DNA star Seqman 使用说明 DNA序列拼接
DNASTAR 使用说明

DNAstar中文使用说明书目录DNAStar的安装与升级 (6)EditSeq的使用方法 (7)打开已有序列寻找开放读框DNA序列翻译遗传密码选择使用遗传密码修改序列的反向互补及反向转换BLAST检索序列信息查看序列校读序列的保存与输出GeneQuest的使用方法 (12)打开已有分析文件GeneQuest的DNA分析方法用分析方法操作方法参数改变结果展示优化Feature注释BLAST检索Entrez Database检索GeneQuest的其他特点保存分析文件MapDraw的使用方法 (18)新酶切图制作过泸器类型应用频率过泸器应用手动过泸器使用过泸器一览表使用Must Cut Here/Don’t Cut Here调色板工具酶信息显示环形展示ORF图显示择选保存,退出MegAlign的使用方法 (23)创建队列文件序列设置Pairwise Alignment使用 Dot Plot Method多序列比较Phylogenetic Tree查看查看队列报告Decorations/Consensi MegAlign文件保存PrimerSelect的使用方法 (28)创建PrimerSelect文件定义引物特点查找引物对浏览其他的引物信息按特征对引物分类引物长度改变在引物中引入突变设计新引物使用寡核苷酸订购表格保存PrimerSelect文件Protean的使用方法 (34)创建蛋白质分析文件Protean’s蛋白质分析方法应用分析方法方法参数改变优化结果显示使用蛋白酶消化与SDS PAGE Feature注释BLAST检索二级结构模拟展示滴定曲线保存分析文件SeqManII的使用方法 (39)输入序列片段Pre-Assembly Options操作检查修整的数据序列装配查看范围和构建结果去除矛盾碱基和缺口手动修改序列末端文件保存与序列输出DNAStar的安装与升级如果您以前已经安装了Lasergene 而且目前有升级和服务联系,您就可以通过英特网来升级您现有的版本,各种模块(module)都是以自解压形式存储的,你可以选择性的下载安装。
seqman使用说明

修改参数命令
关于Seqman的简单介绍就到这里,另外由 于资料准备仓促,如有不完善或不准确的 地方,请大家指出,谢谢! 下期讲座预告 “轻松学用生物软件(3)”,具 体时间及内容将在螺旋课堂上做出预告, 欢迎大家参加!
手工去除侧翼序列
测序准确的峰形图
峰形规则,一般在序列的中部,如下图所示
测序不准确的峰形图
峰形较乱,很难判断是哪个碱基,一般位于序列两端, 如下图所示
轻松学用生物软件(2) 利用SeqMan进行序列拼接
主讲人:huaxing 2008年4月11日
螺旋课堂系列讲座
主要内容
大家好,首先感谢大家关注、支持螺旋网!今天 我们一起一步一步的学习利用Dnastar 6.0中的子 程序Seqman进行序列拼接,希望能对大家有所帮 助。 注意:Dnastar 6.0的安装程序及破解补丁大家可 以到螺旋网的“生物信息学及生物软件交流”版块 免费下载!
手工去除侧翼序列
1. 双击要去除侧翼序列的目标序列 2. 将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标 会变成一个有两个箭头的水平线 3. 按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变 浅,这部分变浅的序列则就被去除,不再参加后面的拼接 4. 此步请将测序不准确或认为是载体的序列用这种方法去 除。
错误的拼接的类型
1. 两条序列的测序结果不一致并明显一条测序质量好而另 一条质量差 处理:直接将该处修改为正确的碱基 2. 3. 两条序列的测序结果不一致并两条测序质量都比较差 处理:重新测序或用新的合适引物重新测定 两条序列的测序结果不一致并明显两条测序质量都好 处理:测序过程出现问题,重新测定
seqman使用说明

seqman使用说明SeqMan使用说明1.概述1.1 背景信息在生物学研究中,序列比对和分析是常见的任务。
SeqMan是一款用于DNA和蛋白质序列分析的软件工具,可以帮助研究人员进行序列比对、剪切、组装和注释等操作。
1.2 目标读者本文档适用于对SeqMan软件感兴趣的研究人员和生物信息学初学者。
1.3 前提条件在使用SeqMan之前,您需要具备一定的生物学和分子生物学知识,并且熟悉基本的计算机操作。
2.安装和配置2.1 SeqMan软件您可以在[SeqMan官方网站]()上最新版本的SeqMan软件。
2.2 安装SeqMan按照的安装包进行安装,根据安装向导的提示进行操作。
2.3 配置SeqMan安装完成后,打开SeqMan软件。
在首次运行时,您需要进行一些配置操作,例如选择默认的比对算法、设置保存结果文件的路径等。
3.序列导入和处理3.1 导入序列文件在SeqMan中,您可以导入各种格式的序列文件,包括FASTA、GenBank、EMBL等。
选择导入菜单,然后选择您要导入的序列文件。
3.2 序列编辑和校正SeqMan提供丰富的序列编辑和校正功能,您可以进行碱基替换、插入、删除等操作,以修正或优化序列。
3.3 序列比对和组装SeqMan可以对导入的序列进行比对和组装操作,以便进行比对结果分析和序列拼接。
4.注释和分析4.1 注释工具介绍SeqMan提供了多种注释工具,包括基因预测、ORF查找、功能注释等。
您可以选择适当的注释工具进行分析。
4.2 序列特征分析SeqMan还提供了序列特征分析功能,可以帮助您查找和分析序列中的特征,如启动子、编码区域、结构域等。
5.结果输出和导出5.1 结果展示SeqMan将分析结果以图表、表格等形式展示,方便您查看和分析。
5.2 结果导出您可以将SeqMan的分析结果导出为各种格式的文件,方便进一步处理和分享。
6.附件本文档不包含附件,请参考SeqMan官方网站获取相关附件。
DNAstar软件的使用(1)Megalign序列比对_唐明智_新浪博客

DNAstar软件的使⽤(1)Megalign序列⽐对_唐明智_新浪博客DNAstar软件共有七个⼩程序(见下图),各⾃执⾏不同的功能,Editseq⽤于序列编辑,Seqman可以去除载体序列和拼接序列,MegAlign则主要执⾏序列⽐对的功能.开启MegAlign软件⾸先需要点击File---New 新建⼀个⼯作⽂件再点击File---Entersequeces 添加需要⽐对的序列,⽀持多种格式的⽂件(.seq;.abi;.pro;.fas等),点Add可添加多个⽂件,点Done导⼊选中的⽂件.这是⽂件导⼊后的界⾯点Align选择序列⽐对的算法,其中多序列⽐对有三种算法:The Jotun Hein method,ClustalV和Clustal W;⼀般选择Clustal W即可。
三种算法的差异如下:“The Jotun Hein method was devised to align sequences that are previously known to be related by descent. The Clust ClustalW is an advancement over ClustalV, and was designed to create more accurate alignments for highly diverged sequ “CLUSTALW是⼀种渐进的多序列⽐对⽅法,先将多个序列两两⽐对构建距离矩阵,反应序列之间两两关系;然后根据距离矩统进化指导树,对关系密切的序列进⾏加权;然后从最紧密的两条序列开始,逐步引⼊临近的序列并不断重新构建⽐对,直到所有序列都被加⼊为⽌”。
红⾊区域表⽰的是相似性最⾼的区域,蓝⾊和绿⾊是同源性较低的区域;多序列⽐对的⼀个缺点就是不会⾃动对序列反向互补后再⽐对,所以反向互补后同源性⾼的序列检测不出来。
如下图中的20.p0和20.p6其实是同⼀模板的两个⽅向的测序结果,像这种需要⽤到两两⽐对(One pair)。
DNAstar软件的使用(1)Megalign序列比对_唐明智_新浪博客

DNAstar软件共有七个小程序(见下图),各自执行不同的功能,Editseq用于序列编辑,Seqman可以去除载体序列和拼接序列,MegAlign则主要执行序列比对的功能.开启MegAlign软件首先需要点击File---New 新建一个工作文件再点击File---Entersequeces 添加需要比对的序列,支持多种格式的文件(.seq;.abi;.pro;.fas等),点Add可添加多个文件,点Done导入选中的文件.这是文件导入后的界面点Align选择序列比对的算法,其中多序列比对有三种算法:The Jotun Hein method,ClustalV和Clustal W;一般选择Clustal W即可。
三种算法的差异如下:“The Jotun Hein method was devised to align sequences that are previously known to be related by descent. The ClustClustalW is an advancement over ClustalV, and was designed to create more accurate alignments for highly diverged sequ“CLUSTALW是一种渐进的多序列比对方法,先将多个序列两两比对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树, 对关系密切的序列进行加权;然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止”。
红色区域表示的是相似性最高的区域,蓝色和绿色是同源性较低的区域;多序列比对的一个缺点就是不会自动对序列反向互补后再比对,所以反向互补后同源性高的序列检测不出来。
如下图中的20.p0和20.p6其实是同一模板的两个方向的测序结果,像这种需要用到两两比对(One pair)。
DNAstar中Seqman拼接序列使用方法
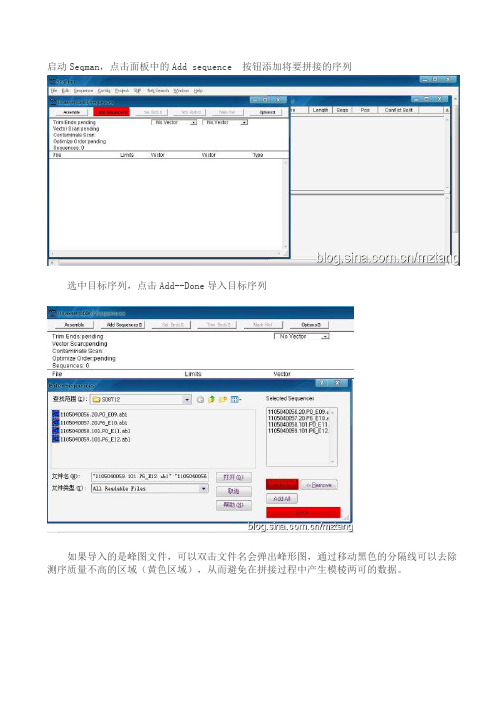
启动Seqman,点击面板中的Add sequence 按钮添加将要拼接的序列
选中目标序列,点击Add--Done导入目标序列
如果导入的是峰图文件,可以双击文件名会弹出峰形图,通过移动黑色的分隔线可以去除测序质量不高的区域(黄色区域),从而避免在拼接过程中产生模棱两可的数据。
数据修正后点击Assemble即拼接,运行结束后会弹出一个新对话框,如果能拼接上,则在Contig一栏有显示。
双击Contig可以看到拼接好的完整序列,以及两个峰图的重叠区域,如果两个峰图的重叠区域完全匹配,则表明拼接的结果可靠;如果两个峰图间有不同碱基,则需要比对两个峰图,选择峰图清晰的作为最终结果。
点击文件前面的三角形可以直接查看序列的峰形图
点击菜单栏的Contig--Save Consense--Single File 可以保存拼接好的序列。
利用SeqMan进行序列拼接ppt课件

12
1. 两条序列的测 序结果不一致 并明显一条测 序质量好而另 一条质量差
处理:直接将该处 修改为正确的碱基
Step5:修改拼接错误 错误拼接的类型13
2. 两条序列的测序结果不 一致并两条测序质量都比 较差
处理:重新测序或用 新的合适引物重新测定
Step5:修改拼接错误 错误拼接的类型14
去除载体序列
26ห้องสมุดไป่ตู้
单击 Scan All按钮,将出现一个report窗口。
• 现在载体栏显示:载体名字前都有一个检 测通过的标志,说明Janus 载体在全部14
去序除列载中体都序已列经检测到了。
• 单击assemble按钮,进行序列拼接。 27
查看末端修整和载体序列去除细节报告
选择Project 菜单的Trim Report打开Trim report窗口。
28
查看修整序列前后的跟踪数据
右键选择6 号样本,然后Show Original Trace Data,打开Trace:Sample 6.abi 窗口
从 5’末端起变淡的部分是载体序列,将不会用于序 列装配,故被清除。
垂直的黑棒出现于修整和未修整的序列之间,根据需 要拖动垂直黑棒,可以调整用于装配的序列末端。
29
点击Vector Catalog选项 自定义实验室常用的载 体序列
自定义载体序列
1、扫描载体插入位点上下500bp 2、多克隆位点位置及兼容位点
30
网 选络定同你源的C序on列tig的序下列载,点和击比N对et Search里的
BLAST Selection 31
网络同源序列的下载和比对
32
DNA测序结果拼接方法
待拼接序列显示区
点击
运用PBIL进行在线拼接
粘贴要拼接的 两个序列
点击
点击Contigs,即可出现 拼接好的序列
运用DNAMAN进行 本地ORF
搜索—开放阅读框
文件--打开--(拼接好的序列)
搜索—开放阅读框--(得到的东西)
蛋白质—翻 译纵览(结 果)
点击(得到的结果)
对序列进行在线ORF
点击
粘贴拼接好 的序列
点击
点击查 看详情
点击
结果
回到NCBI首页,选择BLAST, 下拉,点击TBLASTN
粘贴下载的序列
填写你想拼 接的序 列。。。进 行blast
选择一个序列,进入
将序列下载下来
点击blast
点击
粘贴序列
选择otherS
填写想比对的物种
选入两个待拼接的 序列的保存地址
DNAstar说明书

DNAStar(综合性序列分析软件)全面的生物医学软件,用作DNA和蛋白质序列分析、重叠群拼接和基因工程管理。
包含了7个模块:SeqBuilder - 可视化和序列编辑SeqMan Pro - 序列集结和SNP发现MegAlign - 序列组合PrimerSelect - oligo primer 设计Protean - 蛋白质结构分析和预测GeneQuest - 基因查找EditSeq - 导入特殊文件工具DNAstar中文使用说明书目录DNAStar的安装与升级 (6)EditSeq的使用方法 (7)打开已有序列寻找开放读框DNA序列翻译遗传密码选择使用遗传密码修改序列的反向互补及反向转换BLAST检索序列信息查看序列校读序列的保存与输出GeneQuest的使用方法 (12)打开已有分析文件GeneQuest的DNA分析方法用分析方法操作方法参数改变结果展示优化Feature注释BLAST检索Entrez Database检索GeneQuest的其他特点保存分析文件MapDraw的使用方法 (18)新酶切图制作过泸器类型应用频率过泸器应用手动过泸器使用过泸器一览表使用Must Cut Here/Don’t Cut Here调色板工具酶信息显示环形展示ORF图显示择选保存,退出MegAlign的使用方法 (23)创建队列文件序列设置Pairwise Alignment使用 Dot Plot Method多序列比较Phylogenetic Tree查看查看队列报告Decorations/Consensi MegAlign文件保存PrimerSelect的使用方法 (28)创建PrimerSelect文件定义引物特点查找引物对浏览其他的引物信息按特征对引物分类引物长度改变在引物中引入突变设计新引物使用寡核苷酸订购表格保存PrimerSelect文件Protean的使用方法 (34)创建蛋白质分析文件Protean’s蛋白质分析方法应用分析方法方法参数改变优化结果显示使用蛋白酶消化与SDS PAGEFeature注释BLAST检索二级结构模拟展示滴定曲线保存分析文件SeqManII的使用方法 (39)输入序列片段Pre-Assembly Options操作检查修整的数据序列装配查看范围和构建结果去除矛盾碱基和缺口手动修改序列末端文件保存与序列输出DNAStar的安装与升级如果您以前已经安装了Lasergene 而且目前有升级和服务联系,您就可以通过英特网来升级您现有的版本,各种模块(module)都是以自解压形式存储的,你可以选择性的下载安装。
seqman使用说明
主要内容
大家好,首先感谢大家关注、支持螺旋网!今天 我们一起一步一步的学习利用Dnastar 6.0中的子 程序Seqman进行序列拼接,希望能对大家有所帮 助。 注意:Dnastar 6.0的安装程序及破解补丁大家可 以到螺旋网的“生物信息学及生物软件交流”版块 免费下载!
wwwhelixnetcn手工去除侧翼序列手工去除侧翼序列将鼠标放到测序图谱左边的一个黑色的竖线上此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线那么你就会发现侧翼序列的颜色变浅这部分变浅的序列则就被去除不再参加后面的拼接http
轻松学用生物软件(2) 利用SeqMan进行序列拼接
主讲人:huaxing 2008年4月11日
手工去除侧翼序列
1. 双击要去除侧翼序列的目标序列 2. 将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标 会变成一个有两个箭头的水平线 3. 按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变 浅,这部分变浅的序列则就被去除,不再参加后面的拼接 4. 此步请将测序不准确或认为是载体的序列用这种方法去 除。
错误的拼接的类型
类型1
类型1
类型2
类型3
Step6:导出拼接的序列
可选择合适的格式,导出拼接好的序列
通过以上几步我们就能很快将几个测序片段 进行拼接,大家可以拿着自己的序列试试! 当然如果两个测序片段的拼接片段太短可能 利用默认的参数不能完成拼接,大家可以试 着修改一下拼接参数试试!如降低Match size 及Minimum Match Percentage的值!
手工去除侧翼序列
测序准确的峰形图
峰形规则,一般在序列的中部,如下图所示
seqman使用说明

seqman使用说明SeqMan使用说明一、简介SeqMan是一款功能强大的序列分析软件,适用于DNA和RNA序列的处理与分析。
本文档将详细介绍SeqMan的安装、基本操作和常用功能。
二、安装1、SeqMan安装程序。
2、运行安装程序,按照提示进行安装。
3、安装完成后,启动SeqMan。
三、登陆与用户管理1、打开SeqMan后,在登陆界面输入用户名和密码。
2、如果是首次使用,“注册”按钮进行用户注册。
3、注册完成后返回登陆界面,输入注册时填写的用户名和密码进行登录。
四、主界面SeqMan的主界面由以下几个部分组成:1、菜单栏:提供各种操作和功能选项。
2、工具栏:快速访问常用功能的图标按钮。
3、序列列表:显示打开的序列文件和其基本信息。
4、报告窗口:显示操作的结果和详细信息。
五、打开和保存序列文件1、“文件”菜单,选择“打开”选项。
2、在打开对话框中选择要打开的序列文件。
3、序列文件将显示在序列列表中。
4、若要保存当前文件,“文件”菜单,选择“保存”或“另存为”选项。
六、序列编辑1、选择要编辑的序列文件。
2、“编辑”菜单,选择“编辑序列”选项。
3、在序列编辑器中编辑序列。
支持插入、删除、替换等操作。
4、完成编辑后,“保存”按钮保存修改。
七、序列比对1、选择要比对的序列文件。
2、“分析”菜单,选择“序列比对”选项。
3、在比对设置界面选择比对算法和参数。
4、“开始比对”按钮开始序列比对。
5、比对结果将显示在报告窗口中,并保存为比对报告文件。
八、序列注释1、选择要注释的序列文件。
2、“分析”菜单,选择“序列注释”选项。
3、在注释设置界面选择注释工具和参数。
4、“开始注释”按钮开始序列注释。
5、注释结果将显示在报告窗口中,并保存为注释报告文件。
九、序列分析1、选择要进行分析的序列文件。
2、“分析”菜单,选择“序列分析”选项。
3、在分析设置界面选择分析工具和参数。
4、“开始分析”按钮开始序列分析。
5、分析结果将显示在报告窗口中,并保存为分析报告文件。
利用SeqMan进行序列拼接

谢谢!
SUCCESS
THANK YOU
2019/4/27
• 修整完毕后 Alignment View 中在序列的 左边会有一个黑色的垂直棒,右边有一个 小的黑三角形。
• 要找回修整去掉的序列末端,只需把垂直 棒向序列的两端拖动即可,以前修整去掉 的序列有明亮的黄色背景。
Pre-Assembly Options 操作及序列 装配
• 在拼接前面,可以将所要拼接的片段中清 除载体和污染序列,优化装配顺序,设定 片段末端和标记重复序列
Step5:修改拼接错误
1. 两条序列的测 序结果不一致 并明显一条测 序质量好而另 一条质量差
处理:直接将该 处修改为正确的 碱基
错误拼接的类型
Step5:修改拼接错误
2. 两条序列的测序结 果不一致并两条测序质 量都比较差
处理:重新测序 或用新的合适引物重新 测定
错误拼接的类型
Step5:修改拼接错误
SUCCESS
THANK YOU
2019/4/27
SeqMan进阶
• 手工及自动去除末端 • 消除载体或污染的序列 • 网络比对下载同源相似序列
手动修改序列末端
• SeqMan根据trace数据的质量和载体序列在 装配之前可以自动地进行末端修整。然而 有时候修改的程度难以掌握,下面我们将 用手工的方法找回修整过的末端。测序过程出 现问题,重新测定
类型3
错误拼接的类型
Step6:导出拼接的序列
• 可选择合适的格式,导出拼接好的序列
1
3 2
• 通过以上几步我们就能很快将几个测序片 段进行拼接,大家可以拿着自己的序列试 试!
• 当然如果两个测序片段的拼接片段太短可 能利用默认的参数不能完成拼接,大家可 以试 着修改一下拼接参数试试! 如降低 Match size 及Minimum Match Percentage 的值!
DNAStar使用手册

DNAStar使⽤⼿册DNAStar 中⽂使⽤说明书编者:宋晨⼀、EditSeq (2)三、MapDraw (23)四、MegAlign (32)五、PrimerSelect (42)六、Protean (54)七、SeqMan II开始 (64)⼀、EditSeq打开已有序列我们从⽤苹果计算机打开“TETHIS21MA ”和⽤Windows 打开“tethis21.seq”开始。
假设序列的末尾有载体序列污染。
我们在⽤EditSeq 打开序列的同时,⽤Set Ends 命令去除5’和3’污染序列。
从⽂件菜单(FILE MENU),选择Open。
打开⽂件夹“Demo Sequences”单击选定单击位于对话框右下⾓的序列“TETHIS21”。
Set Ends 按,点钮。
Set Ends 被打开(如右)。
在5’框和3’框中键⼊50和850击OK。
单击Open 打开序列。
当EditSeq 窗⼝打开时,序列长度显⽰在右上⾓。
通过“setting ends,”现在你只有最初序列中的801 bp 的⽚段。
Set Ends 选择在全部Lasergene 应⽤程序中都可以使⽤。
寻找开放读框在这⼊门的⼀部分中,我们将确定序列中最⼤的ORF,并翻译它。
从SEARCH MENU 找到ORF,点击打开会出现右边的对话框。
单击Find Next 寻找第⼀个ORF 的位置。
继续点击Find Next 直到你把ORF 的位置选定在位置183-455。
ORF的坐标会出现在EditSeq 窗⼝的顶端附近。
DNA 序列翻译这⼀节中我们介绍如何翻译我们的ORF,不过任何序列中的读框内部分翻译。
如果你的选择是在三联码的读框内,三联码指⽰棒显⽰为实⼼⿊线(如左图)。
如果都可以⽤下⾯的⽅法进⾏你的选择是不在三联码的读框内,左边的箭头和右⾯的箭头显⽰向左或向右移动⼀个bp,以使所选序列成为三的倍数。
选定ORF,从GOODIES MENU ranslate)。
利用SeqMan进行序列拼接课件PPT

12
Step5:修改拼接错误
1. 两条序列的测 序结果不一致 并明显一条测 序质量好而另 一条质量差
处理:直接将该 处修改为正确 的碱基
2021/3/10
错误拼接的类型 13
Step5:修改拼接错误
2. 两条序列的测序结果 不一致并两条测序质量 都比较差
处理:重新测序或用 新的合适引物重新测定
2021/3/10
2021/3/10
22
Pre-Assembly Options 操作及序列 装配
• 在拼接前面,可以将所要拼接的片段中清 除载体和污染序列,优化装配顺序,设定 片段末端和标记重复序列
2021/3/10
23
去除载体序列
2021/3/10
24
去除载体序列
2021/3/10
25
去除载体序列
• seqman在拼接前,有--点击Unassembled Sequences窗口的右上角的“options“按钮选 择相应功能。默认设定Trim sequence ends, scan for vector,optimize sequence assembly order.。
隆群的处理;基因寻找;蛋白质结构域的
查找;多重序列的比较和两两序列比较; 寡核苷酸设计(PCR引物,测序引物,探 针)。
2021/3/10
2
SeqMan
•SeqMan主要用于多序列的拼接,可以将成千 上万的序列(最多可以支持64000多序列)装 配成contigs,同时在拼接前,可以修整质量 差的序列以及清除自动测序的序列结果中的 污染序列或载体序列。seqman还提供完善的 编辑和输出功能。
报告
• 选择Project 菜单的Trim Report打开Trim report窗口。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
42 SeqMan
笔记本:A电脑
创建时间:2013/12/10 8:35更新时间:2013/12/10 9:07
1.打开lasergene-dnastart-seqman
2.点击add sequences,注意文件格式为.ab1,该文件为测序峰图文件。
3.添加序列文件,本例为16_xxxx.ab1,点击打开,序列添加到Selected sequences窗口。
4.点击done,序列成功加入主程序窗口
5.选中想要拼接的序列,点击assemble,拼接开始。
6.拼接完成后出现,拼接成功提示,creating new contig1:from xxx entering xxx
7.点击窗口右上角,“-”最小化,将拼接提示最小化,回到主窗口。
8. 此时主窗口上方出现拼接好的contig1的信息,574bp,来源于两条序列。
9.双击contig1出现具体的拼接过程窗口。
10.点击16前的黑色三角符号,可以看到序列峰图(注意峰图非常重要,不同颜色代表不同碱基,峰型表示测序可信度)。
11.详细讲一下峰图:
测序反应开始时和结束时的序列是读不准的(测序的原理决定)。
一个测序反应最多能测定500-800个碱基,且测序反应开始和结束的碱基读不准。
ITS45的长度在500bp左右,意味着单向测序末端会读不准。
采用双向测序,在R向峰分辨率极度降低时,F向
正好处在分辨率最高的测序区域,所以这段序列程序会以F向测序结果为准。
seqman在序列拼接的同时,让测序峰图可见,让我们可以判断测序结果的可靠性。
12.接着说拼接完成后如何拷贝拼接好的序列,其实非常简单,选中顶上的consensus中的序列,全选,ctrl+C,拼接好的序列就复制到剪切板中了,可以粘贴到txt中使用。