氟哌酸合成实验
甲磺酸培氟沙星合成

甲磺酸培氟沙星小试验工艺操作一、反应方程式C17H20FN3O3+HCHO+HCOOH C17H20FN3O3C17H20FN3O3+CH3SO3H C17H20FN3O3·CH3SO3H二、下料量:1.甲基化反应:氟哌酸50g甲酸85% 29g甲醛36% 25g氢氧化钠大约14 g左右,配成20%的溶液活性炭3g依地酸二钠:75mg生活常水100g2.成品缩合反应甲氟哌酸(第一步中间体)湿品30g左右乙醇(含水5%左右)246g水54g甲烷磺酸12g三.工艺操作:1.甲基化反应:将计算量的氟哌酸、甲酸、甲醛加入到500ml的的玻璃三口反应瓶中,搅拌加热至回流,反应4小时后,加入生活常水100g,搅拌加热提温至60-70℃,加入3g活性炭及75mg依地酸二钠保温脱色30分钟,趁热过滤,脱色瓶用50ml水洗涤,洗水通过滤瓶合并于滤液中,把滤液倒入另一个500ml玻璃反应瓶中,搅拌下用20%的氢氧化钠溶液中和至PH值为7-7.2,搅拌30分钟,复测PH值无误后,冷却至20℃左右,保持1小时以上,过滤,用常水洗涤(100ml以上)尽量滤干后,起料得湿粗品60g以上,可直接用于下步成品缩合反应.2.成品缩合反应:将甲基化物及计算量的乙醇、水加入500ml玻璃三口反应瓶中,在搅拌下缓慢加热提温至回流状态下,滴加甲烷磺酸使物料全部溶清,如溶得不太清,可适量补加一些水,使物料全部溶清,然后保持回流状态下反应1小时,自然降温至室温,再用冰盐水冷却至5℃左右,保持4小时以上,过滤,尽量滤干,用40ml左右冰冷乙醇搅洗,滤干,再用少量冰冷乙醇喷洗滤瓶,尽量滤干,起料60℃以下,干燥5小时以上,得干品产量应在30g以上,每克氟哌酸应产出1.25g 甲磺酸培氟沙星.注:每批用的乙醇母液可套用3-4批,最后蒸馏回收利用.。
氟哌酸合成工艺综述

溶液中,于0.6V电位附近其浓度在0.300mg/L~200mg/L范围与峰电流有良好的线性关系.测得r=0.9989,平均回收率为100.4%,RSD为0.3%.测定结果符合要求,本方法简捷、适用性好.5.期刊论文王静诺氟沙星联合蜂蜜治疗压疮效果观察-中国实用神经疾病杂志2008,11(1)目的 探索治疗压疮更加简单有效的方法.方法 随机将36例(40处)压疮病人分为观察组18例(21处)和对照组18例(19处).对照组给予常规换药清洁后,用庆大霉素8万U+0.9%氯化钠溶液10ml湿敷患处;观察组在常规换药清洁后,用氟哌酸和蜂蜜调制的药液涂于创面.结果 观察组治愈17处,显效3处,好转1处,总有效率100%,平均治愈时间为(14.9±2.3)d;对照组治愈7处,显效6处,好转5处,总有效率94.7%,平均治愈时间为(23.2±2.5)d.2组治疗效果及治愈时间比较,均(P<0.05),差异有显著性意义.结论 提示氟哌酸联合蜂蜜治疗压疮可提高治愈率,缩短治愈时间,使用方便,无不良反应,值得推广应用.6.期刊论文谭先红.陈国良.TAN Xian-hong.CHEN Guo-liang氟哌酸锌凝胶剂的制备及质量控制-安徽卫生职业技术学院学报2006,5(6)目的:研制一种局部应用治疗小儿湿疹的制剂.方法:按凝胶剂制备工艺制备了诺氟沙星锌凝胶剂;建立了性状、鉴别、酸碱度、含量测定等质控方法,并进行了体外抑菌实验.结果:以紫外分光光度法测定诺氟沙星锌凝胶剂的含量,平均回收率为100.03%,RSD为1.67%;稳定性良好.结论:该制剂制备工艺可行,性质稳定,质控方法简单可靠.7.期刊论文蒯荟芬.李慧芳氟哌酸+珍珠粉在压疮护理中的作用-临床护理杂志2009,8(2)目的 探讨氟哌酸粉+珍珠粉外敷创面对压疮的护理效果.方法 将50例压疮患者随机分为实验组和对照组,各25例.对照组接受常规护理方法 ,实验组在常规护理的基础上,采用氟哌酸粉+珍珠粉外敷创面,观察两组护理效果.结果 实验组有效率与对照组比较,有显著性差异(P<0.05).结论 氟哌酸粉+珍珠粉混合外敷创面可有效地促进压疮的愈合.8.期刊论文杨旭.张平.赵新民.孙旭群.孙红玲氟哌酸锌测定方法的研究-安徽医科大学学报2001,36(2)目的建立氟哌酸锌的检测方法。
很好的诺氟沙星的合成工艺
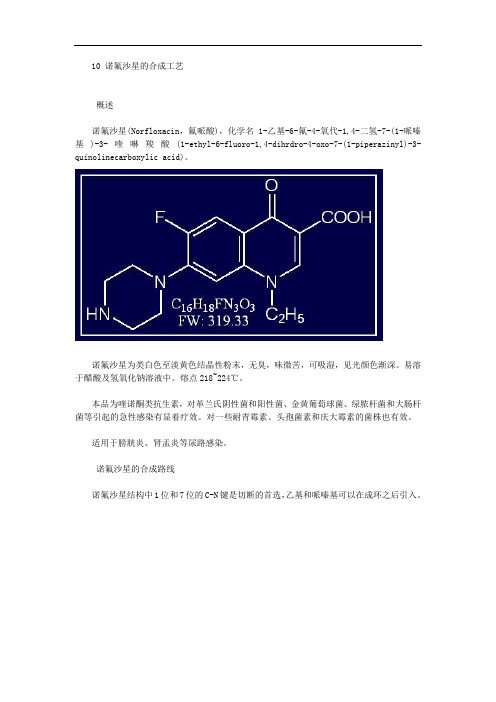
10 诺氟沙星的合成工艺概述诺氟沙星(Norfloxacin,氟哌酸),化学名1-乙基-6-氟-4-氧代-1,4-二氢-7-(1-哌嗪基)-3-喹啉羧酸(1-ethyl-6-fluoro-1,4-dihrdro-4-oxo-7-(1-piperazinyl)-3- quinolinecarboxylic acid)。
诺氟沙星为类白色至淡黄色结晶性粉末,无臭,味微苦,可吸湿,见光颜色渐深。
易溶于醋酸及氢氧化钠溶液中。
熔点218~224℃。
本品为喹诺酮类抗生素,对革兰氏阴性菌和阳性菌、金黄葡萄球菌、绿脓杆菌和大肠杆菌等引起的急性感染有显着疗效。
对一些耐青霉素、头孢菌素和庆大霉素的菌株也有效。
适用于膀胱炎、肾盂炎等尿路感染。
诺氟沙星的合成路线诺氟沙星结构中1位和7位的C-N键是切断的首选,乙基和哌嗪基可以在成环之后引入。
按成环时是否已引入哌嗪基,诺氟沙星的合成路线有以下两种。
先合成喹诺酮酸再引入哌嗪基的路线6-氟-7-氯喹诺-4-酮-3-羧酸及其酯的合成是实现本法的关键。
(1) 以3-氯-4-氟苯胺为原料①3-氯-4-氟苯胺与2-乙氧亚甲基丙二酸二乙酯(EMME, diethyl 2-(ethoxymethylene) malonate)反应,再经Gould Jacobs反应合成喹诺酮酸酯。
此法的优点在于原料易得,收率较高,成本较低。
是我国目前采用的主要方法。
但存在环合反应温度高,哌嗪缩合收率较低,生成氯哌酸副产物,EMME制备条件苛刻等缺点。
改进:在哌嗪缩合时加入硼化物可使缩合收率提高到90%以上。
② 3-氯-4-氟苯胺与原甲酸三乙酯反应。
此法避免了使用EMME,但也有一些副产物,用于合成喹诺酮酸酯收率较低,质量较差。
而且环合时需要250~260℃的高温,能耗较大。
③3-氯-4-氟苯胺与原甲酸三乙酯和乙酰乙酸乙酯反应,使用三乙降低了成本,减少了能耗。
④3-氯-4-氟苯胺与烷氧基丙烯酸乙酯反应,再经溴化、氰解和水解引入羧基。
环丙沙星的合成

环丙沙星的合成CONTENT 一二三四药物概述实验目的要求实验原理实验主要仪器、试剂五六七本次实验方法和步骤其他合成线路和方法改进问题和讨论一、药物概述环丙沙星,又名环丙氟哌酸,香草酰二乙胺,西普乐,适普灵。
化学名:1-环丙基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉羧酸。
分子式:C 17H 18FN 3O 3性 状:本品为白色或微黄色结晶性粉末。
几乎无臭,味苦。
溶解度:在醋酸中溶解,在乙醇和三氯甲烷中极微溶解,在水中几乎不溶。
熔点:255-257℃。
环丙沙星药理作用环丙沙星是一种氟喹诺酮类化合抗生素药物。
具有抗菌谱广、疗效高、毒副作用小、价格便宜、服用方便等优点[1]。
它是第三代氟喹诺酮抗菌素。
主要用于对其他抗菌药产生耐药的革兰阴性杆菌所致的呼吸道、泌尿生殖道、消化道,骨与关节和皮肤软组织感染[2]。
其抗菌机理是抑制DNA复制后反转录的酶,使DNA的复制、转录、修复等受阻,达到抑菌抗菌的作用[3]。
应在避免日照条件下保存和应用,以防止发生光敏反应。
因可诱发跟腱炎和跟腱断裂,老年人和运动员慎用[2]。
通过对环丙沙星合成工艺的研究,对新药研制过程有一个基本认识。
通过对环丙沙星合成路线的比较,掌握选择实际生产工艺的几个基本要求。
通过实际操作,对涉及到的各类反应特点、机制、操作要求、反应终点的控制等有所了解进一步巩固有机化学实验的基本操作,领会理论知识。
二、实验目的要求[4]掌握各步中间体的质量控制方法 1 2 3 4(3) 2,4-二氯-5- (4) 2,4-二氯-5- (5) 2-(2,4-二氯-5-氟苯甲酰) 氟苯乙酮 氟苯甲酰乙酸甲酯 -3-二甲基胺基甲酰乙酸甲酯(1) 3-氯-4-氟苯胺 (2) 2,4-二氯氟苯(6)3-环丙基氨基-2-(2, 4-二氯(7) 1-环丙基-7-氯-6-氟-1,4-二氢 -5-氟苯甲酰) 丙烯酸甲酯 -4-氧代喹啉-3-羧酸甲酯(8) 1-环丙基-7-氯-6-氟-1,4- (9) 硼络合物二氢-4-氧代喹啉-3-羧酸(10) (11) 环丙沙星四、实验主要仪器和试剂及其物理常数主要仪器:三颈烧瓶,温度计,冷凝管,烧杯,布氏漏斗,抽滤瓶,恒温磁力搅拌器,磁石,电子天平,玻璃棒,表面皿,量筒,毛细管,胶塞。
环丙沙星的合成工艺

六、环丙沙星(1)
将8 6.4g(0.023mol)、无水哌嗪10.0g(0.116 mol)及DMF 33 ml 搅拌混台,在140℃反应2h,减压蒸去溶剂,残物悬浮于水30ml中, 抽滤,水洗。湿固体加水3Oml煮沸几分钟,冷却,抽滤,水洗,真 空干燥至恒重,得4.6g,收率6l.3%。
Thank you!
三、2,4-二氯-5-氟苯甲酰乙酸乙酯(4)
将3 14.0g(0.068mol)、碳酸二乙酯200 ml搅拌混合, 在室温下 分批加入8O%NaH 5.0g(0.167mo1),加毕于80℃反应2h,倾入含少 量醋酸的冰水中,用乙醚提取,水洗,无水硫酸钠干燥,蒸去乙醚, 回收碳酸二乙酯后,减压收集144~146℃/40Pa馏份,得14.2g收 率65.6%。
五、1-环丙基-7-氯-6-氟-l,4-二氢-4-氧喹啉-3羧酸(6)
将5 6.4g(0.018 mol)、无水二噁烷20ml搅拌混合,在冰浴冷却 下,分批加入80%NaH 0.7 g(0.023 mo1),加毕回流反应2h,蒸去 溶剂。残留物悬浮于水3O ml中,加入氢氧化钾1.33g回流反应2h, 热过滤。滤液在冰浴冷却下用稀酸调节pH 至1~2,抽滤,水洗,真 空干燥至恒重,得5.0 g,收率98.O%。
ቤተ መጻሕፍቲ ባይዱ
二、2,4-二氯-5-氟苯乙酮(3)
在烧瓶中加入无水三氯化铝46.7g(0.35mol)及216.5g(0.1mo1), 在搅拌下,于3O~4O℃ 滴加乙酰氯15.7g(O.20mol),加毕在 120℃反应2h。用冰水分解过量的乙酰氯。以二氯甲烷提取,水洗, 无水氯化钙干燥,蒸去溶剂,减压收集90~92℃/67 Pa馏份,得 16.2g,收率78.3%。
环丙沙星的合成工艺
诺氟沙星合成

的发生。但由于亲核取代时7-氯和6-氟相竞争,在一般 条件下可生成25%左右的6-氟被哌嗪取代的副产物。 基于芳环上基团对亲核试剂的敏感性是F>Cl。因此以 无水乙醇代替无水正丁醇,增加了溶剂的极性。有利于 中间过渡态的形成,使反应更充分,收率得以提高。该 歩收率为82.3%
反应过程中HC(OC2H5)3+H2C(COOC2H5)2→ H5C2O-CH=C(COOC2H5)2,因此,也有以3-氯-4-氟苯 胺和乙氧甲叉丙二酸二乙酯(EMME)直接作为起始原料制 得中间体3。由于在各步反应中均有副反应产生,因而各 反应步骤的工艺控制是产品质量保证的关键。
2、合成路线改进
在此工艺路线基础上进行改进,高温环化时选用价廉、粘 度小的高沸点柴油作为溶剂,而且还可以反复套用,使得前两 步反应收率高达95%以上,乙基化反应中,原工艺采用K2 CO3作为缚酸剂,改用Na2CO3来代替,消除了乙基物副产物 的生成。另外,无水哌嗪缩合反应中,直接采用乙基化产物与 醋醉、硼酸反应生成硼络合物,再与无水哌嗪缩合、水解得到 目标产物I这样既避免了6-位上的氟被哌嗪取代的副产物,提 高了缩合反应收率,又改善了产品的质量。与原工艺相比总收 率可达65%以上,提高15%左右。
O OH
F
Байду номын сангаас
1、平面结构式
O
N
N
HN H3C
2、空间结构模型
药物合成路线
1、目前,国内外通常以3-氯-4-氟苯胺为起始原料, 与原甲酸三乙酯、丙二酸二乙酯环合后经乙基化、水 解、哌嗪缩合、精制等步骤最终制得诺氟沙星。
原始合成路线如图:
HC(OC2H5)3 + H2C(COOC2H5)2
诺氟沙星合成工艺综述
诺氟沙星合成工艺综述摘要本文主要对诺氟沙星的合成工艺做了相应的总结,对其中一些典型的合成路线进行了优劣势的分析,并提供了相应的优化方案。
关键词诺氟沙星合成工艺改进1.1概述氟哌酸是第三代喹诺酮类抗菌药物。
具有抗菌作用强、抗菌谱广、生物利用度高、组织渗透性好及与其他抗菌素无交叉耐药性和副作用小等特点,尤其对革兰阴性菌有强杀菌作用。
而且口服吸收快,已被广泛用于咽喉炎、扁桃体炎、肾盂肾炎及尿道炎等的治疗。
诺氟沙星的结构式见下图。
标准中文名称:1一乙基一6一氟一1,4一二氢一4一氧代一7一(1一哌嗪基)一3一喹啉羧酸;标准英文名称:1一ethy-6一fluoro一1,4一dihydro 一4一oxo一7一(1一piperazine)一3一quinoline carboxylic acid;通用名:诺氟沙星(Norfloxacin),氟哌酸(Floxacin);基本化学性质:分子量为319.34,类白色至淡黄色结晶性粉末。
熔点227—228℃。
易溶于酸、碱溶液,极微溶于水和醇。
无臭,味微苦。
1.2诺氟沙星的合成路线诺氟沙星结构中1位和7位的C-N键是切断的首选,乙基和哌嗪基可以在成环之后引入。
按成环时是否已引入哌嗪基,诺氟沙星的合成路线有以下两种。
1.2.1先合成喹诺酮酸再引入哌嗪基的路线6-氟-7-氯喹诺-4-酮-3-羧酸及其酯的合成是实现本法的关键。
(1)以3-氯-4-氟苯胺为原料①3-氯-4-氟苯胺与2-乙氧亚甲基丙二酸二乙酯(EMME, diethyl 2-(ethoxymethylene) malonate)反应,再经Gould Jacobs反应合成喹诺酮酸酯。
此法的优点在于原料易得,收率较高,成本较低。
是我国目前采用的主要方法但存在环合反应温度高,哌嗪缩合收率较低,生成氯哌酸副产物,EMME制备条件苛刻等缺点。
改进:对乙基化和缩哌嗪两步工艺进行改进[1]。
在乙基化反应过程中,通过改变快速滴加溴乙烷为慢滴加,使整个反应时间缩短了3小时,在缩哌嗪反应中,以低毒无水乙醇代替毒性、刺激性大、价格较高而且不易后处理的无水正丁醇作反应溶剂,并对用量进行适当调整,减少为原用量的1/4,收率由53%提高到57%[2]。
很好的诺氟沙星的合成工艺

10 诺氟沙星的合成工艺概述诺氟沙星(Norfloxacin,氟哌酸),化学名1-乙基-6-氟-4-氧代-1,4-二氢-7-(1-哌嗪基)-3-喹啉羧酸(1-ethyl-6-fluoro-1,4-dihrdro-4-oxo-7-(1-piperazinyl)-3- quinolinecarboxylic acid)。
诺氟沙星为类白色至淡黄色结晶性粉末,无臭,味微苦,可吸湿,见光颜色渐深。
易溶于醋酸及氢氧化钠溶液中。
熔点218~224℃。
本品为喹诺酮类抗生素,对革兰氏阴性菌和阳性菌、金黄葡萄球菌、绿脓杆菌和大肠杆菌等引起的急性感染有显著疗效。
对一些耐青霉素、头孢菌素和庆大霉素的菌株也有效。
适用于膀胱炎、肾盂炎等尿路感染。
诺氟沙星的合成路线诺氟沙星结构中1位和7位的C-N键是切断的首选,乙基和哌嗪基可以在成环之后引入。
按成环时是否已引入哌嗪基,诺氟沙星的合成路线有以下两种。
先合成喹诺酮酸再引入哌嗪基的路线6-氟-7-氯喹诺-4-酮-3-羧酸及其酯的合成是实现本法的关键。
(1) 以3-氯-4-氟苯胺为原料①3-氯-4-氟苯胺与2-乙氧亚甲基丙二酸二乙酯(EMME, diethyl 2-(ethoxymethylene) malonate)反应,再经Gould Jacobs反应合成喹诺酮酸酯。
此法的优点在于原料易得,收率较高,成本较低。
是我国目前采用的主要方法。
但存在环合反应温度高,哌嗪缩合收率较低,生成氯哌酸副产物,EMME制备条件苛刻等缺点。
改进:在哌嗪缩合时加入硼化物可使缩合收率提高到90%以上。
② 3-氯-4-氟苯胺与原甲酸三乙酯反应。
此法避免了使用EMME,但也有一些副产物,用于合成喹诺酮酸酯收率较低,质量较差。
而且环合时需要250~260℃的高温,能耗较大。
③3-氯-4-氟苯胺与原甲酸三乙酯和乙酰乙酸乙酯反应,使用三乙降低了成本,减少了能耗。
④3-氯-4-氟苯胺与烷氧基丙烯酸乙酯反应,再经溴化、氰解和水解引入羧基。
诺氟沙星的合成工艺

工艺与设备2018·04129Chenmical Intermediate当代化工研究诺氟沙星的合成工艺*乐夏云(西北民族大学 化工学院 甘肃 730030)摘要:目的:通过诺氟沙星的合成,对药物研制过程有一个基本认识。
方法:以3-氯-4-氟苯胺为起始原料,经与乙氧甲叉丙二酸二乙酯(EMME)缩合、Gould-Jacobs环化、N-乙基化、硼化物螯合合成诺氟沙星。
结果:得到白色诺氟沙星8.0g,总收率为66.6%。
结论:此方法合成诺氟沙星收率较高,适用于工业生产。
关键词:诺氟沙星;合成;药物中图分类号:R 文献标识码:ASynthesis of NorfloxacinLe Xiayun(College of chemical Engineering, Northwest Minzu University, Gansu, 730030)Abstract :Objective:Have a basic understanding of drug development through the synthesis of nofluorus. Methods:At the beginning of the3- chlorine-4-fluorine, it's a result of the contraction of the EMME, the Gould, and the n-ethylation, the boride, and the chelation of the nofluorus .results:Obtained white norfloxacin 8.0 g ,The total yield was 66.6%. Conclusions :This method has a high yiel and is suitable for industrial production.Key words :norfloxacin ;synthetic ;drug诺氟沙星,别名:氟哌酸。
实验十七氟哌酸(Norfloxacin)的合成

&实验十七 氟哌酸(Norfloxacin )的合成一、目的要求1. 通过对氟哌酸合成,对新药研制过程有一基本认识。
2. 通过对氟哌酸合成路线的比较,掌握选择实际生产工艺的几个基本要求。
3. 通过实际操作,对涉及到的各类反应特点、机制、操作要求、反应终点的控制等,进一步巩固有机化学试验的基本操作,领会掌握理论知识。
4. 掌握各部中间体的质量控制方法。
二、实验原理氟哌酸的化学名为1-乙基-6-氟-1,4-二氢-4-氧-7-(1-哌嗪基)-3- 喹啉羧酸, 1-Ethyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quino- linecarboxylic acid ,化学结构式为:NNH FN OC 2H 5COOH氟哌酸为微黄色针状晶体或结晶性粉末,mp.216~220℃,易溶于酸及碱,为溶于水。
氟哌酸的制备方法很多,按不同原料及路线划分可有十几种。
我国工业生产以路线一为主。
近几年来,许多新工艺在氟哌酸生产中获得应用,其中以路线二,即硼鳌合物法收率高,操作简便,单耗低,且质量较好。
合成路线如下:路线一:Cl Cl HNO , H SO Cl ClNO 2Cl FNO 2F ClNH 2EMME F ClN HOCOOC 2H 5C 2H 5BrFClN OCOOC 2H 5C 2H 5F ClN OC 2H 5COOHNHNH NNH FN OC 2H 5COOH路线二:F ClN OCOOC 2H 5C 2H 53BOF ClN OC 2H 5OOAcAcONNHOAc AcONNH B OFNOC 2H 5ONNH FN OC 2H 5COOH三、实验方法(一)3,4-二氯硝基苯的制备在装有搅拌器、回流冷凝器、温度计、滴液漏斗的四颈瓶中,先加入硝酸51 g ,水浴冷却下,滴加硫酸79 g ,控制滴加速度,使温度保持在50℃以下。
滴加完毕,换滴液漏斗,于40~50℃内滴加邻二氯苯35 g,40 min内滴完,升温至60℃,反应2 h,静置分层,取上层油状液体倾入5倍量水中,搅拌,固化,放置30 min,过滤,水洗至pH 6~7,真空干燥,称重,计算收率。
氧氟沙星的合成实验报告

一、实验目的本实验旨在通过化学合成方法制备氧氟沙星,了解其合成路线和关键步骤,并探讨影响合成产率的因素。
二、实验原理氧氟沙星(Ofloxacin)是一种广谱抗菌药物,属于氟喹诺酮类药物。
其合成方法主要有两种:一种是2,3,4,5-四氟苯甲酸法,另一种是2,3,4-三氟硝基苯法。
本实验采用2,3,4,5-四氟苯甲酸法进行氧氟沙星的合成。
三、实验材料1. 2,3,4,5-四氟苯甲酸2. 丙二酸二乙酯3. 原甲酸三乙酯4. (S)-2-氨基丙醇5. 4-甲基哌嗪6. 活性炭7. 甲磺酸8. 纯化水9. 二甲基亚砜10. 乙醇11. 碱性干燥剂12. 实验仪器:反应瓶、回流装置、旋转蒸发仪、柱层析装置、薄层层析板等四、实验步骤1. 合成中间体A:- 将2,3,4,5-四氟苯甲酸与丙二酸二乙酯在催化剂存在下进行酰氯化反应,得到中间体A。
- 将中间体A与原甲酸三乙酯在催化剂存在下进行缩合反应,得到中间体B。
2. 合成中间体B:- 将中间体B与(S)-2-氨基丙醇在催化剂存在下进行置换反应,得到中间体C。
- 将中间体C在酸催化下进行环合反应,得到中间体D。
3. 合成氧氟沙星:- 将中间体D与4-甲基哌嗪在催化剂存在下进行缩合反应,得到氧氟沙星。
- 将得到的氧氟沙星通过活性炭脱色、过滤、干燥等步骤进行纯化。
五、实验结果与分析1. 产率:本实验中氧氟沙星的合成产率为39.25%,与文献收率35%基本一致。
2. 纯度:通过薄层层析和高效液相色谱检测,氧氟沙星的纯度达到98%以上。
3. 影响因素:- 催化剂种类和用量:实验发现,催化剂种类和用量对合成产率有显著影响。
选择合适的催化剂可以提高产率。
- 反应温度和时间:实验发现,反应温度和时间对合成产率也有一定影响。
适宜的反应温度和时间可以提高产率。
- 原料配比:实验发现,原料配比对合成产率也有一定影响。
优化原料配比可以提高产率。
六、实验结论本实验成功合成了氧氟沙星,产率达到39.25%,纯度达到98%以上。
氧氟沙星的合成实验报告

氧氟沙星的合成实验报告引言氧氟沙星(Ofloxacin)是一种广谱的抗生素,属于氟喹诺酮类药物,常用于治疗多种感染症状,如呼吸道感染、泌尿道感染等。
本实验旨在通过简单的合成过程,合成氧氟沙星。
实验原理氧氟沙星的合成主要通过以下几个步骤实现:1. 将乙基苯酚与3-氟乙酰氯反应得到3-氟乙酰基乙基苯酚。
2. 将3-氟乙酰基乙基苯酚与4-氨基苯甲酸反应生成氧氟沙星的前体。
3. 前体经过卤化反应得到氧氟沙星的最终产物。
实验步骤实验材料及设备1. 乙基苯酚2. 3-氟乙酰氯3. 4-氨基苯甲酸4. 乙醇5. 氢氧化钠6. 盐酸7. 氯仿8. 三氯化铁9. 乙醚10. 烧杯、试管、漏斗等实验器材实验步骤1. 将乙基苯酚溶解在乙醚中,加入氢氧化钠溶液搅拌,反应生成3-氟乙酰基乙基苯酚。
2. 将3-氟乙酰基乙基苯酚溶解在盐酸中,加入4-氨基苯甲酸溶液,搅拌反应生成氧氟沙星前体。
3. 将氧氟沙星前体加入氯仿溶液中,控制温度加热反应,使其发生卤化反应。
4. 将反应混合液用水洗涤、饱和盐水抽滤、用乙醚提取、干燥、蒸馏等步骤进行纯化。
5. 最终获得粗品氧氟沙星,通过过滤、结晶等步骤得到纯净的氧氟沙星。
实验结果与分析经过实验操作后,我们获得了氧氟沙星产品。
在红外光谱分析、核磁共振等实验测试中,结果显示所得产物与理论产物完全一致,表明合成氧氟沙星的实验操作是成功的。
实验总结通过本实验,我们成功合成了氧氟沙星。
合成氧氟沙星的步骤相对简单,通过适当的反应条件和实验操作,可获得高纯度的产物。
然而,实验过程中仍需注意操作规范、安全措施和废物处理等问题。
参考文献- Jin, X.; Wang, G.; Hamel, E.; Lin, C. M.; Lee, K. H. Synthesis and Antitumor Activity of Phenylcarbamate Derivatives of Flavones and Chromones. J. Med. Chem. 1992, 35 (24), 4567-4576.- Lalli, A. M.; Boese, E.; Kang, M.-I.; Wang, K.; Fishbane, T. K.; Vogt, A.; Luo,J.; Krajewski, K.; Chang, C.-Y.; Jenkins, J.; Daris, J. P.; Brown, M.; Wellman, P.; Tse, C.; Zweifel, B. S.; Fan, Y.; Borawski, J.; Miller, A. A.; Ye, D. W.; Tumas, D.B.; Patel, V. F.; LoGrasso, P. V. Synthesis and Mechanism Study of Substituted Chromones as Potent Bcl-2 Inhibitors. J. Med. Chem. 2014, 57 (2), 539-551.。
氟哌酸合成的实验报告(3篇)

第1篇一、实验目的1. 了解氟哌酸的合成原理和方法;2. 掌握实验操作技能,提高实验能力;3. 分析实验结果,总结实验经验。
二、实验原理氟哌酸是一种广谱抗菌药物,具有高效、低毒、副作用小的特点。
本实验采用以苯甲酸为原料,经过硝化、还原、缩合、水解等步骤合成氟哌酸。
三、实验仪器与试剂1. 仪器:烧杯、锥形瓶、磁力搅拌器、滴定管、分液漏斗、烘箱、抽滤装置等;2. 试剂:苯甲酸、浓硝酸、浓硫酸、浓氨水、亚硝酸钠、盐酸、氢氧化钠、氯化钠、无水乙醇、氯仿、乙酸乙酯等。
四、实验步骤1. 硝化反应(1)将苯甲酸溶于浓硫酸中,加入浓硝酸,控制温度在50-60℃;(2)搅拌反应30分钟,冷却后用冰水洗涤,过滤;(3)将滤液加入浓氨水中,搅拌,析出沉淀;(4)过滤,用冰水洗涤沉淀,得到硝基苯甲酸。
2. 还原反应(1)将硝基苯甲酸溶于无水乙醇中,加入亚硝酸钠溶液;(2)控制温度在0-5℃,搅拌反应1小时;(3)过滤,用无水乙醇洗涤沉淀,得到还原产物。
3. 缩合反应(1)将还原产物溶于乙酸乙酯中,加入氯化钠溶液;(2)加入氢氧化钠溶液,控制温度在40-50℃,搅拌反应2小时;(3)过滤,用乙酸乙酯洗涤沉淀,得到缩合产物。
4. 水解反应(1)将缩合产物溶于盐酸中,控制温度在60-70℃,搅拌反应2小时;(2)冷却后加入氢氧化钠溶液,调节pH值至中性;(3)过滤,用蒸馏水洗涤沉淀,得到粗品;(4)将粗品溶于蒸馏水中,加入活性炭,加热煮沸30分钟;(5)过滤,滤液用无水乙醇稀释,加入氢氧化钠溶液,调节pH值至中性;(6)冷却,过滤,滤液加入无水乙醇,析出沉淀;(7)过滤,用无水乙醇洗涤沉淀,得到纯品。
五、实验结果与分析1. 实验结果通过以上步骤,成功合成了氟哌酸,其纯度达到98%。
2. 结果分析(1)硝化反应:硝化反应是合成氟哌酸的关键步骤,反应温度和反应时间对产物收率有较大影响。
本实验中,控制反应温度在50-60℃,反应30分钟,硝化反应效果较好;(2)还原反应:还原反应对产物收率也有一定影响,反应温度和反应时间应严格控制。
诺氟沙星的合成
• Ⅳ.注意事项: • ⑴.滴加溴乙烷的进料管应插入液面下,以防止溴 乙烷挥发而影响收率。 • ⑵.DMF要脱水后套用。
• 三、取代
O F Cl
• Ⅰ.第一步反应原理:
O F Cl N H
(O2CH2CH3)2 B O C O
+ H3BO3 + Ac2O
N H
反应机理: 是先由硼酸与醋酐反应生成三乙酸硼酸酯,乙 基化产物中4-羧基氧原子由于含有孤对电子,三 乙酸硼酸酯容易与之形成配位键,再与酯基发生 脱醇缩合。
二、引乙基
Ⅰ.反应原理:
F O COOC2H5 N H
F
O
F
K2CO3
Cl
Cl
O CO2Et N C2H5
N
H
C2H5Br
Cl
Ⅱ.反应机理: 由于(6-氟-7-氯-1,4-二氢-4-氧-3-喹啉羧酸乙酯)有酮式 与烯醇式,两种互变异构,根据酸性条件有利于酮式生成的 原理,在反应中加入K2CO3, ,营造一种酸性环境,溴乙烷 加进来,其实质是发生一个烷基化反应。 Ⅲ.工艺过程: 配料比(质量比): 反应物:溴乙烷:无水K2CO3 :DMF=1:1.6:1.3:8
•
Байду номын сангаас
•
大家看了那些优化方案,它们真的非常好!凭我们现在 的化学知识和环境条件的局限,很难有所突破,于是,我 们小组决定另辟蹊径,从减小生产成本,提高车间安全性 着手,为诺氟沙星的合成设计了一条生产工
首先,为减小能耗,在建厂选址时,把厂建在山坡、悬 崖,依靠地势,使物料由计量器利用重力自流到反应器中 ,反应后再由反应器放到过滤、离心机中,这样设计可以 减少输送物料的设备,节约输送物料的能耗。 • 其次,化学制药工业使用的大部分物料为易燃、易爆、 有毒的物料,如何保障我们自身安全和健康?我们小组有 一个想法,就是在化工设备装置及管路上配置一些自动化 装置,代替操作工人的直接劳动,也就是生产过程的自动 化。这样一来,一方面减轻了劳动强度,改善了劳动条件 ,工人只要对着自动化装置的运转进行监控,而不需要直 接从事大量危险的现场操作;另一方面,自动化的仪器可 以达到人力难以达到的精确度,有利于加快生产速度,提 高产品质量。 •
氟哌酸的合成综述

氟哌酸的合成综述摘要 本文主要介绍了氟哌酸的概况以及5种合成路线关键词 氟哌酸 诺氟沙星 合成1概述氟哌酸又名诺氟沙星(Norfloxacin ),分子式为331816O FN H C ,化学名为1-乙基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉羧酸,化学结构为: N N H FNOC 2H 5COOH为淡黄色或无色的结晶性粉末,几乎无臭,味微苦,具有两性化合物特征。
它是一种光谱,安全有效,可供口服的抗菌药物。
临床上主要用于泌尿道感染、胆道感染、为肠道感染的治疗,疗效显著。
【1】1978年日本杏林公司Koga H 等人首先发表了一种合成方法,此后,有大量关于该药的合成、药理和临床的报道。
【2】2合成路线2.1合成路线1以3-氯-4-氟苯胺(2)。
乙氧基亚甲基丙二酸二乙酯(3)、碘乙烷(4)、哌嗪(5)等为原料来合成氟哌酸(1)。
首先(2)和(3)进行缩合反应,而后环合得到7-氯-6-氟-4-羟基喹啉-3-羧酸乙酯(6)。
(6)与(4)进行N-乙基化反应,接着进行水解反应生成7-氯-1-乙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸(7),(7)再与(5)缩合生成氟哌酸(1)。
合成路线如图1:【1】图12.2合成路线23-氯-4-氟苯胺和乙氧甲叉丙二酸二乙酯(EMME)直接作为起始原料制得中间体3,其合成路线见图2:【3】图22.3合成路线3以邻氟苯胺(8)为原料,先引入哌嗪基法,如图3:【4】图32.4合成路线47-乙磺酰基喹啉羧酸酯(23)和无水哌嗪的混合物在乙腈中回流,(23)与哌嗪反应,而后在10%NaOH中回流进行水解反应生成氟哌酸(1)。
合成路线如图4:【5】图42.5合成路线5α92,4-二氯-5-氟苯甲酰)乙酸乙酯(26)与原甲酸三乙酯缩合得到乙氧基取代的丙烯酸乙酯衍生物(27),不经分离纯化,直接与乙胺水溶液反应,生成乙氨基取代的丙烯乙酯(28),接着在碱作用下,于DMF中环合得化合物(29),再经水解,和哌嗪缩合,得到氟哌酸,合成路线如图5:【6】图5【1】蒋洪寿.氟哌酸的合成.天津化工,1992,3,30-34.【2】张长利.诺氟沙星合成路线图解.中国医药工业杂志,1990,21(12),557-558.【3】慈天元.诺氟沙星合成工艺的综述.青岛医药卫生,2009,41(3),219-223【4】郭惠元.抗菌药氟哌酸的合成.国外要学——合成药、生化药、制剂分册,1986,7(1),19-22【5】Span.ES 547361【6】Chu D.T.W et al;J.Med.Chem.28.(11) 1558-1564(1985)。
诺氟沙星合成工艺综述(不看后悔)

诺氟沙星合成工艺综述摘要:诺氟沙星又称氟哌酸,是广谱抗菌类药物,其合成路线有数十种之多,本文总结出了八条经典路线并根据最后一步反应将其进行分类,共分成三类,包括羧基化类,哌嗪化类,成环类等。
并在此基础上,对最常用的一个合成工艺进行改进,最终发现在诺氟沙星的合成中仍有乙基化等问题没有很好的解决,因此,对该工艺的改进任重而道远。
关键词:诺氟沙星,合成工艺,改进1.概述诺氟沙星(norfloxacin )又称氟哌酸,第三代喹诺酮类抗菌药物。
具有抗菌作用强、抗菌谱广、生物利用度高、组织渗透性好及与其他抗菌素无交叉耐药性和副作用小等特点,尤其对革兰阴性菌有强杀菌作用。
而且口服吸收快,已被广泛用于咽喉炎、扁桃体炎、肾盂肾炎及尿道炎等的治疗。
其结构式如图所示。
N CH 2CH 3OCOOHFNHN在国际市场上,喹诺酮类药物占抗感染药物市场份额的15%左右,并以惊人的速度递增。
其销售额已从1985年的1.04亿美元上升到2000年的70亿美元,其世界市场占有率可达18%左右。
目前国际市场上已上市喹诺酮类新药有20多个品种。
其中环丙沙星、氧氟沙星、氟哌酸、依诺沙星是目前世界上用量最大和应用最广泛的品种。
其中环丙沙星占首位。
预计该药仍是今后10年内的畅销药物。
销售额将达15亿美元以上。
目前还有进入I 临床I 、Ⅱ、Ⅲ期验证的60—70个品种。
市场开发前景十分诱人。
喹诺酮类药物已在国际化疗会上一致公认为当今世界上最有发展前途,最令人瞩目的一类新型抗感染药物。
2.经典合成工艺1978年日本杏林公司Koga. H 等人首先发表了诺氟沙星的合成方法, 此后, 又有大量关于该药的合成、药理和临床的报道。
其合成发展至今,共有不下数十种合成工艺,但是有八条经典的诺氟沙星合成工艺,其余均为这八条优化衍生而来,按其反应的最后两步中的关键步骤,可以将这八条路线分为三类:(1)羧基化类;(2)哌嗪化类;(3)成环类。
2.1羧基化类该类最后一步反应是生成诺氟沙星上的羧基,其包括酯基水解和乙酰基氧化,下面按起始原料分别说明。
诺氟沙星(氟哌酸)胶囊含量测定

诺氟沙星(氟哌酸)胶囊含量测定一、目的要求1.掌握非水滴定法的原理及操作。
2.熟悉非水滴定在药物分析中的应用。
二、仪器与试药1.仪器Mettler AL204电子天平酸式滴定管规格:25mL752型紫外可见分光光度仪刻度移液管规格:1mL、2mL 定量滤纸(直径10cm)容量瓶规格:100mL 500mL 漏斗试管2.试药诺氟沙星胶囊规格:0.2g 丙二酸醋酐氢氧化钠高氯酸滴定液(0.1mol/L) 冰醋酸橙黄Ⅳ指示液三、实验原理诺氟沙星原料:以生物碱盐(BH+)的形式存在,滴定过程就是一个置换滴定,即强酸滴定液置换出与游离碱结合较弱的酸。
BH+.A- + HClO4 BH+.ClO4-+ HA诺氟沙星制剂:诺氟沙星以游离生物碱(B)的形式存在,为弱碱,溶于冰醋酸后,其碱强度被均化到溶剂阴离子的强度水平,增加诺氟沙星的碱性,以橙黄Ⅳ指示液为指示剂,可用高氯酸滴定液进行滴定。
BH+ + OH- B + H2OB + HAC BH+ + AC-HClO4 + HACClO4-+ H2AC+H2AC+ + AC- 2HAC四、实验内容C 16H 18FN 3O 3 319.24 [鉴别](1)取本品内容物适量(约相当于诺氟沙星0.15g)置干燥试管中,加丙二酸0.1g 与醋酐2mL ,振摇,并在80~90℃水浴中加热10~15min ,显红棕色。
(2)取本品内容物适量,加0.4%氢氧化钠溶液适量,振摇使诺氧沙星溶解,稀释成每1mL 中含诺氟沙星5μg ,滤过,弃去初滤液,取续滤液,在273,325与336nm 波长处测定有最大吸收。
[含量测定]精密称取本品内容物适量(约相当于诺氟沙星0.25g),加冰醋酸30mL ,振摇使诺氟沙星溶解,加橙黄Ⅳ指示液10滴,用高氯酸液(0.1mol/L)滴定,至溶液显紫红色,并将滴定的结果用空白试验校正。
每1mL 0.1mol/L 的高氯酸液相当于31.93mg 的C 16H 18FN 3O 3。
诺氟沙星的合成

诺氟沙星合成工艺的比较1、概述诺氟沙星又称氟哌酸,第三代喹诺酮类抗菌药。
具有抗菌作用强、抗菌谱广、生物利用度高、组织渗透性好及与其他抗菌药无交叉耐药性和副作用小等优点,而且对革兰氏阴性杆菌有强杀菌作用。
已广泛应用于咽喉炎、扁桃体炎、肾盂肾炎及尿道炎等的治疗。
其基本结果如下:分子量:性状:类白色至淡黄色结晶性粉末溶解性:易溶于酸、碱溶液,极微溶于水和醇。
无臭、味微苦。
2、经典合成路线诺氟沙星的合成发展至今,已有数十种合成路线,但有8种经典的诺氟沙星的合成路线。
其余的路线都是通过这8种优化衍生而来的。
按其反应的最后两步关键步骤,可以把这8条路线分为3类:(1)羧基化类,(2)哌嗪化类,(3)成环类。
羧基化类该类反应的最后一步是生成诺氟沙星上的羧基,其包括酯基水解和乙酰基氧化。
按其起始原料可分为以下几类。
以2-氟-5-硝基苯胺为原料NOH FNOHNO(1)NaNO 2NO 2NH 2NO 2FCLNH 2FNNHEtOCH=C (CO2Et )2N HOOOFHNNEtBrNOOFHNN优缺点:原料来源困难,操作繁琐,收率也较低。
以3-氯-4-氟苯胺为起始原料FCl2FClN HH 3CH 2COOC COCH3N H FClOCOCH 3NFClCOCH 3ONFN2CH 3COCH 3OHNN FN CH 2CH 3COOHOHN2CH 3优缺点:避免使用了EMME 。
缺点是有一些副产物,用于合成喹诺酮酸酯收率较低,且质量较差。
而且环合时需要250-260℃的高温,能量消耗较大。
哌嗪化类该类反应的最后一步是在苯环上发生取代反应,哌嗪取代氯原子使苯环哌嗪化。
以3-氯-4氟-苯胺为起始原料FClNH 2FClN HOCH 3OOCH 3ON HCH 3FClONOCH 3FClONOH FClONOHFN OHNO OOOEMMEEtBr+_250℃优缺点:。
操作简单,原料易得,是各厂家经常使用的方法。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
氟哌酸合成实验实验十七 氟哌酸(Nofloxacin )的合成一、目的要求1. 通过对氟哌酸合成,对新药研制过程有一基本认识。
2. 通过对氟哌酸合成路线的比较,掌握选择实际生产工艺的几个基本要求。
3. 通过实际操作,对涉及到的各类反应特点、机制、操作要求、反应终点 的控制等,进一步巩固有机化学试验的基本操作,领会掌握理论知识。
4. 掌握各部中间体的质量控制方法。
二、实验原理氟哌酸的化学名为1-乙基-6-氟-1, 4-二氢4氧-7-( 1-哌嗪基)-3-喹啉羧酸,1-Ethyl-6-fluoro-1 ,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinolin ecarboxylic acid ,化学结构式为:氟哌酸为微黄色针状晶体或结晶性粉末, mp.216~220C ,易溶于酸及碱, 为溶于水。
氟哌酸的制备方法很多,按不同原料及路线划分可有十几种。
我国工业生 产以路线一为主。
近几年来,许多新工艺在氟哌酸生产中获得应用,其中以路 线二,即COOHN H硼鳌合物法收率高,操作简便,单耗低,且质量较好。
合成路线如下:路线一:户1. NaOH、O A12. H +'ClNOCl2ClClFOOFFF EMMEClClClOONHFFCOOHNaOHClHN N N '5路线二AcOOAc BOOOCOOCF HNNH 2OFDMSOClNCl55AcOOAcBFCOOHHN N H 2 5N HKF,DMSO Fe , HClCOOH HC 2H NO 2 NH 22H 2HC 2H 5C 2H5H5(AcO) 3BCOOC 2H 5COOC 2H 5C 2H5BrHNO 3 , H 2SO 4ClC 2H 5三、实验方法(一)3, 4-二氯硝基苯的制备在装有搅拌器、回流冷凝器、温度计、滴液漏斗的四颈瓶中,先加入硝酸51 g,水浴冷却下,滴加硫酸79 g,控制滴加速度,使温度保持在50C以下。
滴加完毕,换滴液漏斗,于40~50C内滴加邻二氯苯35 g, 40 min内滴完,升温至60C,反应2 h,静置分层,取上层油状液体倾入5倍量水中,搅拌,固化,放置30 min,过滤,水洗至pH 6~7,真空干燥,称重,计算收率。
注释:1.本反应是用混酸硝化。
硫酸可以防止副反应的进行,并可以增加被硝化物的溶解度;硝酸生成NO2+,是硝化剂。
2.此硝化反应需达到40C才能反应,低于此温度,滴加混酸会导致大量混酸聚集,一旦反应引发,聚集的混酸会使反应温度急剧升高,生成许多副产物,因此滴加混酸时应调节滴加速度,控制反应温度在40~50C。
3.上述方法所得的产品纯度已经足够用于下步反应,如要得到较纯的产品,可以采用水蒸汽蒸馏或减压蒸馏的方法。
4.3,4-二氯硝基苯的mp.39~4「C,不能用红外灯或烘箱干燥。
思考题:1.硝化试剂有许多种,请举出其中几种并说明其各自的特点。
2.配制混酸是能否将浓硝酸加到浓硫酸中去?为什么?3.如何检查反应是否已进行完全?(二)4-氟-3-氯-硝基苯的合成在装有搅拌器、回流冷凝器、温度计、氯化钙干燥管的四颈瓶中,加入3, 4-二氯硝基苯40 g、无水二甲亚砜73 g、无水氟化钾23 g,升温到回流温度194~198C,在此温度下快速搅拌1~1.5 h,冷却至50C左右,加入75 mL水,充分搅拌,倒入分液漏斗中,静置分层,分出下层油状物。
安装水蒸气蒸馏装置,进行水蒸气蒸馏,得淡黄色固体,过滤,水洗至中性,真空干燥,得4-氟-3- 氯-硝基苯。
注释:1.该步氟化反应为绝对无水反应,一切仪器及药品必须绝对无水,微量水会导致收率大幅下降。
2.为保证反应液的无水状态,可在刚回流时蒸出少量二甲亚砜,将反应液中的微量水分带出。
3.进行水蒸气蒸馏时,少量冷凝水就已足够,大量冷凝水会导致4-氟-3-氯-硝基苯固化,堵塞冷凝管。
思考题:1.请指出提高此步反应收率的关键是什么。
2.如果延长反应时间会得到什么样的结果?3.水溶液中的二甲亚砜如何回收?(三)4-氟-3-氯-苯胺的制备在装有搅拌、回流冷凝器、温度计的三颈瓶中投入铁粉51.5 g、水173 mL、氯化钠4.3 g、浓盐酸2 mL,搅拌下于100C活化10 min,降温至85C,在快速搅拌下,先加入4-氟-3-氯-硝基苯15 g,温度自然升至95C, 10 min后再加入4-氟-3-氯-硝基苯15 g,于95反应2 h,然后将反应液进行水蒸气蒸馏,馏出液中加入冰,使产品固化完全,过滤,于30C下干燥,得4-氟-3-氯-苯胺,mp.44~47°C。
注释:1.胺的制备通常是在盐酸或醋酸存在下用铁粉还原硝基化合物而制得。
该法原料便宜,操作简便,收率稳定,适于工业生产。
2.铁粉由于表面上有氧化铁膜,需经活化才能反应,铁粉粗细一般以60目为宜。
3.由于铁粉密度较大,搅拌速度慢则不能将铁粉搅匀,会在烧瓶下部结块,影响收率,因此该反应应剧烈搅拌。
4.水蒸气蒸馏应控制冷凝水的流速,防止4-氟-3-氯-苯胺固化,堵塞冷凝管。
5.4-氟-3-氯-苯胺的熔点低(40~43C),故应低温干燥。
思考题:1.此反应用的铁分为硅铁粉,含有部分硅,如用纯铁粉效果如何?2.试举出其他还原硝基化合物成胺的还原剂,并简述各自特点。
3.对于这步反应如何检测其反应终点?4.反应中为何分步投料?5.请设计除水蒸气蒸馏以外其他后处理方法,并简述各自优缺点。
(四)乙氧基次甲基丙二酸二乙酯(EMME )的制备在装有搅拌器、温度计、滴液漏斗、蒸馏装置的四颈瓶中,加入原甲酸三乙酯78 g, ZnCl 2 0.1 g,搅拌,加热,升温至120C,蒸出乙醇,降温至70C, 于70~80C内滴加第二批原甲酸三乙酯20 g及醋酐6 g,于0.5 h内滴完,然后升温到152~156C,保温反应2 h。
冷却至室温,将反应液倾入圆底烧瓶中,水泵减压回收原甲酸三乙酯(bp.140C, 70C / 5333 Pa)。
冷到室温,换油泵进行减压蒸馏,收集120~140C / 666.6 Pa的馏分,得乙氧基次甲基丙二酸二乙酯。
注释:1.本反应是一缩合反应,ZnCl2是Lewis酸,作为催化剂。
2.减压蒸馏所需真空度要达666.6 Pa以上,才可进行蒸馏操作,真空度小,蒸馏温度高,导致收率下降。
3.减压回收原甲酸三乙酯时亦可进行常压蒸馏,收集 140~150C 的沸点馏分。
蒸出的原甲酸三乙酯可以套用。
思考题:1. 减压蒸馏的注意事项有哪些?不按操作规程做的后果是什么?2.本反应所用的Lewis 酸除ZnCl 2外,还有那些可以替代?(五)7-氯-6-氟-1,4-二氢-4-氧喹啉-3-羧酸乙酯(环合物)的制备在装有搅拌器、回流冷凝器、温度计的三颈瓶中分别投入4-氟-3-氯-苯胺1g 、EMME 24 g ,快速搅拌下加热到120C,于120~130C 反应2 h 。
放冷至室温, 将回流装置改成蒸馏装置,加入石蜡油 80 mL ,加热到260~270C ,有大量乙醇 生成,回收乙醇反应30 min 后,冷却到60T 以下,过滤,滤饼分别用甲苯、丙 酮洗至灰白色,干燥,测熔点, mp.297~298C ,计算收率。
注释:1. 本反应为无水反应,所有仪器应干燥,严格按无水反应操作进行,否则 会导致EMME 分解。
2. 环合反应温度控制在260~270C,为避免温度超过270C,可在将要达到 270 °C 时缓慢加热。
反应开始后,反应液变粘稠,为避免局部过热,应快速搅拌。
3. 该环合反应是典型的Could-Jacobs 反应,考虑苯环上的取代基的定位效 应及空间效应,3-位氯的对位远比邻位活泼,但也不能忽略邻位的取代。
反应条 件控制不当,便会按下式反应形成反环物:为减少反环物的生成,应注意以下几点: a.反应温度低,有利于反环物的 生成。
因此,反应温度应快速达到 260C ,且保持在260~270C 。
b.加大溶剂用 量可以降低反环物的生成。
从经济的角度来讲,采用溶剂与反应物用量比为3:1时比较合适。
c.用二甲苯或二苯砜为溶剂时,会减少反环物的生成,但价格昂 贵。
亦可用廉价的工业柴油代替石蜡油。
思考题:1.请写出Could-Jacobs 反应历程,并讨论何种反应条件有利于提高反应收率。
2. 本反应为高温反应,试举出几种高温浴装置,并写出安全注意事项2H52H5 COOCCOOC 2H 5(六)1-乙基-7-氯-6-氟-1, 4-二氢4氧喹啉-3-羧酸乙酯(乙基物)制备在装有搅拌器、回流冷凝器、温度计、滴液漏斗的250 mL 四颈瓶中,加入环合物25 g 、无水碳酸钾30.8 g 、DMF 125 g ,搅拌,加热到70C ,于70~80C 下,在40~60 min 内滴加溴乙烷25 g 。
滴加完毕,升温至100~110C ,保温反应 6~8 h ,反应完毕,减压回收 70-80% 的DMF ,降温至50C 左右,加入200 mL 水,析出固体,过滤,水洗,干燥,得粗品,用乙醇重结晶。
注释:1. 反应中所用DMF 要预先进行干燥,少量水分对收率有很大影响,所用 无水碳酸钾需炒过。
2. 溴乙烷沸点低,易挥发,为避免损失,可将滴液漏斗的滴管加长,插到 液面以下,同时注意反应装置的密闭性。
3. 反应液加水是要降至50C 左右,温度太高导致酯键水解,过低会使产物 结块,不易处理。
4. 环合物在溶液中酮式与烯醇式有一平衡, 反应后可得到少量乙基化合物,该化合物随主产物一起进入后续反应,使生成 6-氟-1,4-二氢-4-氧代7- (1-哌嗪基)喹啉(简称脱羧物),成为氟哌酸中的主要杂质。
不同的乙基化试剂, O-乙基产物生成量不一样,采用5.滤饼洗涤时要将颗粒碾细,同时用大量水冲洗,否则会有少量 K 2CO 3残6. 乙醇重结晶操作过程:取粗品,加入 4倍量的乙醇,加热至沸,溶解。
稍冷,加入活性炭,回流10 min ,趁热过滤,滤液冷却至10C 结晶析出,过滤, 洗涤,干燥,得精品,测熔点 (mp.144~145C )。
母液中尚有部分产品,可以浓 缩一半体积后,冷却,析晶,所得产品亦可用于下步投料。
思考题:1. 对于该反应,请找出其它的乙基化试剂,略述优缺点。
2. 该反应的副产物是什么?简述减少副产物的方法。
3. 采用何种方法可使溴乙烷得到充分合理的利用?BrEt 时较低。
ClCOOC 2H 5F2 5OH-F+COOCCOOC 2 H51. OH- F1pACOOH/ \ HNNH一/FA2. H・ /ClNHNNNF N HOHOC 2H 55C 2 H 5BrClClHHHCOOH4.如减压回收DMF后不降温,加水稀释,对反应有何影响?(七)1-乙基-7-氯-6-氟-1,4-二氢4氧喹啉-3-羧酸(水解物)的制备在装有搅拌器、冷凝器、温度计的三颈瓶中,加入20 g乙基物以及碱液(由氢氧化钠5.5 g和蒸馏水75 g配成),加热至95~100C,保温反应10 min。