据。
其次,需要开发高效的算法和工具来解析这些复杂的网络关系,并挖掘其中隐藏的模式和规律。
最后,需要通过实验证实预测的结果,验证其准确性和可靠性。
近年来,许多研究者通过整合和分析大量的蛋白质互作数据,开发了一系列功能预测算法和工具。
蛋白质互作网络分析在疾病发病机制和药物靶点发现中的应用

蛋白质互作网络分析在疾病发病机制和药物靶点发现中的应用近年来,随着生物学、生物信息学和计算机技术的飞速发展,蛋白质互作网络分析成为一种重要的方法,可以帮助人们深入了解蛋白质之间的互作关系,并揭示其在生物学中的重要作用。
在疾病发病机制和药物靶点发现中,蛋白质互作网络分析也得到了广泛应用。
一、蛋白质互作网络分析的基础蛋白质互作网络是由多个蛋白质相互作用组成的复杂网络模型,其中蛋白质和蛋白质之间通过复杂的互作关系形成网络。
蛋白质互作网络分析主要通过挖掘蛋白质结构、功能和相互作用等信息,来深入了解蛋白质之间的互作关系,并揭示其在生物学中的重要作用。
二、蛋白质互作网络分析在疾病发病机制中的应用1、疾病基因鉴定蛋白质互作网络分析可以帮助人们确定患者患有的疾病与哪些基因相关联,从而对疾病进行准确的诊断和治疗。
这种方法已被广泛应用于多种疾病的研究中,例如乳腺癌、帕金森病、肝癌和白血病等。
同时,通过分析蛋白质互作网络,人们还能够找到潜在的新的致病基因,为研究和治疗疾病提供新的思路和方法。
2、药物靶点发现蛋白质互作网络分析可以帮助人们发现新的药物靶点,从而为药物研发提供新的思路和方法。
通过分析蛋白质之间的互作关系,可以找到具有重要生物学功能的关键蛋白质,进而研究它们的结构和功能特点,为药物靶点的开发提供有力支持。
三、蛋白质互作网络分析在药物研发中的应用1、药物靶点鉴定和优化蛋白质互作网络分析可以帮助人们确定药物的靶点,进而预测药物的效果和安全性。
同时,通过分析蛋白质之间的互作关系,可以找到靶点的关键残基,从而设计优化更有效的药物。
2、药物作用机制研究蛋白质互作网络分析可以帮助人们研究药物的作用机制,揭示药物与蛋白质之间的相互作用关系。
通过分析药物与蛋白质之间的互作关系,可以发现药物靶点的结构和功能特点,进而优化药物的结构和活性,提高药物的疗效。
四、蛋白质互作网络分析面临的挑战和展望蛋白质互作网络分析在疾病发病机制和药物靶点发现中的应用不仅拓展了我们对生物学的认识,也提供了新的思路和方法来研究和治疗疾病。
阿尔兹海默症的病理生理学变化与疾病机制

阿尔兹海默症的病理生理学变化与疾病机制阿尔茨海默症(Alzheimer's disease)是一种常见的神经退行性疾病,其主要特征是进行性的认知功能障碍与记忆受损。
本文旨在探讨阿尔茨海默症的病理生理学变化与疾病机制。
通过对病理学异常变化的深入研究,我们可以更好地理解该疾病的进展和治疗方法的探索。
1. 蛋白质异常沉积阿尔茨海默症的主要特征是脑内β-淀粉样蛋白(β-amyloid)斑块和Tau蛋白异常沉积。
β-淀粉样蛋白在正常情况下通过蛋白质代谢路径得到清除,但在阿尔茨海默症患者中,其过度聚集形成斑块。
这些斑块的积累会刺激神经元炎症反应和氧化应激,导致神经元的损伤和死亡。
另一方面,Tau蛋白的异常磷酸化会导致其在神经元内异常聚集形成神经原纤维缠结,进一步加剧病理学改变。
2. 神经递质异常在阿尔茨海默症患者脑组织中,乙酰胆碱水平显著下降。
乙酰胆碱是一种重要的神经递质,与学习记忆等认知功能密切相关。
其丢失会导致神经元之间的通信障碍,进而影响到认知能力。
此外,谷氨酰胺和多巴胺等其他神经递质也可能与阿尔茨海默症的发病机制有关,但具体机制尚不完全清楚。
3. 炎症和免疫反应在阿尔茨海默症患者的大脑中,可以观察到神经元周围胶质细胞的活化和炎症反应。
这些炎症细胞产生促炎细胞因子,如肿瘤坏死因子α(TNF-α)等,进一步激活炎症反应。
免疫细胞也参与了阿尔茨海默症的发展过程,如导致神经元损伤的β-淀粉样蛋白诱导B细胞和T细胞的异常激活。
4. 氧化应激和线粒体功能障碍氧化应激是指细胞内活性氧(ROS)生成过多而导致的一种紊乱状态。
在阿尔茨海默症患者中,由于β-淀粉样蛋白斑块的沉积和炎症反应的增加,氧化应激水平显著升高。
氧化应激可以引发线粒体功能障碍,导致能量代谢紊乱,进而损伤神经元。
此外,纤维化和神经元的线粒体DNA损伤也是阿尔茨海默症病理生理学变化的重要组成部分。
5. 基因和遗传风险阿尔茨海默症具有一定的遗传倾向,APOE基因ε4等多个基因缺陷与该疾病的发病风险密切相关。
生物信息学中的蛋白质相互作用预测研究

生物信息学中的蛋白质相互作用预测研究蛋白质相互作用在生物学中起着至关重要的作用。
它们在细胞信号传递、代谢调控、基因调控等生物过程中发挥着调控和协调等功能。
蛋白质相互作用预测是生物信息学领域的重要研究方向之一,其目的是准确预测蛋白质之间的相互作用,从而揭示细胞内复杂的分子网络。
为什么研究蛋白质相互作用预测如此重要?首先,蛋白质的相互作用对于疾病的发生和发展具有重要影响。
许多疾病的发生是由于蛋白质相互作用异常引起的。
例如,癌症的发展常常涉及异常的细胞信号传递和调控。
其次,蛋白质相互作用预测可以帮助科学家理解细胞内多种生物过程背后的机制。
最后,蛋白质相互作用预测有助于发现新的药物靶点和治疗方法。
通过预测蛋白质相互作用,科学家可以设计靶向这些相互作用的新药物,从而提高药物的疗效和减少副作用。
那么,如何进行蛋白质相互作用预测呢?目前,常用的预测方法可以分为实验方法和计算方法两类。
实验方法是通过实验手段直接检测蛋白质相互作用的发生。
例如,双杂交技术(yeast two-hybrid)、免疫沉淀、共沉淀等实验手段被广泛应用于蛋白质相互作用研究中。
这些方法可以从全局或局部角度鉴定蛋白质间的相互作用关系。
然而,实验手段的繁琐和昂贵使得其在大规模蛋白质相互作用预测中受到限制。
计算方法是利用生物信息学技术进行蛋白质相互作用预测的一种快速和经济的策略。
计算方法主要分为基于序列和基于结构的方法。
基于序列的预测方法主要是依据蛋白质序列之间的相似性,通过比对和模式识别等手段来推断蛋白质之间的相互作用。
这些方法包括蛋白质相似性搜索算法、蛋白质特征提取和机器学习等方法。
基于序列的方法具有计算复杂度低和适用于大规模预测等优点。
然而,这些方法在准确性和可靠性上有一定的限制,特别是当蛋白质序列相似度低时。
基于结构的预测方法则利用蛋白质的三维结构信息推断蛋白质之间的相互作用。
这些方法主要包括结构基因组学和分子模拟等技术。
结构基因组学方法通过对比蛋白质结构中的相互作用模式来推断未知蛋白质之间的相互作用。
蛋白质相互作用预测方法的研究

蛋白质相互作用预测方法的研究蛋白质相互作用预测是生物信息学领域的重要问题之一。
蛋白质之间的相互作用在生物体内发挥着至关重要的作用,与许多疾病的发生和发展密切相关。
因此,预测蛋白质之间的相互作用对于理解生物过程和药物研发具有重要意义。
本文将介绍常用的蛋白质相互作用预测方法及其优缺点,并讨论未来的研究方向和展望。
蛋白质是生命活动的基本单位,其相互作用在细胞信号转导、代谢调节和疾病发生等方面起着至关重要的作用。
因此,预测蛋白质之间的相互作用对于理解生物过程和疾病治疗具有重要意义。
随着生物技术的发展,蛋白质相互作用预测方法已经成为生物信息学领域的研究热点之一。
该方法主要是利用基因组学和进化学分馏技术,寻找与目标蛋白质相互作用的蛋白质。
具体实现过程包括以下几个步骤:通过基因组学方法确定目标蛋白质的基因序列;利用进化学分馏技术对该基因序列进行分馏,得到进化树;根据进化树上的信息,确定与目标蛋白质相互作用的蛋白质。
该方法的优点是可以找到与目标蛋白质相互作用的潜在蛋白质,缺点是需要大量的计算资源和时间。
该方法主要是通过分析蛋白质的相互作用口袋,预测不同蛋白质之间的相互作用。
相互作用口袋是指蛋白质在相互作用时暴露出来的疏水性氨基酸口袋,可以通过计算蛋白质表面氨基酸的亲/疏水性比值和溶剂可及性来进行预测。
该方法的优点是可以较为准确地预测蛋白质之间的相互作用,缺点是需要手动设定口袋特征和机器学习模型,且对于未知蛋白质之间的相互作用难以预测。
为了评估上述两种预测方法的准确性和可靠性,我们采用已知的蛋白质相互作用数据集进行实验。
实验结果表明,基于相互作用口袋的分析方法相比基于基因组学的方法具有更高的预测准确性和可靠性。
具体来说,基于相互作用口袋的分析方法对于已知蛋白质相互作用的预测准确率可以达到70%,而基于基因组学的预测方法准确率仅为40%。
本文介绍了常用的蛋白质相互作用预测方法及其优缺点,并对其准确性和可靠性进行了实验评估。
基于网络药理学预测白藜芦醇治疗阿尔茨海默症的关键潜在靶点

基于网络药理学预测白藜芦醇治疗阿尔茨海默症的关键潜在靶点田晓燕 江思瑜 张睿 许顺江 李国风*【摘要】目的通过网络药理学预测白藜芦醇(resveratrol,RSV)治疗阿尔茨海默症(Alzheimer's disease,AD)的关键靶点。
方法 利用TCMSP数据库检索含RSV的中药,并对其性味、归经和功效进行归纳分析。
利用SwissTargetPrediction、SEA、HERB数据库预测RSV作用靶点;利用GeneCards、OMIM、TTD、DisGeNRT 数据库检索AD靶点;取RSV的作用靶点与AD靶点的交集为潜在治疗靶点。
利用DAVID数据库进行潜在治疗靶点的GO分析。
利用STRING数据库获取潜在治疗靶点的KEGG富集分析和蛋白质交互作用(protein-protein interaction, PPI),并用Cytoscape绘制PPI网络图。
AlzData数据库验证AD关键靶点变化。
SwissDock网站对RSV与关键蛋白进行分子对接。
结果含RSV中药的性味为苦味最多;归经中入肝经最多;功效中清热解毒功效最多。
RSV预测靶点388个,AD靶点1624个,交集靶点119个。
KEGG富集通路中的阿尔兹海默症通路共富集到27个蛋白。
AlzData数据库分析发现AD患者表达发生变化的蛋白。
分子对接结果发现,RSV与丝氨酸/苏氨酸激酶(serine/threonine kinase 1, AKT1)、白介素-6(interleukin-6, IL-6)、连环蛋白-1(β-catenin, CTNNB1)、肿瘤坏死因子(tumor necrosis factor, TNF)均有较好的结合能力。
结论网络药理分析结果显示RSV对AD的治疗是多靶点、多通路的,可为后续研究方向提供参考。
【关键词】 网络药理学;白藜芦醇;阿尔兹海默症;分子对接中图分类号 R285文献标识码 A 文章编号1671-0223(2023)24-1879-08Predicting the key potential targets of resveratrol in the treatment of Alzheimer's disease based on network pharmacology Tian Xiaoyan, Jiang Siyu, Zhang Rui, Xu Shunjiang, Li Guofeng. Chengde Medical University, Chengde 067000, China【Abstract】Objective Key targets of resveratrol (RSV) in the treatment of Alzheimer's disease (AD) are predicted by network pharmacology. Methods The traditional Chinese medicines which contain RSV were searched by the TCMSP database, and their property and flavor, meridian distribution and phamacologic action were summarized and analyzed. The targets of RSV were predicted by SwissTargetPrediction, SEA and HERB databases. The targets of AD were retrieved using GeneCards, OMIM, TTD and DisGeNRT databases. The intersection targets of RSV and AD were taken as the potential therapeutic targets.Analysis gene ontology (GO) annotations of potential therapeutic targets by biological information annotation database (DAVID). Did KEGG cluster analysis and protein interactions (PPIs) of potential therapeutic targets in STRING database, and mapped PPI networks in Cytoscape. Verified changes of AD key targets in AlzData database.Docking RSV and key proteins in SwissDock website. Results The most Tropism of taste of the traditional Chinese medicines that contain RSV: bitter, cold, in the liver. And the main phamacologic action is clearing away heat and toxic materials.There are 388 predicted targets of RSV,1624 targets of AD, 119 intersection targets. Alzheimer's pathway in KEGG enriched pathway was enriched to 27 proteins. The proteins which expression changed of AD patients was analysised in AlzData database. The results of molecular docking showed that RSV had good binding ability with AKT1, IL-6, CTNNB1 and TNF. Conclusion The results of network pharmacological analysis show that the treatment of AD by RSV is multi-target and multi-pathway, which can provide reference for subsequent research directions.【Key words】 Network pharmacology; Resveratrol; Alzheimer's disease; Molecular docking作者单位:067000 河北省承德市,承德医学院研究生学院 (田晓燕、李国风);河北医科大学第一医院中心实验室(江思瑜、张睿、许顺江);河北省疾病预防控制中心药物研究所(李国风)*通讯作者现代科学研究认为,阿尔兹海默症(Alzheimer disease,AD)是一种不可逆的退行性神经疾病,临床上多以记忆力障碍、执行能力障碍以及人格变化等为特征,是老年性痴呆的最主要因素。
蛋白质相互作用的预测方法

蛋白质相互作用的预测方法蛋白质相互作用在生物体内发挥着至关重要的作用,它们参与调控细胞的生长、分化和凋亡等过程。
预测蛋白质相互作用有助于揭示生命现象的本质,为疾病诊断和治疗提供理论依据。
本文将详细介绍几种蛋白质相互作用的预测方法。
一、基于生物信息学的方法1.同源比对法:该方法通过比较不同物种中同源蛋白质的相互作用信息,预测目标物种中蛋白质的相互作用。
同源比对法具有较高的准确性,但受限于已知相互作用数据的多少。
2.融合蛋白质组学法:该方法将多个蛋白质相互作用数据集进行整合,通过概率模型预测蛋白质相互作用。
融合蛋白质组学法可以提高预测的准确性,但计算复杂度较高。
3.神经网络法:神经网络法通过学习已知相互作用数据,构建预测模型,对未知相互作用进行预测。
该方法具有较高的预测准确性,但需要大量训练数据。
二、基于实验的方法1.酵母双杂交法:该方法利用酵母细胞中的转录激活因子,检测两个蛋白质是否能够相互作用。
酵母双杂交法操作简单,但可能存在假阳性。
2.免疫共沉淀法:通过特异性抗体捕获目标蛋白质,检测与其相互作用的蛋白质。
免疫共沉淀法具有较高的可靠性,但实验操作复杂。
3.分子对接法:该方法利用计算机模拟技术,预测两个蛋白质之间的结合模式。
分子对接法可以预测蛋白质之间的相互作用强度和结合位点,但计算过程耗时较长。
三、综合方法综合方法将多种预测方法进行融合,以提高预测准确性。
例如,将生物信息学方法与实验方法相结合,利用实验数据校正预测模型,从而提高预测的可靠性。
总结:蛋白质相互作用预测方法众多,各有优缺点。
在实际应用中,研究者可根据具体需求选择合适的方法,或结合多种方法提高预测准确性。
阿尔兹海默症基本机制-概述说明以及解释

阿尔兹海默症基本机制-概述说明以及解释1.引言1.1 概述阿尔茨海默症(Alzheimer’s disease)是一种慢性进行性脑部退化性疾病,是老年痴呆的最常见形式。
它以认知和记忆功能的丧失为主要表现,进一步导致个体的日常生活能力丧失。
随着世界人口老龄化的加剧和平均寿命的增加,阿尔茨海默症已成为全球重大公共卫生问题之一。
尽管已经有多年的研究,但阿尔茨海默症的确切病因和发病机制仍然不完全清楚。
阿尔茨海默症的发病机制是多因素复杂的,其中包括遗传、环境和生活方式等多种因素共同作用。
大多数阿尔茨海默症患者患有早发性阿尔茨海默症,这种病情通常与基因突变有关。
APOE基因的ε4等位基因与阿尔茨海默症的发病风险显著相关。
此外,一些环境因素如心血管疾病、高血压、高胆固醇和糖尿病等也被认为与阿尔茨海默症的发病有关。
在神经病理学上,阿尔茨海默症的主要特征是β淀粉样蛋白斑块和tau 蛋白神经纤维缠结的形成。
这些异常蛋白聚集在大脑中特定的区域,如海马和顶叶皮质,进而导致神经元功能紊乱和死亡。
此外,炎症反应和神经递质的紊乱也与阿尔茨海默症的发病机制密切相关。
对于阿尔茨海默症机制的深入了解,不仅有助于揭示其病因学和发病机制,还为阿尔茨海默症的治疗提供了新的思路。
目前,许多研究致力于寻找针对阿尔茨海默症的疾病修复和干预的方法,以改善患者的生活质量。
未来,我们需要进一步研究阿尔茨海默症的基本机制,寻找更精确的诊断标志物和治疗策略,以缓解这个全球性挑战带来的健康、社会和经济压力。
1.2 文章结构文章结构部分的内容应该包括对整篇文章的结构进行简要介绍,让读者能够了解该篇文章的组成部分和篇章安排。
文章结构部分的内容可以参考以下例子进行编写:在本篇长文中,将介绍阿尔兹海默症的基本机制。
文章主要分为引言、正文和结论三个部分。
引言部分将首先给出对阿尔兹海默症的概述,包括其定义和特征。
接下来,将简要说明本文的结构和各个部分所涉及的内容。
最后,将明确本文的目的,为读者提供文章阅读的指导和预期结果。
蛋白质相互作用网络的构建与分析

蛋白质相互作用网络的构建与分析蛋白质相互作用是细胞内重要的分子交互方式,在维持生命活动和调控生物系统中起着重要的作用。
蛋白质相互作用网络是研究蛋白质功能和互作机制的重要工具。
本文将介绍蛋白质相互作用网络的构建与分析的方法和应用。
一、蛋白质相互作用网络的构建方法1. 实验方法实验方法是构建蛋白质相互作用网络的重要手段。
传统的实验方法包括酵母双杂交、共免疫沉淀等。
酵母双杂交方法利用酵母细胞中两个融合蛋白之间的相互作用来识别蛋白质间的相互作用关系。
共免疫沉淀方法通过抗体特异性地沉淀目标蛋白及其相互作用的蛋白,从而实现蛋白质间的相互作用检测。
2. 预测方法预测方法是通过计算模型来预测蛋白质间的相互作用。
常用的预测方法包括基于结构预测、基于序列预测和基于机器学习的方法。
结构预测方法通过模拟蛋白质的结构和动态过程来预测蛋白质间的相互作用。
序列预测方法通过分析蛋白质序列中的保守特征来预测蛋白质的相互作用。
机器学习方法则是通过从已知相互作用数据中学习模式,来预测未知蛋白质间的相互作用。
二、蛋白质相互作用网络的分析方法1. 网络可视化与分析网络可视化是将蛋白质相互作用网络以图的形式展现出来,便于直观地观察和分析网络中的相互作用关系。
常用的网络可视化工具包括Cytoscape、Gephi等。
通过网络可视化,可以发现网络中的关键节点和模块,进而研究蛋白质间的相互作用机制。
2. 功能注释与富集分析功能注释与富集分析是对蛋白质相互作用网络中的蛋白质进行功能注释和功能富集分析的方法。
通过将蛋白质与已知的功能数据库进行比对和匹配,可以了解蛋白质的功能及其在生物过程中的作用。
同时,利用富集分析方法可以发现网络中富集的功能模块,从而揭示蛋白质相互作用网络的生物学意义。
3. 拓扑分析与网络特征拓扑分析与网络特征是研究蛋白质相互作用网络中拓扑结构和关键节点的方法。
通过计算网络中节点的度、聚集系数、介数中心性等网络特征,可以揭示网络的拓扑结构特征和关键节点。
基因调控网络和蛋白质相互作用网络的构建及其生物学意义

基因调控网络和蛋白质相互作用网络的构建及其生物学意义基因调控网络和蛋白质相互作用网络是生命科学领域中的两个重要概念。
它们的构建是帮助我们理解和探究生命现象的重要手段。
同时,也从不同层次帮助我们理解细胞和生物的复杂机制。
本文将讨论这两个概念及其生物学意义。
基因调控网络(Gene Regulatory Network, GRN)是指由一组调控基因表达的信号通路组成的网络。
基因表达的过程中,会产生许多RNA和蛋白质,从而影响细胞的各种功能。
这一过程涉及到上游因子、转录调节因子、miRNA等,构成一个复杂的调控网络。
GRN的构建能够帮助我们了解个体发育、生长、分化、细胞命运决策等生物学过程。
GRN构建的关键在于了解基因之间的相互作用和调节,这就需要对基因表达过程的生物学机制有深入的理解。
一些高通量实验技术,如转录组测序和染色质免疫共沉淀(ChIP),可以用来确定基因调控关系。
同时,一些数学模型也可以用来描述和预测GRN中基因之间的丰富关系。
通过对基因调控网络的重构和分析,生物学家们可以更好地了解基因之间的相互作用,深入探究细胞与生命现象的本质。
与GRN不同的是,蛋白质相互作用网络(Protein-Protein Interaction Network, PPI)是描述蛋白质之间相互作用的一类网络。
蛋白质是生物体内大量功能重要的分子。
除了直接参与信号传导、代谢、转录等生物学过程外,它们也能够相互作用,形成复杂的PPI网络。
这种网络的构建有助于探究不同蛋白质之间的作用、类比和相似性,以及生物体内各种物质代谢、信息传递方式的本质。
在构建PPI网络时,需要在实验数据和计算模型的帮助下,确定蛋白质之间的相互作用。
现有的PPI网络,可以帮助研究人员更好地了解生物体内各种复杂的生物学过程,如信号传递、代谢调节、组织发生与分化等。
同时,PPI网络也可以用于构建分析因果鉴别模型,来预测蛋白质复合物及其功能,并预测新型药物及其作用机制,具有广泛的应用前景。
生命科学中的蛋白质互作网络分析

生命科学中的蛋白质互作网络分析在生物学研究中,互作网络分析(Interactome Network Analysis)被广泛应用于解释蛋白质互作的复杂性质和功能。
蛋白质互作网络是指由蛋白质相互作用所形成的图(Graph)结构,其中每个蛋白质作为一个节点,蛋白质之间的互作作为边进行表示。
通过对这些互作关系的分析,我们可以深入了解蛋白质之间的相互作用和功能。
这对于揭示疾病发生机制、发现新的药物靶点和开发治疗方法有着重大意义。
蛋白质互作网络分析的基本方法包括:网络构建、网络拓扑分析、蛋白质功能注释和功能模块发现。
1. 网络构建互作网络的构建是网络分析的首要步骤。
目前,蛋白质互作数据的来源主要有两种:实验数据和基于计算的预测数据。
实验技术包括酵母双杂交、免疫共沉淀、质谱法等。
计算的预测技术使用蛋白质序列数据和结构信息来预测蛋白质互作关系。
构建的网络数据集不但要全面、准确,还应具有一定的可重复性和可比性。
2. 网络拓扑分析网络拓扑分析主要涉及网络的结构和特性:节点度数、网络密度、平均路径长度、聚类系数、介数中心性和特异性等。
节点度数表示某一个蛋白质与其他蛋白质互作的数量,它是一个节点在网络中重要性的一个潜在指标;网络密度表示网络中节点之间的连接紧密程度;平均路径长度是指整个网络中所有节点之间的平均路径长度;聚类系数表示网络中节点之间的聚类程度,即它们互相之间互联的概率;介数中心性则表示一个节点作为经过所有网络上路径的桥梁的可能性。
特异性反映了一个蛋白质在其互作伙伴之间的特别地位。
他们的结合可能是高度特异的,而不是随机的。
网络拓扑分析是互作网络研究的核心部分,它可以帮助我们深入了解蛋白质互作网络的性质和规律。
3. 蛋白质功能注释蛋白质功能注释是对每一个蛋白质进行功能分类的过程。
可以根据蛋白质的序列和结构特征来预测其功能,并根据蛋白质在网络中的拓扑特征,如度数和特异性等,进一步预测和验证其功能。
目前,蛋白质功能分类主要是基于Gene Ontology(GO)数据库进行的。
疾病相关蛋白质的折叠与聚集

疾病相关蛋白质的折叠与聚集近年来,越来越多的科学家们开始探究疾病相关蛋白质的折叠与聚集机制。
这类蛋白质在人体中很普遍,它们的正常功能对于人体的生命活动至关重要。
但是,当它们出现异常时,就会引发一系列的疾病。
因此,深入了解这些蛋白质的折叠与聚集机制,对于疾病的预防和治疗有着重要的意义。
一、疾病相关蛋白质的折叠蛋白质是组成人体的基本物质之一,它对人体的生命活动有着巨大的影响。
蛋白质的折叠是指蛋白质在生物体内摆动、振动、扭曲和旋转等过程中,由其自身的物理和化学性质自动形成的一个空间结构。
这个结构决定了蛋白质的功能和相互作用。
如果蛋白质能够正确地折叠,那么它就可以发挥正常的功能。
如果折叠错误,那么蛋白质就没有正常的功能,甚至会产生危害。
例如,阿尔茨海默病(Alzheimer's disease)是一种神经系统疾病,它的发病机制与蛋白质的折叠错误有关。
阿尔茨海默病患者脑结构中出现了β淀粉样蛋白物(β-amyloid)的团块,这些团块会堵塞神经元之间的通信管道,导致神经元逐渐死亡。
研究表明,β淀粉样蛋白物最初存在于单体状态,然后通过积累、聚集和变形,逐渐转化为稳定的草酸盐形态。
这个过程中,随着蛋白质的折叠错误不断积累,团块的大小和数量不断增加,最终导致阿尔茨海默病的发生和发展。
二、疾病相关蛋白质的聚集除了折叠错误之外,疾病相关蛋白质的聚集也是引发疾病的另一个主要原因。
一些疾病相关蛋白质会在细胞内形成异常的集聚物,如聚集、纤维或原花生球蛋白(amyloid fibril)等,这些集聚物对细胞和身体健康产生了很大的不利影响。
例如,帕金森病(Parkinson's disease)就是与蛋白质聚集有关的疾病之一。
帕金森病患者脑内黑质的细胞退化,导致了一系列运动功能失调的症状,如震颤、僵硬、动作迟缓等。
研究发现,帕金森病患者脑内有α-突触核蛋白(α-synuclein)的团块。
这些团块导致麻烦的、复杂的即刻复制过程,即聚集依赖的蛋白质聚集(Aggregate-dependent neuronal cell death)。
蛋白质折叠异常与阿尔茨海默病的关系

蛋白质折叠异常与阿尔茨海默病的关系在当今的医学研究领域,阿尔茨海默病(Alzheimer's disease,简称AD)一直是备受关注的焦点。
这是一种渐进性的神经退行性疾病,其特征表现为认知能力下降、记忆力减退以及行为和人格的改变。
随着全球老龄化进程的加速,AD 患者的数量不断增加,给家庭和社会带来了沉重的负担。
虽然目前对于 AD 的病因和发病机制尚未完全清楚,但越来越多的研究表明,蛋白质折叠异常在 AD 的发生和发展中扮演着至关重要的角色。
要理解蛋白质折叠异常与 AD 的关系,首先需要了解什么是蛋白质折叠。
蛋白质是生命活动的重要执行者,它们由氨基酸组成。
在细胞内,新合成的蛋白质需要通过折叠形成特定的三维结构,才能发挥其正常的生理功能。
这个折叠过程受到多种因素的精确调控,包括分子伴侣、环境条件等。
如果蛋白质在折叠过程中出现错误,就可能导致异常的结构和功能,从而引发一系列疾病。
在 AD 患者的大脑中,有两种标志性的蛋白质病变:β淀粉样蛋白(Amyloidβ,Aβ)斑块和tau 蛋白缠结。
Aβ 是由淀粉样前体蛋白(Amyloid Precursor Protein,APP)经过一系列酶切反应生成的。
正常情况下,APP 会被切割成无害的片段。
然而,在某些因素的影响下,APP 的加工过程出现异常,产生大量的Aβ 。
Aβ 具有很强的聚集倾向,它们会错误折叠并聚集形成寡聚体和纤维状的斑块,沉积在大脑的神经元之间,干扰神经元的正常功能。
Tau 蛋白是一种微管相关蛋白,在正常情况下,它能够稳定神经元的微管结构,促进物质运输和信息传递。
但在 AD 患者的大脑中,tau蛋白会过度磷酸化,导致其折叠异常,失去与微管的结合能力,并聚集形成神经纤维缠结,进一步破坏神经元的结构和功能。
那么,是什么导致了这些蛋白质的折叠异常呢?目前的研究认为,遗传因素、环境因素以及细胞内的氧化应激等都可能参与其中。
一些基因突变,如APP、早老素1(Presenilin 1)和早老素2(Presenilin 2)等基因的突变,会增加Aβ 的产生和聚集。
蛋白质聚集机制对阿尔茨海默病发展影响因素分析

蛋白质聚集机制对阿尔茨海默病发展影响因素分析阿尔茨海默病(Alzheimer's disease,AD)是一种神经系统退行性疾病,通常表现为认知功能的退化和记忆力的丧失。
随着人口老龄化趋势的加剧,AD已成为全球负担最重的疾病之一。
研究表明,蛋白质的异常聚集是AD发展过程中的一个关键因素。
在AD患者的大脑中,淀粉样β-蛋白(Aβ)和tau蛋白是主要的蛋白质聚集物。
这些异常聚集物在形成“斑块”和“缠结”等结构时,会干扰神经元的正常功能,导致细胞死亡和神经网络连接的破坏。
首先,影响蛋白质聚集机制的因素之一是突变。
AD的早起病例中约有5%与基因突变有关,这些突变主要影响Aβ前体蛋白的切割和清除通路,导致Aβ产生过剩和聚集。
蛋白质控制质押(proteostasis)的突变也可能导致蛋白质的异常聚集,进而影响AD的发展。
此外,炎症反应也被认为是影响蛋白质聚集机制的重要因素。
炎症反应是机体对损伤或感染的自我保护反应,但过度的炎症反应会导致蛋白质处理机制的紊乱,破坏蛋白质的清除和降解过程。
研究显示,过度激活的炎症反应会刺激Aβ的产生和聚集,促进AD的发展。
此外,氧化应激也被认为是影响蛋白质聚集机制的重要因素之一。
氧化应激是指体内自由基和氧化物质积累过多,超过体内抗氧化能力的情况。
氧化应激可以引起细胞内蛋白质的结构和功能异常,进而导致蛋白质的异常聚集。
一些研究发现,氧化应激可增加tau蛋白的磷酸化和聚集,加速AD的发展。
此外,蛋白质的清除和降解通路的紊乱也是AD发展过程中的重要因素。
研究发现,酶体和自溶酶体途径的功能障碍会导致蛋白质的异常聚集和聚集物的堆积。
此外,自噬途径的紊乱也与AD的发展密切相关。
自噬是细胞中蛋白质降解的主要途径之一,它通过降解蛋白质聚集物来保持细胞内蛋白质正常水平。
然而,在AD的早期过程中,自噬途径可能受到抑制,导致蛋白质的异常聚集。
最后,环境因素和生活方式也对蛋白质聚集机制和AD的发展起到重要的影响。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
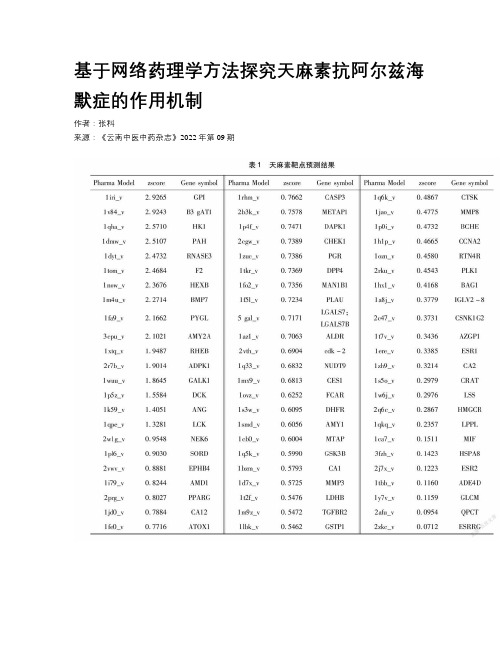
第42卷 第5期2006年9月南京大学学报(自然科学)JOURNAL OF NANJING U NIVERSITY(NAT URA L SCIENCES)Vo l.42,No.5Sept. 2006Alzheimer s疾病相关蛋白质相互作用网络构建及其相互作用预测*蒋雄飞1,2,杨 洁1**,王 炜2(1.南京大学生物医药技术国家重点实验室,南京,210093;2.南京大学物理系,南京,210093)摘 要: 依据无标度网络的相关理论,提出一种预测蛋白质-蛋白质相互作用的算法,并预测潜在的作用位点。
收集了所有与阿尔兹海默疾病(A lzheimer s disease)相关疾病的人类蛋白,并找出与之能发生相互作用的蛋白质,绘制一个有关人类AD相关蛋白质相互作用网络。
分析和计算了该网络的一些详细属性,并对网络进行生物信息学分析,找出网络中的蛋白质调控路径,该路径可能涉及到Apoptosis antag onizing tr anscript ion factor(A A T F)蛋白对于阿尔兹海默疾病的抑制作用。
几个潜在与AA T F蛋白作用的位点被自己开发的程序预测出,将它们的潜在作用位点做了比对,最后得到一个高度收敛的序列模体,其正则表达式为I-x(0,1)-E-x(2)-[A EN T]-x-[EK].关键词: 阿尔茨海默氏病,蛋白质相互作用,复杂网络,无标度网络,相互作用预测,序列模体中图分类号: Q615The Complex Network of Protein Protein Interaction of Alzheimer s Disease Associated Proteins and an Interaction PredictingJ iang X iong F ei1,2,Yang J ie1,Wang Wei2(1.State Key L abo rato ry o f Pharmaceutical Bio technolog y,Nanjing U niver sity,N anjing,210093,China; 2.D epar tment o f Physics,Nanjing U niver sity,N anjing,210093,China)Abstract: Pr oteins potentially asso ciated w ith the patholog y o f A lzheimer s disease w ere g ather ed into o ur database,and w ere then mapped into a pr otein inter action netw or k.H ere,w e repor ted a nov el appro ach to predict pr otein pro tein interactio ns and their po tential inter action sit es,based on the character istics of t he free scale netw or k.M eanwhile,t he det ailed pr operties of netwo rk wer e analyzed,some pro teins meditative pathw ays wer e also fo und,which may be invo lv ed in the antago nizat ing P AW R mediated induction o f aber rant amy lo id peptide pr oduction in Alzheimer s disease.T hen sever al potential pro tein interactio ns and their sites were predicted thro ug h pr og ram compiled by o ur selves.O ne interactio n betw een O96018and P05067confir med in ex periment w as r e pr edicted thro ug h the P PIP.Pending pro teins pr edicted to interact with A po pto sis antag onizing transcription factor pro tein w ith hig h po ssibilit y w ere picked o ut,and their potential interactio n sequences wer e in alig nment.A well conservat ive pattern* **基金项目:国家自然科学基金(30171094,30271497)收稿日期:2006-07-01通讯联系人,E mail:yangjie@n (mot if)ma ybe invo lv ed in mo lecular mechanism of interactio n w ith A poptosis antago nizing transcriptio n factor pr otein w as g enerated,w ith a reg ular ex pression of I-x(0,1)-E-x(2)-[A ENT]-x-[EK]Key words: Alzheimer s disease,pr otein inter act ion,com plex netwo rk,free scale netw or k,int eractio n predictio n,motif阿尔茨海默氏病是中枢神经系统退行性疾病中的一种,病理表现主要为神经元纤维缠结老年斑和颗粒空泡变性,其内在机制到目前尚不非常清楚.但很显然,它的产生是由于某些蛋白质异常行为而引起的[1~3],很多报道指出其与 淀粉样蛋白(APP)密切相关[4,5].到目前为止,生物学研究一直侧重在对单个蛋白的研究,即研究蛋白个体的特征及能够和其发生相互作用的其他蛋白。
现面临的课题是如何将这些已研究过的基于单体的实验信息整和起来,以更为系统和全面的眼光来看待生物体内在分子水平上的相互作用及其调控路径.为此,我们选择以复杂网络作为工具来研究蛋白分子水平上的整体特征.对于蛋白质相互作用网络的研究不仅具有理论上的重要意义,也具有潜在的实际应用前景.在新药研发时,它可使药物指向特定靶点,对网络进行相应的调控或者阻碍操作,而取代以往简单的以给定的蛋白质功能为目标靶点.对于细胞系统明显的网络特性和代谢途径中普遍存在的瓶颈步骤,当前发现药物方法都仍依赖于对确定的单个目标进行破坏,这本身就有局限性[6].迄今为止,有500种人类的蛋白已被确认可作为药物作用的靶子,这些蛋白代表了基因功能和更高水平的细胞表型之间直接的一对一关系[7].同时,预测蛋白的高级结构及其功能一直是计算生物学中的一个核心的问题,一套好的预测方法能给实验人员以有效的理论指导,如何预测蛋白之间是否存在着相互作用也是其中一项任务,很多预测两个蛋白或者亚基契合的算法被开发出来,比如Jones和Thornton曾使用包括疏水性、表面区域(accessible surface area)、形状以及残基偏好性等来比对结构特性[8].近来的研究集中在使用残基疏水性来预测相互作用的接触位点,如Kor n和Burnett使用疏水分析来预测在二聚体蛋白相互作用中的作用表面位置[9],Yo ung进一步发展他们的方法,提出基于蛋白簇残基(clusters o f residues)疏水分析的自动预测算法[10].根据Anfinsen的蛋白质序列决定其高级结构理论,在蛋白质之间存在着相互作用时,可以看作是两个蛋白的结合区域的三维结构正好相契,所以当有另一蛋白的某一区域的三维结构与其中某一蛋白的结构相似,那么,它就很有可能与另一蛋白发生作用.以此思想为基础,通过比对,即可能从已有的相互作用序列片断中找出未知蛋白是否可能与靶蛋白发生作用,若有,可计算出大致发生的位置.以此方法,我们开发程序预测未知蛋白会与某些AD (A lzheimer s disease)相关蛋白发生作用,从而将其纳入人类AD相关蛋白相互作用网络.1 蛋白质相互作用网络1.1 构建蛋白质相互作用网络 首先,有关所有中枢神经系统退行性疾病的相关蛋白被收集起来,建立CNSNDD数据库(Central Nerv ous System N eurodeg enerative Diseases Database),而AD则是CN S退行性疾病中典型的一种疾病.CNSNDD中的蛋白相互作用数据来源于IntA ct[11],基于网络规模,考虑三度以内的蛋白,数据均可以在CNSNDD方便的查询到.为更有效的分析网络性质,在开始只选取与AD相关的人类蛋白构建网络,未推广至所有的模式动物蛋白.一共确定119个蛋白与480南京大学学报(自然科学) 第42卷AD 相关,其中有70个人类蛋白.以该70个蛋白作为骨架,获取其三度以内的相互作用蛋白,最后构建了人类AD 相关蛋白相互作用网络.利用可视化网络构建软件,将整个网络图画出来.如图1所示.深色点均是70个CNSNDD 中的部分蛋白,都已在实验上证实与AD 的发病机理有关,其中圆点代表引起或促进AD 发病的蛋白,三角形点代表抑制AD 发病的蛋白;而浅色点表示它们暂时仅仅确定是深色点的相互作用蛋白,而没有包含于CNSNDD 数据库中,即尚未在实验上发现与AD 的发病有关联。
由于实验的限制,在该70个蛋白中,还有部分蛋白尚无相互作用蛋白,而无法联入网络.虚线表明为预测的相互作用,将在后面叙述.图1 人类AD 相关蛋白质相互作用图Fig.1 Illustration showing protein protein interaction of hum an s Alzheimer sdisease associated proteins481 第5期蒋雄飞等:A lzheimer s 疾病相关蛋白质相互作用网络1.2 蛋白相互作用网络(Scale free Network)的性质 这里,首先列举出一些在工作中要使用的量[12]:(1)网络是由节点(Node)与它们两两之间的连线(Connection)构成.在蛋白相互作用网络中,节点代表着蛋白,而线则意味着它们之间有相互作用.(2)度(Degree):一个节点所联结的所有的线,即某蛋白所能作用的蛋白数量,以k表示,度分布P(k)则意味着拥有某个度k的节点在所有的可能的k中的可能性.(3)平均度(M ean Deg ree):网络中所有节点的k的平均值,以<k>表示.无标度网络[12,13]具有一些奇特的性质,这里只关注那些与本文工作密切相关的性质.有两个机制主导着无标度的性质,一、网络的生长是新的节点连接到网络原有的节点上的过程,二、也是最重要的,在大多数真实的网络中,拥有大的连接数,即度(k)大的节点拥有更大的几率连接新的节点,这就是优先连接(prefer ential attachment)[12,13]原则.在无标度网络中,一个具有大的连接度的节点(k <k>)从统计角度说要比其他的节点重要的多,这些节点被称为中心节点(H ub),网络的属性在很大程度上由这些只占很小部分的中心节点决定.一个重要的后果是无标度网络具有抗随机扰动能力,但对加在中心节点上的作用很敏感.换言之,随机节点的故障不会破坏网络的完整性,然而,如有目的的将中心节点切除,则网络结构会迅速瓦解[14,15].这个特点对于生物系统尤为重要,因为它反映了生物医学网络对于随机突变的鲁棒性[16].1.3 AD相关蛋白相互作用网络的属性特征的详细分析 整个网络中,共有91个节点(不包含孤立蛋白),119个连接,平均度,<k>= 1.31.7个蛋白被选择为中心蛋白,它们与AD 的发病机理密切相关,分别是P05067、P29353、Q14192、O75369、P10636、Q9NY61和Q8IZP0 (表1),度分别为15、3、23、11、22、6和8,除P29353只有3,其他均远大于平均度<k>=1.31,而P29353的入选是因为其重要的连接作用.尤其P05067,Amy loid beta A4pro tein,即APP蛋白,有6个深色,即实验上证实于AD相关的蛋白与其直接相连.如果考虑3度以内,即3步以内,很明显整个网络可以被分割为两个主要的子网络.第一个子网络是以APP为中心,而在此3度以内的深色节点迅速上升到14个,占到27个深色节点的55.6%.从某种程度上说,APP 在整个网络中的地位是最重要的.最近的研究普遍认为,APP的聚集在AD的发病机理起着至关重要的作用[17~19].所以,可以看出,无论是实验,还是本文的生物信息学方法,发现APP 确实在AD的发病机理中十分关键.另一个子网络的中心蛋白为apopto sis antag onizing tr anscription facto r(AAT F),即网络右边的三角形点(图1),聚集在它周围的深色节点则为4个.从宏观角度看,AD相关蛋白相互作用网络就是通过上面提到的两组蛋白质的相互作用.Q9NY61,也就是AAT F,最近被确认是DA P like kinase(Dlk)-DAP(death associated pr otein)kinase家族的成员之作用伴侣.AATF会阻抗PAWR(Q96IZ0)诱导的细胞凋亡,这表明AATF直接或间接的参与PAWR活性的调节.AATF的表达会阻碍由PAWR诱导的APP异常表达和分泌,同时AAT F/PA WR的复合系于AAT F对A PP异常分泌的抑制作用起着最基本的作用.这些结果意味着AAT F是PAWR活性和APP在凋亡环境下异常表达、分泌的内生性阻抗物[20].然而,尚无实验报道AATF与PAWR能够与APP直接发生相互作用.所以,根据以上的推理,我们认为从AATF到APP之间应该存在着某条调控路径,通过调控路径上的蛋白级联作用而最后使得AAT F对APP的异常表达产生抑制作用,并对AD起到一定的阻止或抑制作用.从网络中,得出几条潜在的调控路径,如下:1.Q9NY61(AATF)!Q99547!P60866482南京大学学报(自然科学) 第42卷!O75369!P29353!P05067(APP)2.Q9NY61(AATF)!P10636!Q71U36 !Q14192!P29353!P05067(APP)3.Q9NY61(AATF)!P10636!P05218 (or Q08211,or P11940,or P46940)!O75369 !P29353!P05067(APP)(注:Q99547:M-phase phosphoprotein 6;P60866:40S ribo som al protein S20; O75369:Filamin-B;P29353:SH C tr ansform ing protein1;P10636:tau protein; Q71U36:Tubulin alpha-3chain;Q14192: Four and a half LIM domains protein2; P05218:Tubulin beta-2chain;Q08211: ATP-dependent RNA helicase A;P11940: Poly adeny late binding protein1;P46940:Ras GT Pase activating like protein IQ GAP1)分析第一条路径,首先AATF,作为细胞凋亡阻抗因子,该过程与磷酸化密切相关,所以紧随其后的是Q99547,M相磷酸蛋白;然后与核糖体蛋白40s亚基发生作用;接下来与O75369发生作用,该蛋白与FLNA结合后将允许成神经细胞从心室区转移至大脑皮层,这一步的含义并不是很清楚;最后通过P29353,实际上每条路径最后都是通过该蛋白与APP 发生作用,该蛋白起到了将激活生长因子受体与信号通路耦合的信号适配作用.同理也可发现,P29353、Q14192和O75369这几个蛋白的位置也是很重要的,它们在左右两个子网络之间起到了桥梁作用.H.Jeo ng等研究了一个真实的酵母的蛋白相互作用网络[21].通过酵母蛋白的随机移除,发现酵母基因组的随机突变不会影响到整个网络的拓扑结构.同时作为对比,当最大联结的那些蛋白被移除时,网络迅速解体.换言之,那些拥有大联结数目的蛋白时细胞的生存时最重要的.所以,一些在我们网络中高联结数的蛋白也被挑选了出来.它们对于AD网络是所至关重要的,决定着网络的属性,在某种程度上.如果即使在现在它们还未被实验证实与AD相关,但从网络的性质可推断,很可能将是会相关,像P29353和Q92870,至少在AD的调控上起着重要作用(在表1中,被标记上星号).如上所述,P05067、P29353、Q14192、O75369、P10636、Q9NY61、Q8IZP0和Q06481被挑选为中心节点蛋白.由表1可见,除Q14192和P29353之外,其他的中心节点都多少的和神经功能相关,同时都在神经元或者大脑神经干细胞中表达.所以,它们确实与神经系统相关,而从组织表达的特异性上说,与AD的发病机制也许相关.2 蛋白质相互作用预测2.1 预测算法 鉴于实际计算能力,不可能对所有的蛋白进行预测,所以,如前文所述,中心节点蛋白在网络中的作用要远远的超出一般的蛋白,且由于优先连接(preferential attachment)规则的介入,使得那些具有大k的节点,即节点蛋白更倾向于接受新的蛋白相互作用,尽管它们只是占了少数,所以优先检测中心节点蛋白的相互作用蛋白.预测方法是基于序列比对。