Real-time PCR 定量 HBV DNA using 自动 sample preparation and murine cytomegalovirus internal control
实时荧光定量PCR与PCR-ELISA在乙型肝炎病毒DNA定量检测中的应用比较

(上接第 98 页)
标本编号 1 2 3 4 5 6 7 8 9 10 11
COBAS Amplicor 系统 2.18×105 6.31×104 4.27×105 9.33×105 3.24×105 7.41×104 1.23×104 5.01×104 1.62×104 1.62×103 9.55×103
Light Cycler 仪器的实验结果的灵敏度要低于 COBAS Amplicor,但是在临床应用过程当中仍然可以满足人 们的需求。COBAS Amplicor 系统的 CV 较低,所以具 有较好的重复性 [5]。在与 COBAS Amplicor 对比的过 程当中也有较好的相关性,所以通过国产试剂配合进 口仪器进行实时荧光定量 PCR 的检测具有一定的临 床价值。但是在应用过程当中一定要注意,由于国产 的试剂盒不同制造批次所表现出来的质量也不同,所 以在选择的时候一定要做好相关的管理和判断。另 外还要注意的是乙性肝炎病毒的发展程度所受到的 相关因素影响比较多,例如患者自身的遗传因素、免 疫状态以及病毒变异情况等,患者在感染乙型肝炎病 毒之后整体的疾病发展是进行状态中的,所以临床的 相关医护人员也要注意 [6]。
4.0~8.5 (40±1)℃保存 1 h、2 h、3 h,恢复至室温后未出现油水分离现象
-5~-10 ℃保存 1 h、2 h、3 h,恢复至室温后未出现油水分离现象 3 000 r/min 离心,未出现油水分离现象
检查结果 1 符合 符合 符合 5.2 符合 符合 符合
检查结果 2 符合 符合 符合 5.2 符合 符合 符合
实时荧光定量PCR检测HBV-DNA与化学发光磁微粒法检测乙肝五项的关系

实时荧光定量PCR 检测HBV-DNA 与化学发光磁微粒法检测乙肝五项的关系马晶晶孟雨(遵义市第一人民医院,遵义医科大学第三附属医院,贵州遵义563000)为主占56.4%,与有关报道基本一致[1,2]。
排名前三位依次为大肠埃希菌、粪肠球菌、肺炎克雷伯菌,其中大肠埃希菌检出率远高于其他细菌占24.1%,与卜黎红等人的研究报道基本一致[3]。
分离的革兰阳性菌中以粪肠球菌和凝固酶阴性的表皮葡萄球菌、溶血葡萄球菌最为多见,由于肠球菌对氨基糖苷类和头孢菌素等药物天然耐药,没有出现对万古霉素、利奈唑胺耐药的菌株。
轻症患者可以选择环丙沙星、左氧氟沙星来治疗,重症时选用万古霉素、利奈唑胺。
表皮葡萄球菌、溶血葡萄球菌等凝固酶阴性葡萄球菌虽然毒力和致病力较弱,但由于泌尿系统侵袭性操作、患者抵抗力等原因,可导致感染性疾病[4]。
凝固酶阴性葡萄球菌对抗生素的耐药性高,经验用药可选择万古霉素、利奈唑胺。
分离的革兰阳性菌中大肠埃希菌对哌拉西林/他唑巴坦、亚胺培南、阿米卡星和呋喃妥因耐药率在10%以下。
肺炎克雷伯菌、奇异变形杆菌对氨苄西林、氨苄西林/舒巴坦、环丙沙星耐药率达55%以上。
该院大肠埃希菌、肺炎克雷伯菌、奇异变形杆菌的ESBLs 发生率分别为58.1%,53.8%,45%,产生原因有:①三代头孢类抗生素药物在临床的广泛应用;②对于临床上产生的多重耐药菌株未做好隔离消毒措施。
药敏结果显示肠杆菌科对厄他培南、亚胺培南耐药率低,碳青霉烯类抗菌药物依然是治疗大肠埃希菌等肠杆菌细菌引起泌尿系统感染最有效的药物[5]。
真菌中的白色念珠菌是该院真菌尿路感染最常见的病原体,占6%,与相关研究报告大致相同[6]。
药敏结果显示对白色念珠菌耐药率最高的是伊曲康唑,对两性霉素和5-氟胞嘧啶、伏立康唑、氟康唑有很高的敏感性。
综上所述,泌尿外科医师应了解尿路感染病原菌的分布和耐药情况变化,合理使用抗生素,减少耐药菌株的产生,提高诊疗效果。
参考文献[1]何雁,高婷,王岚.尿路感染患者病原菌分布及药敏试验结果[J].中国卫生检验杂志,2011,21(2):434-436.[2]丁厚文,刘周,吴园园,等.1127株尿培养病原菌分布及耐药性分析[J].国际检验医学杂志,2018,39(4):477-480。
实时定量PCR测定治疗性HBV_DNA疫苗工程菌的质粒拷贝数

广东药科大学学报Journal of Guangdong Pharmaceutical University Sep,2023,39(5)实时定量PCR测定治疗性HBV DNA疫苗工程菌的质粒拷贝数陈孟璋,何星垚,林汉森(广州广药益甘生物制品股份有限公司,广东广州511495)摘要:目的比较不同的定量方法和总DNA制备方法对实时定量PCR(real-time quantitative PCR,qPCR)测定治疗性HBV DNA疫苗工程菌的质粒拷贝数(plasmid copy number,PCN)的影响,为建立快速简便的治疗性HBV DNA 疫苗工程菌的PCN测定方法提供参考。
方法通过试剂盒提取法、Triton X-100煮沸法和培养基煮沸法分别制备总DNA样品用于qPCR检测,使用绝对定量方法和相对定量方法计算不同制备方法样品的PCN,并对测定结果进行统计学分析。
结果试剂盒提取法制备的样品经绝对定量和相对定量计算PCN,结果分别为43.10±4.02和42.92±3.98,显著低于Triton X-100煮沸法的结果(69.32±6.51和68.58±6.42)以及培养基煮沸法的结果(75.73±7.46和76.15±7.50)。
任一样品制备方法,比较其绝对定量和相对定量结果,差异均无统计学意义。
结论qPCR的绝对定量和相对定量计算方法都适用于治疗性HBV DNA疫苗工程菌PCN测定,Triton X-100煮沸法更适合于总DNA模板制备。
关键词:HBV疫苗;DNA疫苗;实时定量PCR;质粒拷贝数中图分类号:R392.1文献标识码:A文章编号:2096-3653(2023)05-0087-06DOI:10.16809/ki.2096-3653.2023051703Determination of plasmid copy number of therapeutic HBV DNA vaccine engineered strain by real-time quantitative PCRCHEN Mengzhang*,HE Xingyao,LIN Hansen(Guangzhou Guangyao Yigan Biological Products Co.,Ltd.,Guangzhou511495,China)*Corresponding author Email:**************Abstract:Objective To compare the effects of different quantitative methods and total DNA preparation methods on the plasmid copy number(PCN)determination of therapeutic HBV DNA vaccine engineered strain by real-time quantitative PCR(qPCR),so as to provide reference for the establishment of a rapid and simple method for the PCN determination of therapeutic HBV DNA vaccine engineered strain.Methods Total DNA samples were prepared for qPCR by kit extraction method,Triton X-100boiling method and medium boiling method, respectively.The PCN of samples prepared by different methods was calculated by absolute and relative quantification methods,and the results were statistically analyzed.Results The absolute and relative quantification results of PCN of samples prepared by kit extraction method were43.10±4.02and42.92±3.98, respectively,which were significantly lower than those obtained by Triton X-100boiling method(69.32±6.51) and medium boiling method(75.73±7.46).There was no significant difference between the absolute and relative quantitative results of any sample preparation method.Conclusion Both the absolute and relative quantitative methods of qPCR are suitable for the determination of PCN of therapeutic HBV DNA vaccine engineered strain, and the Triton X-100boiling method is more suitable for the preparation of total DNA template.Key words:HBV vaccine;DNA vaccine;real-time quantitative PCR;plasmid copy number收稿日期:2023-05-17基金项目:广东省重大科技专项(2012A080204009)作者简介:陈孟璋(1985-),男,硕士,制药工程师,主要从事生物医药研发和临床试验研究,Email:**************。
real-time pcr原理

实时聚合酶链式反应(Real-time PCR)是一种分子生物学技术,用于定量测量DNA或RNA的含量。
它可以用于检测、量化和分析特定基因的表达水平,也可以用于检测病原体、基因突变和许多其他应用。
下面是实时PCR的基本原理:1. **选择性扩增目标DNA/RNA**:实时PCR的第一步是选择性扩增目标DNA或RNA片段。
这通常涉及到设计一对特异性引物(引导片段)来诱导DNA合成。
引物是两个短的DNA片段,它们会结合到目标序列的两端。
2. **荧光探针**:为了实时检测PCR反应的进展,一种叫做荧光探针的特殊DNA分子被引入反应混合物中。
荧光探针是一个含有荧光染料的短链DNA片段,它的一端与一个荧光染料分子紧密相连,另一端与一个荧光阻尼器分子相连。
在初始PCR反应中,这两个分子紧密靠在一起,从而抑制了荧光的发射。
3. **PCR反应**:PCR反应开始后,DNA引物将目标序列进行扩增。
如果目标序列存在,PCR会不断复制它,导致DNA量指数级增加。
4. **荧光信号检测**:在每个PCR循环的末尾,荧光探针靠近的情况会改变。
当PCR过程中荧光探针结合到目标序列时,它被降解,荧光染料与阻尼器分离,导致荧光信号的释放。
荧光信号的强度与PCR反应中目标序列的数量成正比。
5. **实时监测**:荧光信号会在PCR过程中实时检测和记录。
这种监测可以通过特殊的仪器完成,该仪器测量荧光信号的强度并将其显示在一个图表中。
荧光信号的强度与PCR反应中目标序列的初始数量成比例。
因此,通过比较样本中的荧光信号与标准曲线或对照样本,可以定量测量目标DNA或RNA的含量。
总的来说,实时PCR允许科学家在PCR反应进行的同时实时监测和记录DNA或RNA的数量,因此它是一种非常有用的技术,用于定量和检测基因表达、基因变异、病原体等多种生物学研究和诊断应用中。
乙肝病毒DNA检测2种不同核酸提取方法的性能验证情况分析

乙肝病毒DNA检测2种不同核酸提取方法的性能验证情况分析董剑;许小华【摘要】目的:对乙肝病毒DNA(HBV-DNA)检测的2种不同核酸提取方法,即手工法(煮沸法)和全自动核酸提取仪法(磁珠法),在使用相同的检测试剂和核酸扩增仪的条件下进行性能验证,以评价其验证结果.方法:依据《医学实验室质量和能力认可准则》《全国临床检验操作规程》等相关文件,由同一操作人员应用中山达安公司生产的HBV-DNA检测试剂盒和ABI7500实时荧光核酸扩增仪,分别采用手工法和全自动核酸提取仪法提取HBV-DNA进行临床样本的批内精密度、批间精密度、正确度、线性范围性能验证实验.结果:手工法的高值血清批内精密度为2.18%,低值血清批内精密度为3.76%;高值质控品批间精密度为3.29%.全自动核酸提取仪法的高值血清批内精密度为1.78%,低值血清批内精密度为2.44%;高值质控品批间精密度为2.84%.正确度验证分别取浓度为2.0×103、2.0×104、2.0×105、2.0×106copies/ml的可溯源标准品进行验证,手工法偏倚分别为-4.88%、-2.51%、-2.46%,-2.12%,全自动核酸提取仪法偏倚分别为-2.37%、-0.95%、0.74%、-1.33%.线性范围验证中,手工法和全自动核酸提取仪法的相关系数分别为0.9660、0.985 8,均能满足临床检测的需求.结论:全自动核酸提取仪法较手工法提取核酸有利于简化HBV-DNA提取流程,实现分子生物实验室自动化提取核酸.【期刊名称】《医疗卫生装备》【年(卷),期】2019(040)006【总页数】4页(P40-43)【关键词】乙型肝炎病毒DNA定量检测;手工(煮沸法);全自动核酸提取仪法(磁珠法);性能验证;血清【作者】董剑;许小华【作者单位】联勤保障部队第909医院检验科,厦门大学附属东南医院检验科,福建漳州363000;联勤保障部队第909医院检验科,厦门大学附属东南医院检验科,福建漳州363000【正文语种】中文【中图分类】R318;R446.10 引言乙型肝炎也被称为乙肝,是目前世界上最常见的一类传染病,据不完全统计[1] ,在全世界范围每年至少有100万以上的患者死于乙肝。
经典的Real-time PCR定量的四种方法
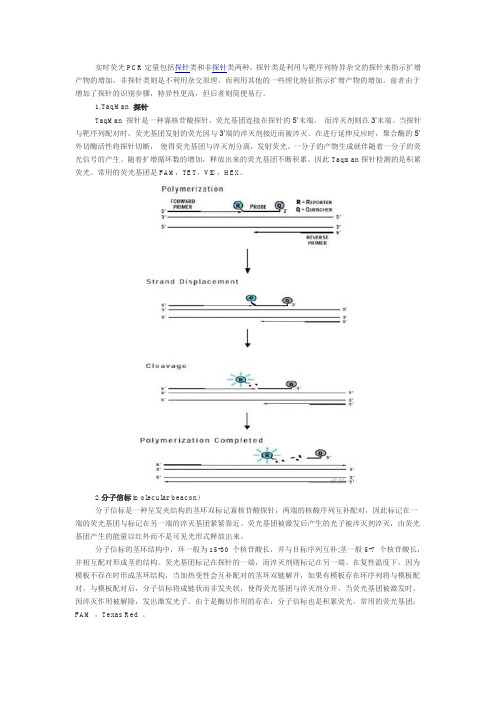
实时荧光PCR定量包括探针类和非探针类两种,探针类是利用与靶序列特异杂交的探针来指示扩增产物的增加,非探针类则是不利用杂交原理,而利用其他的一些理化特征指示扩增产物的增加。
前者由于增加了探针的识别步骤,特异性更高,但后者则简便易行。
1.TaqMan 探针TaqMan 探针是一种寡核苷酸探针,荧光基团连接在探针的5’末端,而淬灭剂则在3’末端。
当探针与靶序列配对时,荧光基团发射的荧光因与3’端的淬灭剂接近而被淬灭。
在进行延伸反应时,聚合酶的5’外切酶活性将探针切断,使得荧光基团与淬灭剂分离,发射荧光。
一分子的产物生成就伴随着一分子的荧光信号的产生。
随着扩增循环数的增加,释放出来的荧光基团不断积累。
因此Taqman探针检测的是积累荧光。
常用的荧光基团是FAM,TET,VIC,HEX。
2.分子信标(molecular beacon)分子信标是一种呈发夹结构的茎环双标记寡核苷酸探针,两端的核酸序列互补配对,因此标记在一端的荧光基团与标记在另一端的淬灭基团紧紧靠近。
荧光基团被激发后产生的光子被淬灭剂淬灭,由荧光基团产生的能量以红外而不是可见光形式释放出来。
分子信标的茎环结构中,环一般为15-30 个核苷酸长,并与目标序列互补;茎一般5-7 个核苷酸长,并相互配对形成茎的结构。
荧光基团标记在探针的一端,而淬灭剂则标记在另一端。
在复性温度下,因为模板不存在时形成茎环结构,当加热变性会互补配对的茎环双链解开,如果有模板存在环序列将与模板配对。
与模板配对后,分子信标将成链状而非发夹状,使得荧光基团与淬灭剂分开。
当荧光基团被激发时,因淬灭作用被解除,发出激发光子。
由于是酶切作用的存在,分子信标也是积累荧光。
常用的荧光基团:FAM ,Texas Red 。
3.SYBR Green ISYBR Green I 是一种能与双链DNA 结合发光的荧光染料。
其与双链DNA结合后,荧光大大增强。
因此,SYBR Green I的荧光信号强度与双链DNA的数量相关,可以根据荧光信号检测出PCR体系存在的双链DNA数量。
realtimepcr简易操作流程

Real-time PCR(实时聚合酶链反应)是一种用于检测和定量DNA或RNA的技术。
它可以在PCR(聚合酶链反应)进行的实时监测PCR 产物的累积量。
real-time PCR技术适用于病毒检测、基因表达分析、SNP检测等许多领域。
real-time PCR技术的操作流程相对简单,但需要一定的实验操作技巧和严格的操作规范。
以下是real-time PCR的简易操作流程:1. 样品准备- 从实验对象中收集所需的组织样品或细胞样品。
- 样本的处理和提取要遵循标准的生物安全操作规程,确保样品的纯度和完整性。
2. DNA/RNA提取- 根据实验需求选择合适的DNA/RNA提取试剂盒和方法,从样品中提取目标DNA或RNA。
- 保证提取的DNA/RNA质量和浓度满足real-time PCR的要求。
3. Primer和探针设计- 根据目标基因序列设计引物和探针,确保其特异性和有效性。
- 引物和探针的设计应遵循一定的原则和标准,可以使用专业的设计软件或委托专业实验室进行设计。
4. PCR体系设置- 根据实验设计和引物/探针的特性设置PCR反应体系,包括引物和探针的最佳浓度、PCR反应缓冲液、酶和模板DNA/RNA的加入量等。
- 严格按照实验方案中的要求和比例进行PCR反应体系的设置,确保反应的准确性和稳定性。
5. PCR反应- 将PCR反应体系加入到real-time PCR仪器中,根据实验方案设置好PCR程序。
- 在PCR过程中,实时监测PCR产物的累积量,记录和分析反应曲线。
6. 数据分析- 根据实验目的和方法选择合适的real-time PCR数据分析软件,对实验数据进行处理和分析。
- 分析数据并计算目标基因的相对表达量或浓度,进行统计学分析和结果解读。
7. 结果呈现- 将实验结果以图表或表格的形式清晰呈现出来,配以必要的文字解释。
- 结果的呈现应简洁明了,突出实验的关键发现和结论。
real-time PCR技术的操作流程虽然相对简单,但在实际操作中仍然需要严格遵循操作规范和注意实验细节。
实时荧光定量pcr原理

实时荧光定量pcr原理实时荧光定量PCR(real-time quantitative PCR)是一种用于检测和定量DNA或RNA的技术。
它结合了PCR技术和荧光探针技术,能够实时监测PCR反应过程中的荧光信号变化,从而实现对目标核酸的定量分析。
本文将介绍实时荧光定量PCR的原理及其在科研和临床中的应用。
实时荧光定量PCR的原理基于PCR技术,PCR是一种体外扩增DNA的方法,通过DNA聚合酶酶链反应(PCR)使目标DNA序列在体外得到扩增。
而实时荧光定量PCR则在PCR反应中引入了荧光探针,利用荧光信号的变化来监测PCR反应的过程。
在实时荧光定量PCR中,通常使用的荧光探针包括SYBR Green和TaqMan探针。
SYBR Green是一种DNA结合染料,它能够与PCR反应中产生的双链DNA结合并发出荧光信号,因此可以用来监测PCR反应的进程。
TaqMan探针则是一种双标记探针,包括一个荧光素和一个荧光淬灭器,它能够在PCR反应中特异性结合目标DNA序列并发出荧光信号,因此可以用来定量PCR反应中的目标DNA。
实时荧光定量PCR的原理可以简单概括为,首先,将待扩增的DNA样品与引物和荧光探针一起加入PCR反应体系中;然后,利用热循环装置对PCR反应体系进行多次循环加热和降温,使目标DNA序列得到扩增;在PCR反应过程中,荧光探针与目标DNA结合并发出荧光信号,这些信号会被实时监测和记录下来;最后,通过分析监测到的荧光信号的变化,可以计算出PCR反应体系中目标DNA的起始量,并进行定量分析。
实时荧光定量PCR在科研和临床中有着广泛的应用。
在科研领域,实时荧光定量PCR可以用于基因表达分析、病原微生物检测、基因型分析等方面,其高灵敏度和高特异性使其成为了研究人员进行生物学研究的重要工具。
在临床诊断中,实时荧光定量PCR可以用于疾病的早期诊断、疾病的病原体检测、药物代谢酶基因型分析等方面,其快速、准确和可靠的特点使其成为了临床诊断中不可或缺的手段。
实时荧光定量PCR检测乙型肝炎病毒DNA性能验证的临床应用

实时荧光定量PCR检测⼄型肝炎病毒DNA性能验证的临床应⽤《激光杂志》2014年第35卷第10期LASERNAL (V ol.35.No.10.2014)·激光医学(论⽂与临床报告)·摘要:⽬的:探讨⼀种适合于临床实际应⽤的荧光定量PCR 检测HBV-DNA 性能验证⽅法。
⽅法:参考美国临床实验室标准化协会(CLSI )性能验证的相关⽂件,应⽤实验室检测病⼈标本,室内质控数据,能⼒验证结果,按⽅法学性能评价指标设计验证试验,通过对实验结果的统计学分析,评价检测⽅法的性能是否符合临床预期⽤途。
结果:本⽂荧光定量PCR 检测HBV-DNA 批内CV ⾼、中、低⽔平分别为2.9%、2.6%和9.7%;批间CV⾼、低⽔平分别为3.7%和4.5%;实验室结果与参考靶值⽐较的回归⽅程为:y=0.9909x+0.0008;r 2=0.9945;线性范围验证为7.30E2~2.51E7,线性回归⽅程为:y=1.0247x-0.2232;r 2=0.9941;溶⾎时⾎红蛋⽩≤5g/l 对本实验结果⽆影响。
结论:经验证本实验荧光定量PCR 检测HBV-DNA 性能指标满⾜临床预期要求,常规应⽤结果满意;荧光定量PCR 的性能验证是保证其检验质量的必要⼿段,选择适合的验证⽅案将会⼤⼤提⾼性能验证的临床应⽤。
关键词:性能验证;聚合酶链反应;⼄型肝炎病毒;核酸检测中图分类号:R446.6;R512.6+2⽂献标识码:A DOI 编码:10.14016//doc/4c4e75a002020740bf1e9b08.html ki.jgzz.2014.10.131The application of the performance verification of the real-time quantitative PCR of thedetection in HBV-DNAXIA Ji-rong ,CAO Ju ,YANG Shuang-shuang(The first affiliated hospital of Chongqing medical university,Chongqing 400016,China )Abstract :Objective:To establish a new way of evaluating performance verification tests in detectingHBV-DNA by real-time PCR clinically.Methods:According to reference documents from Clinical and Laboratory Standards Institute (CLSI),we designed verification tests from the performance evaluation methodology using pa-tient samples,internal quality control data and proficiency test results,and evaluated if performance verification tests could be carried out in routing clinical tests.Results:The high,medium and low level of within-run precision CV in detecting HBV-DNA by real-time PCR is 2.9%、2.6%and 9.7%,respectively,and the high and low level of be-tween-run precision CV is 3.7%and 4.5%,respectively.By calculating test data and target values,one-dimensional linear regression equation is y=0.9909x+0.0008;r2=0.9945;linearity range is 7.30E2~2.51E7;equation of linear re-gression is y=1.0247x-0.2232;r2=0.9941;when hemoglobin is≤5g/l,hematolysis has no effects on HBV-DNA data.Conclusion:Performance verification indexes could be applied to routing clinical tests and satisfied clinical expecta-tions.Performance verification test is essential for the quality of real-time PCR,and appropriate performance verifi-cation test could improve clinical laboratory work.Key words:Performance verification;Polymerase chain reaction;HBV;DNA tests实时荧光定量PCR 检测⼄型肝炎病毒DNA 性能验证的临床应⽤收稿⽇期:2014-07-28基⾦项⽬:本⽂受国家临床重点专科建设项⽬经费资助,财社〔2010〕305号夏吉荣,曹炬,杨双双(重庆医科⼤学附属第⼀医院检验科,重庆400016)夏吉荣:实时荧光定量PCR 检测⼄型肝炎病毒DNA 性能验证的临床应⽤实时荧光定量PCR 检测⼄型肝炎病毒核酸(HBV-DNA),具有⾼敏感性和特异性,已被临床⼴泛应⽤于⼄型肝炎患者的诊断和疗效判断[1-5]。
Real-time PCR

实时PCR实时PCR(real-time PCR),属于定量PCR(Q-PCR)的一种,以一定时间内DNA的增幅量为基础进行DNA的定量分析。
real time PCR 的定量使用荧光色素,目前有二种方法。
一种是在ds DNA中插入特异的荧光色素,例如SYBR Green荧光染料;另一种使用一种能与增幅DNA序列中特定寡核酸序列相结合的一种荧光探针(probe),如Taqman 探针。
、两种方法原理不同。
SYBR Green荧光染料游离时不发光,当反应体系中存在双链DNA时,SYBR Green与双链DNA结合,此时才发光。
Taqman探针带有一个荧光基团和一个淬灭基团,荧光基团连接在探针的5’末端,而淬灭基团则在3’末端。
PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,此时荧光基团发出的荧光信号可以被检测到。
real time PCR 与 reverse transcription PCR 相结合,能用微量的RNA来找出特定时间、细胞、组织内的特别表达的遗传基因。
这两种RT PCR的组合又被称之为“定量RT-PCR(quantitative RT-PCR)”反应体系oligo: 多聚体,相当于mRNA引物AMV RT:禽类成髓细胞瘤病毒逆转录酶MMLV RT:莫洛尼鼠白血病病毒逆转录酶dNTPs:脱氧核苷酸RNase:RNA酶抑制剂PCR Buffer:RT-PCR缓冲液MgCl2:2价镁离注意事项:绝对定量:几种可能:1. 你的cDNA浓度是不是通过紫外分光测定的?紫外的干扰因素很多,只能作为浓度参考,也就是可能会导致你的内参Ct有差异,正常现象。
2. 反转录的效率,如果你的内参扩增产物在靠近mRNA的3'端影响不大,如果在里面或者5'端,Ct值存在差异正常。
实时荧光定量PCR法在乙肝病毒DNA及血清标志物检测中的应用价值

实时荧光定量PCR法在乙肝病毒DNA及血清标志物检测中的应用价值张艳奇【摘要】目的:探讨实时荧光定量PCR法在乙肝患者乙肝病毒(HBV)DNA及血清乙肝病毒标志物检测中的应用价值.方法:选取我院收治的327例乙肝患者,均先采用酶联免疫吸附法(ELISA)对HBV-M模式进行定性检测,再采用实时荧光定量PCR法对HBV-DNA进行定量检测.统计检测结果.结果:327例乙肝患者不同HBV-M模式下大三阳模式的HBV-DNA阳性检出率为96.43%(108/112)、⑤模式[依次检测乙型肝炎病毒表面抗原(HBsAg)(+)、乙型肝炎e抗原(HbeAg)(+)]HBV-DNA 阳性检出率为81.82%(18/22),高于其他模式(P<0.05);大三阳模式HBV-DNA水平高于其他模式(P<0.05).结论:实时荧光定量PCR法应用于乙肝患者HBV-DNA 及血清标志物检测中,HBV-DNA阳性检出率高,对疾病的早期治疗具有指导意义.【期刊名称】《黑龙江医药》【年(卷),期】2019(032)001【总页数】2页(P204-205)【关键词】乙肝;实时荧光定量PCR法;乙肝病毒DNA;血清标志物【作者】张艳奇【作者单位】安阳市第三人民医院检验科河南安阳 45500【正文语种】中文【中图分类】R512.6+2乙肝即乙型肝炎是一种发病率较高且较严重的慢性传染性疾患,相关文献资料显示我国乙肝患者高达2000万之多,居传染性疾病第二位,且近年有逐年上升趋势[1-2]。
该病具有病程长、病情迁延难愈等特点,可进展为肝硬化、肝细胞癌,对患者生命安全造成极大威胁。
早发现、早治疗为控制乙肝病情进展、延长患者生存时间、改善其生活质量的关键。
但乙肝患者症状表现较为复杂,仅从症状表现上评估诊断难度较大,不利于疾病的早期治疗;ELISA为HBV感染常用检测手段,具有操作简单、灵敏度高、检测迅速等优势,但对HBV的复制及感染状况不敏感,存在一定局限性[3]。
实时荧光定量pcr检测原理

实时荧光定量pcr检测原理实时荧光定量PCR(Quantitative Real-time PCR)是一种在DNA扩增反应中,以荧光化学物质测每次聚合酶链式反应(PCR)循环后产物总量的方法。
这种方法通过内参或者外参法对待测样品中的特定DNA序列进行定量分析。
Real-timePCR是在PCR扩增过程中,通过荧光信号,对PCR进程进行实时检测。
由于在PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系,所以成为定量的依据。
实时荧光定量PCR的基本原理是利用DNA聚合酶的5’-3’外切酶活性,在DNA合成过程中检测荧光信号的变化。
当DNA聚合酶与特定荧光染料标记的DNA引物结合时,荧光染料会被标记在引物的3’端。
在PCR反应过程中,每当DNA聚合酶添加一个核苷酸到引物3’端时,聚合酶的外切酶活性将荧光染料从引物上切割下来,释放出荧光。
通过实时检测荧光信号的变化,可以实时监测DNA的合成过程。
实时荧光定量PCR的定量原理是利用PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系。
在PCR扩增的指数时期,随着循环次数的增加,DNA产物的量呈指数增长。
在这个阶段,每个循环的DNA产物量与上一个循环的DNA产物量成比例。
因此,通过实时检测荧光信号的变化,可以确定PCR进程中的DNA产物量。
由于每个模板的起始拷贝数不同,因此不同模板的Ct值也不同。
通过比较不同模板的Ct值,可以确定模板的起始拷贝数。
实时荧光定量PCR具有许多优点,如高灵敏度、高特异性和高自动化程度。
它可以用于多种类型的样本检测,包括血液、组织、细胞培养液等。
此外,实时荧光定量PCR还可以用于基因表达分析、突变检测和病原体鉴定等应用。
实时荧光定量PCR是一种非常有用的技术,可以用于多种类型的样本检测和分析。
它的基本原理是利用DNA聚合酶的外切酶活性和荧光染料标记的引物来实时监测DNA的合成过程。
通过比较不同模板的Ct值,可以确定模板的起始拷贝数,从而实现定量分析。
实时荧光定量PCR检测乙型肝炎病毒DNA的研究

实时荧光定量PCR检测乙型肝炎病毒DNA的研究发表时间:2016-06-08T13:39:54.490Z 来源:《医师在线》2016年2月第4期作者:李亚萍[导读] 全世界超过20亿人感染过乙肝病毒,大约3. 5亿人为乙型肝炎患者,每年约有100万人死于乙型肝炎病毒相关的肝硬化和肝癌。
李亚萍(无锡市江阴利港医院超声科;江苏无锡 214444)【摘要】探讨:乙型肝炎血清标志物和乙型肝炎病毒DNA量之间的关系,分析荧光定量PCR在乙型肝炎检测中的作用。
HBVDNA定量是目前诊断乙型肝炎、判断病毒复制和评价患者肝脏病变、预后及药物疗效的重要指标之一,但在指导诊疗时仍需结合血清标记物、肝损害指标乃至HBV基因分型。
方法:PCR核酸探针杂交法、RELISA、竞争定量PCR、实时荧光定量PCR,本文综述定量检测HBVDNA意义及PCR检测方法。
结论: 实时荧光定量PCR检测乙型肝炎病毒DNA是反映HBV复制水平的可靠指标,有助于疾病的诊断及预后判断。
【关键词】乙型肝炎病毒核糖核酸实时荧光定量PCR全世界超过20亿人感染过乙肝病毒,大约3. 5亿人为乙型肝炎患者,每年约有100万人死于乙型肝炎病毒相关的肝硬化和肝癌。
而我国更是全球乙型肝炎的高发国家之一,人群乙型肝炎病毒HbsAg的携带率高达10%。
多年以来临床诊断HBV感染情况通常采用酶联免疫吸附试验(ELISA)检测乙肝病毒血清标志物模型(HBV -M,俗称两对半),以了解和判断乙型肝炎病毒在体内的复制水平、传染性强弱等,对乙型肝炎的诊断、抗病毒疗效观察及预后判断均具有重要意义。
实时荧光定量PCR是近几年发展起来的,可以定量检测HBV-DNA,由于其灵敏、快速、假阴、假阳性率极低,该技术已被广泛应用于临床检测,结合ELISA检测能够更为精确地了解HBV的感染情况。
以下就对HBVDNA定量在诊断治疗上的定量原理和意义、定量方法作综述,探讨乙型肝炎血清标志物和乙型肝炎病毒DNA数量之间的关系,分析荧光定量PCR在乙型肝炎检测中的作用。
real-time pcr原理

real-time pcr原理
实时聚合酶链反应(real-time PCR)是一种广泛应用于分子生
物学研究中的技术,用于检测和定量DNA或RNA的存在。
与传统聚合酶链反应(PCR)相比,实时PCR能够提供更快、更准确的结果。
实时PCR基于聚合酶链反应原理,通过扩增目标DNA或
RNA序列来检测其存在。
实时PCR使用一对特异性引物,即
前向引物和反向引物,与目标序列的两侧结合。
在反应过程中,DNA聚合酶会复制模板DNA,并在每个引物的结合位点依次
合成新的DNA链。
然而,与传统PCR不同的是,实时PCR在反应混合物中加入
了一种叫做探针的荧光探测剂。
这种探针通常由一个引物和一个荧光信号发生器组成。
当探针与目标序列结合时,引物会选择性地与模板DNA结合,并将荧光信号发生器离开。
在PCR
反应进行的同时,荧光信号会产生,并且可以实时监测到
PCR反应的进程。
实时PCR设备一般配备了一个光学系统,可以记录PCR反应
过程中产生的荧光信号。
光学系统能够定量检测荧光信号的强度,并将其转化为DNA或RNA的相对数量。
这使得实时
PCR能够定量目标DNA或RNA序列在样本中的存在量。
总的来说,实时PCR结合了PCR的增幅特性和荧光技术的快
速检测特性,可以在PCR反应进行的同时实时监测和定量目
标DNA或RNA序列的存在量。
这使得实时PCR成为现代分子生物学研究中的重要工具。
定时定量PCR

实时定量PCR (Real-time quantitative PCR )[ 文章来源: | 文章作者: | 发布时间:2007-09-14| 字体: [大 中 小]实时定量PCR (Real-time quantitative PCR )Vs 普通PCR ,RT-PCR 的优势采用封闭的检测模式,因此扩增产物导致污染的可能性比普通PCR 要小得多 。
扩增产物的检测在PCR 扩增过程中同时进行,并且数据的采集、分析全部由仪器自动完成,因此整个检测所需的时间比普通PCR 要节省许多。
检测模式功能强大,具备定性、定量、突变、多项目等检测功能。
而普通PCR 要完成上述项目需采用不同的技术平台。
进行定量检测时,其定量线性范围(5-6 logs )比普通PCR (2-3 logs )要宽得多。
Real-time 定量PCR 仪是在普通PCR 仪的基础上再配备一个激发光源和荧光信号检测系统的装置。
通过PCR 反应的数学函数关系,加入标准品,可以对待测样品中的目标基因进行准确定量。
Real-Time PCR热循环仪(PCR 仪)荧光检测系统计算机及软件系统回顾:PCR基本原理PCR是在试管中进行DNA复制反应,基本原理与体内相似在模板、引物、4种dNTP和耐热DNA聚合酶等存在的条件下,特异扩增位于两段已知序列之间的DNA 区段的酶促合成反应。
基本步骤变性:加热使双链DNA变为单链退火:降温使引物和互补模板在局部形成杂交链延伸:耐热DNA聚合酶按5′→3′方向催化以引物为起始点的延伸反应示意图Real-time定量PCR仪是在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
在real-time Q-PCR中,对整个PCR反应扩增过程进行了实时的监测和连续地分析扩增相关的荧光信号,随着反应时间的进行,监测到的荧光信号的变化可以绘制成一条曲线。
在PCR反应早期,产生荧光的水平不能与背景明显地区别,为了便于对所检测样本进行比较,在real-time Q-PCR反应的指数期,首先需设定一定荧光信号的域值,一般这个域值(threshold)是以PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3~15个循环的荧光信号的标准偏差的10倍。
实时定量PCR在乙型肝炎(HBV)诊断中的应用

实时定量PCR在乙型肝炎(HBV)诊断中的应用实时定量PCR是一种高效、敏感、特异性强的检测方法,可以在非常短的时间内检测出样品中的分子数量。
在乙型肝炎(HBV)的诊断中,实时定量PCR被广泛应用。
本文将从HBV的病因、临床诊断、传统检测方法以及实时定量PCR的优势等方面,对其在乙型肝炎诊断中的应用进行探讨。
乙型肝炎是由乙型肝炎病毒(HBV)引起的肝脏疾病。
乙型肝炎在全球范围内广泛流行,特别是在亚洲地区。
HBV通过血液、体液、母婴传播。
慢性乙型肝炎可引起肝硬化和肝癌等严重后果,给人类健康带来了严重威胁。
因此,准确和及时的乙型肝炎的诊断至关重要。
传统的HBV诊断方法包括放射免疫、酶联免疫、PCR等。
这些方法都具有一定的优缺点。
其中,PCR技术在HBV检测中表现出了明显的优势。
PCR技术可以检测微量的HBV DNA,即使是在临床表现症状不明显时也能够在血液中检测到HBV DNA。
但是,传统PCR在检测HBV时也存在一些不足。
例如,难以区分HBV DNA的真正来源,难以确定HBV DNA的数量。
实时定量PCR可以弥补传统PCR的这些不足之处。
实时定量PCR的检测过程比传统PCR更简单、更快和更准确。
实时定量PCR可以定量检测HBV DNA的数量,这是传统PCR所无法做到的。
同时,实时定量PCR可以根据多个样本中的HBV DNA数量,准确地评估慢性乙型肝炎患者疾病的发展和疗效。
实时定量PCR检测结果真实可信,能够有效指导医学决策。
实时定量PCR在HBV慢性感染的诊断中具有更高的敏感性和特异性,可大大提高治疗慢性HBV感染的成功率。
总之,实时定量PCR在乙型肝炎(HBV)的诊断中有着广泛的应用前景。
与传统PCR方法相比,实时定量PCR的优势在于检测速度更快、准确性更高、对HBV DNA数量的定量检测更为精准,能够更好地指导医学决策。
随着技术的进一步完善、价格的进一步降低,实时定量PCR在临床免疫学和分子诊断的应用前景将会更加广阔。
简述实时定量pcr的原理和应用

简述实时定量PCR的原理和应用1. 实时定量PCR简介实时定量PCR(quantitative real-time PCR, qPCR)是一种用于检测和定量DNA 或RNA的技术。
相比传统PCR技术,实时定量PCR可以快速、准确、高灵敏地测量靶分子的数量。
该技术广泛应用于生物医学研究、基因表达、疾病诊断等领域。
2. 实时定量PCR原理实时定量PCR基于传统PCR技术,通过引入荧光探针来实时监测PCR反应的进行。
其基本原理如下:•步骤1:DNA样本的准备与处理。
从待检测的样本中提取DNA,并对其进行纯化和定量。
•步骤2:引物设计和荧光探针制备。
根据目标DNA序列设计特异性引物和荧光探针。
•步骤3:PCR反应的进行。
将DNA模板与引物、荧光探针和PCR反应液混合,通过一系列的温度循环对DNA进行扩增。
•步骤4:荧光信号检测与分析。
在PCR反应过程中,荧光探针与目标DNA结合,并发出荧光信号。
荧光信号的强度与目标DNA的数量成正比,通过荧光信号的实时监测和分析,可以推断出DNA的起始量。
3. 实时定量PCR的应用实时定量PCR广泛应用于许多领域,包括:3.1 基因表达分析实时定量PCR可以定量检测基因在不同组织、不同时期、不同条件下的表达水平变化。
通过相对定量和绝对定量分析,可以研究基因的功能、调控机制及调控因子。
3.2 病原体检测实时定量PCR对病原体的检测具有高度灵敏性和特异性。
通过特异性引物和荧光探针的设计,可以快速、准确地检测病毒、细菌、真菌等病原体。
3.3 疾病诊断实时定量PCR在疾病诊断方面有广泛应用。
例如,它可以检测癌症相关基因的突变、染色体重排等变异,从而为癌症的早期诊断和治疗提供重要参考。
3.4 转基因检测实时定量PCR可以用于转基因生物的快速检测。
利用特异性引物和荧光标记的转基因探针,可以检测食品、农作物中是否存在转基因成分。
3.5 环境监测实时定量PCR可以用于环境中微生物的监测和定量分析。
两种全自动核酸检测系统对乙肝核酸检测性能的比较

两种全自动核酸检测系统对乙肝核酸检测性能的比较乙肝病毒(HBV)感染是一种常见的严重传染病,是全球范围内的公共卫生问题。
乙肝核酸检测是诊断和监测乙肝病毒感染的重要手段。
近年来,随着技术的不断发展,出现了许多全自动核酸检测系统。
本文将比较两种全自动核酸检测系统对乙肝核酸检测性能的差异。
第一种全自动核酸检测系统是荧光定量PCR(qPCR)系统。
这种系统利用荧光探针来测量PCR扩增产物的含量。
qPCR具有高灵敏度和高特异性的优势,是目前乙肝核酸检测的标准方法。
它可以精确地定量HBV病毒DNA的含量,从而评估病毒的复制水平。
qPCR还可以检测HBV基因的变异情况,帮助研究乙肝病毒的进化和耐药性。
第二种全自动核酸检测系统是基因芯片技术。
这种技术利用微阵列芯片上固定的探针来检测多个核酸序列。
它具有高通量、高灵敏度和高特异性的特点。
基因芯片技术可以通过一次实验检测多个核酸标志物,不仅可以检测HBV病毒DNA,还可以检测其他乙肝相关基因的表达水平。
这种多目标检测的能力使得基因芯片技术在乙肝的早期诊断和分型上具有潜力。
两种全自动核酸检测系统在乙肝核酸检测性能上存在一些差异。
qPCR系统具有更高的灵敏度。
它可以检测到非常少量的HBV病毒DNA,可以帮助早期诊断乙肝病毒感染。
相比之下,基因芯片技术的灵敏度稍低,可能会错过一些低水平的感染。
两种系统在特异性上也存在差异。
qPCR系统具有非常高的特异性,可以准确区分HBV 和其他相关病毒的核酸序列。
而基因芯片技术可能存在一定的交叉反应,可能会导致一些假阳性结果。
两种系统的成本和操作复杂性也有所不同。
qPCR系统相对简单易用,操作便捷,但成本较高。
而基因芯片技术的成本相对较低,但操作复杂,需要专业技能人员进行操作。
两种全自动核酸检测系统在乙肝核酸检测性能上存在一些差异。
选择适合的系统应根据实际需求和资源情况进行综合考虑。
qPCR系统适用于需要高灵敏度和高特异性的临床实验室,而基因芯片技术适用于大规模筛查和研究领域。
实时荧光PCR定量检测乙型肝炎病毒脱氧核糖核酸的检测及临床意义

实时荧光PCR定量检测乙型肝炎病毒脱氧核糖核酸的检测及临床意义摘要】目的:探讨乙型肝炎患者体内乙型肝炎病毒(HBV)的实时荧光PCR定量方法及临床意义。
方法:对60例临床血清标本用荧光PCR定量检测的操作方法及临床意义。
结果:乙型肝炎大三阳患者血清HBV DNA检出率为100% ,HBV DNA的对数值为7.2±1.8;小三阳患者血清HBV DNA检出率为64.5% , HBV DNA的对数值为4.9±1.3。
结论:HBV DNA是HBV感染后病毒血症最直接的标志,是判断病毒复制最灵敏的指标,同时也是判断慢性乙型肝炎是否具有活动性的重要指标。
【关键词】实时荧光PCR定量检测;乙型肝炎;病毒脱氧核糖核酸;临床意义【中图分类号】R51 【文献标识码】A 【文章编号】1007-8231(2017)28-0171-02实时荧光PCR定量检测乙型肝炎病毒脱氧核糖核酸(HBV DNA)通常分为二步:首先从标本中提取HBV DNA,然后采用实时荧光PCR扩增 HBV DNA。
目前荧光定量聚合酶链反应(FQ-PCR)检测技术逐渐应用于临床实验诊断[1]。
本实验采用实时荧光定量PCR方法(PCR FQ-PCR)测试了60份临床血清标本结果进行分析。
1.材料与方法1.1 标本来源收集本院住院及门诊收治的乙肝患者血清60份,健康体检者血清60分,其中男40例,女20例,年龄7~80岁。
1.2 标本采集、保存与运送无菌采集静脉血3ml,收集于无菌离心管中(不抗凝或采用EDTA、枸橼酸钠抗凝),室温放置不超过2h,1 600g离心20min,分离血清或血浆,转入无菌离心管中备用。
分离的血浆或血清在4℃保存不应超过24h;-20℃保存不超过3个月;-70℃以下长期保存。
应避免反复冻融。
采用冰壶加冰或泡沫箱加冰密封进行运输。
1.3 方法主反应混合物的配制,从试剂盒中取出HBV PCR反应液、 Taq/UNG酶、HBV荧光探针和MgCl2,室温融化并振荡混匀后,2000r/min离心10s。
乙型肝炎病毒核酸的实时荧光定量PCR检测及其临床意义

乙型肝炎病毒核酸的实时荧光定量PCR检测及其临床意义王旭东;张冬雷;李立人;施健;崔之础;王惠民【期刊名称】《现代检验医学杂志》【年(卷),期】2006(021)003【摘要】目的建立实时荧光定量PCR(real-time fluorescence quantitative PCR,RFQ-PCR)检测HBV-DNA含量的方法,探讨其在乙型肝炎预防、诊断、治疗及判断病情等方面的临床应用价值.方法在HBV S基因高保守区设计了特异性的引物和探针,利用TaqMan探针的基本原理,PCR扩增目的片段并实时检测产物的荧光强度,根据标准品建立的标准曲线,由软件自动计算出待测样本中HBV-DNA的准确含量.结果 220例临床样本的检测结果显示,乙肝大三阳患者HBV-DNA阳性率(98.8%)显著高于其它各组(P<0.01),病毒平均含量为7.52±0.43 copies/ml;小三阳患者HBV-DNA阳性率低于大三阳组(P<0.01),但平均病毒含量与大三阳组无差异(P>0.05),高达7.41±0.26 copies/ml;单独HBsAg阳性和单独HBsAb阳性组HBV-DNA阳性率分别为56.4%,19.2%,平均病毒含量分别为5.35±0.32 copies/ml,4.02±0.31 copies/ml;80例献血员血清仍有3例HBV-DNA阳性,其病毒含量低于其它各组.结论 RFQ-PCR检测HBV-DNA含量比ELISA法具有更好的灵敏度和特异性,能准确反映HBV在体内的复制情况,对临床上抗病毒药物的选择和判断疾病的预后意义重大.【总页数】5页(P15-19)【作者】王旭东;张冬雷;李立人;施健;崔之础;王惠民【作者单位】南通大学附属医院检验医学中心,江苏南通,226001;南通大学附属医院酶学研究室,江苏南通,226001;南通大学附属医院消化内科,江苏南通,226001;南通大学附属医院酶学研究室,江苏南通,226001;南通大学附属医院酶学研究室,江苏南通,226001;南通大学附属医院检验医学中心,江苏南通,226001【正文语种】中文【中图分类】Q503;R512.6+2【相关文献】1.实时荧光定量PCR检测乙型肝炎病毒核酸载量的测量不确定度构成分析 [J], 余南;毕晓云;崔进;陈小坚;李汛2.实时荧光定量PCR检测TMPRSS4mRNA在41例肺腺癌中的表达及临床意义[J], 李受南;吴正球;卢强昌;雷敬富;罗德丰3.实时荧光定量PCR检测胃癌SOX2的表达及其临床意义 [J], 黄正接;江龙;尤俊;曾岳岳;吴炳霖;陈百胜;冯庆钊;罗琪4.乙型肝炎病毒核酸实时荧光定量PCR检测在临床诊断中的价值 [J], 黄四爽;熊勤5.实时荧光定量PCR检测人巨细胞病毒感染的临床意义分析 [J], 谭天照; 李学仿; 黄广荣因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Journal of Virological Methods126(2005)207–213Real-time PCR quantitation of hepatitis B virus DNA using automated sample preparation and murine cytomegalovirus internal control J.A.Garson a,b,∗,P.R.Grant b,U.Ayliffe a,R.B.Ferns a,R.S.Tedder a,ba Centre for Virology,Department of Infection,University College London,London,UKb Department of Virology,UCL Hospitals NHS Foundation Trust,London,UKReceived16December2004;received in revised form24February2005;accepted3March2005Available online2April2005AbstractQuantitation of circulating hepatitis B virus(HBV)DNA is important for monitoring disease progression and for assessing the response to antiviral therapy.Several commercial and‘in house’assays for HBV DNA quantitation have been described but many of these have limitations of relatively low sensitivity and limited dynamic range.This study describes the development and evaluation of a FRET-based real-time PCR assay designed to overcome these limitations and to provide accurate quantitation of DNA from all eight genotypes of HBV(A–H).The assay employs a fully automated nucleic acid extraction system permitting high-sample throughput with minimal‘hands-on’time and incorporates a murine cytomegalovirus(mCMV)internal control to prevent false negative results and under-reporting due to unrecognised problems with viral lysis,DNA purification or PCR amplification.Sensitivity,assessed by Probit analysis at the95% detection level,was24.4IU/ml,associated with an extremely wide dynamic range(∼9log10).Coefficients of variation were low for both intra-assay and inter-assay variability(CV%,7–11%)and quantitative data correlated well(R2=0.97)with the Digene hybrid capture assay. This assay provides an ideal system for therapeutic monitoring and for studying the relationship between HBV viral load and stage of disease.©2005Elsevier B.V.All rights reserved.Keywords:Hepatitis B virus;HBV DNA;Chronic hepatitis;Real-time PCR;Automation;Therapeutic monitoring1.IntroductionHepatitis B virus(HBV)has infected2billion people across the globe and given rise to at least350million per-sistently infected carriers.Carriers are at increased risk of developing cirrhosis and hepatocellular carcinoma,and as a consequence HBV is now estimated to be the10th lead-ing cause of death worldwide(Lavanchy,2004).In recent years,measurement of the circulating HBV DNA level has come to be regarded as the most direct and reliable means of monitoring HBV infection and has been widely used in pre-treatment evaluation and clinical staging(Mommeja-Marin et al.,2003;Chu et al.,2002).Quantitative HBV DNA assays have also been employed for assessing infectivity,for mon-∗Corresponding author.Tel.:+442076799490;fax:+442075805896.E-mail address:j.garson@(J.A.Garson).itoring antiviral therapy and for detecting the emergence of drug resistance(Berger et al.,2001;Corden et al.,2003).Numerous commercial and‘in house’molecular assays for HBV DNA using a variety of hybridisation technolo-gies including dot blot,hybrid capture,branched DNA sig-nal amplification and polymerase chain reaction(PCR)have been described over the past decade(Zaaijer et al.,1994; Hendricks et al.,1995;Poljak et al.,2001)but many of these have relatively low sensitivity and limited dynamic range. The recent introduction offluorescence resonance energy transfer(FRET)-based real-time PCR(Ho et al.,2003;Weiss et al.,2004;Stelzl et al.,2004;Sum et al.,2004)has been par-ticularly advantageous for HBV DNA quantitation because it provides high sensitivity with a much broader dynamic range (typically≥7log10)than alternative assay types.In addition, real-time PCR assays offer greater accuracy,are more rapid and pose less risk of amplicon contamination than other PCR0166-0934/$–see front matter©2005Elsevier B.V.All rights reserved. doi:10.1016/j.jviromet.2005.03.001208J.A.Garson et al./Journal of Virological Methods126(2005)207–213formats because they permit simultaneous amplification and detection in a closed-vessel system.We describe here a novel real-time quantitative assay for HBV DNA which utilises high-throughput robotic sam-ple preparation and a murine cytomegalovirus(mCMV)in-ternal control.The assay has been designed to enable ac-curate quantitation of all eight genotypes of HBV(A–H) and to monitor the efficiency of viral lysis,DNA ex-traction,PCR amplification and signal detection for each sample.2.Materials and methods2.1.Clinical samples and controlsResidual EDTA plasma and serum from clinical samples referred for diagnostic HBV marker analysis were stored at−20◦C.These included samples from32patients with chronic HBV infection who had previously had their serum HBV DNA levels quantified by Digene Hybrid Capture®II assay and samples from38patients with HBeAg pos-itive chronic HBV infection who were being treated with Lamivudine.A consecutive series of256HBV DNA posi-tive plasma samples from patients of known e antigen sta-tus was also analysed(181HBeAg positive and75anti-HBe positive).Controls included HCV-RNA positive plasma sam-ples from32patients with chronic hepatitis C infection and 48samples from healthy blood donors.Samples from blood donors were provided by the National Blood Service and the HBV DNA proficiency panel was obtained from Quality Control for Molecular Diagnostics().Geno-typed HBV samples(genotypes confirmed by dideoxy se-quencing,Norder et al.,1993)were kindly donated by S.Ijaz (Health Protection Agency,London,UK).For assay calibra-tion,the WHO HBV International Standard97/746(1million International Units per millilitre(IU/ml))was supplied by the UK National Institute for Biological Standards and Control (Saldanha et al.,2001).For use as an internal control,the Smith strain of murine cytomegalovirus(American Type Cell Collection,ATCC No.VR-1399)was cultured in the murine fibroblast cell line C1217(Selgrade et al.,1981).Culture su-pernatant was harvested,clarified and stored in aliquots at −70◦C.The titre of the clarified mCMV supernatant was estimated by the limit dilution/Poisson distribution method (Simmonds et al.,1990)using a nested PCR primer set de-scribed previously(Tedder et al.,2002a).2.2.Automated nucleic acid extractionViral DNA for quantitation by real-time PCR was ex-tracted automatically on a Qiagen BioRobot9604utilising QIAamp96Virus Kit reagents(Qiagen,Hilden,Germany) according to the manufacturer’s protocol.Ten microlitres of mCMV supernatant(at500copies/l)was added to each millilitre of lysis buffer(AL buffer),equivalent to approx-imately1000copies of mCMV per200l of plasma/serum sample or∼250copies of mCMV DNA per PCR reaction. Briefly,200l of plasma/serum was added to200l of the mCMV-containing viral lysis buffer and incubated at56◦C for10min,after which230l of100%ethanol were added. The mixture was passed through a96-well QIAamp vacuum plate and washed twice.DNA was eluted from the matrix in 86l of nuclease-free water.2.3.Real-time PCR primers and TaqMan probesOne hundred and twenty-six sequences representing all eight HBV genotypes(A–H)were downloaded from the GenBank nucleotide database and aligned using the program ClustalX v1.81(Thompson et al.,1997).A highly conserved region of the S gene was selected for the design of real-time PCR primers and afluorogenic5 -nuclease assay probe(Taq-Man probe).A single base mismatch in genotype B was ac-commodated by inclusion of a mixed nucleotide(G/T)in the probe sequence.Sequences of the HBV primers and dual labelled TaqMan probe are as follows:HBVTAQ1(sense,5 -GTG TCT GCG GCG TTT TAT CA);HBVTAQ2(antisense, 5 -GAC AAA CGG GCA ACA TAC CTT);HBVTAQPR (sense,5 -FAM-CCT CT(T/G)CAT CCT GCT GCT ATG CCT CAT C-TAMRA).Murine cytomegalovirus primers and TaqMan probe were based on GenBank accession number U68299:mCMVTAQ1 (sense,5 -AAC CCG GCA AGA TTT CTA ACG,nucleotides 95916–95936);mCMVTAQ2(antisense,5 -ATT CTG TGG GTC TGC GAC TCA,nucleotides96004–95984);mCMV-TAQPR(sense,5 -VIC-CTA GTC ATC GAC GGT GCA CAT CGG C-TAMRA,nucleotides95952–95976).Primers and probes were synthesised by MWG Biotech(UK)Ltd. BLAST searches(Altschul et al.,1990)predicted that the HBV and mCMV primer/probe sets would be specific for their respective targets and would not be expected to cross react with any other microbial or human sequences.2.4.Real-time PCR amplification and detectionViral DNA extracted from plasma/serum was amplified and quantified in an ABI Prism7000sequence detection sys-tem(Applied Biosystems,Foster City,CA,USA).The re-action was prepared using25l TaqMan®Universal PCR Master Mix(containing AmpliTaq Gold®DNA polymerase, AmpErase®UNG,dNTPs with dUTP,passive reference dye and optimized buffer components;Applied Biosystems, product number4304437)with0.4M primers,0.2M probes and20l extracted DNA(equivalent to approximately 50l of plasma/serum)in a50l total reaction volume. An initial2min incubation at50◦C to activate uracil-N-glycosylase(UNG)and destroy potential carry-over ampli-cons was followed by10min at95◦C to activate the ther-mostable DNA polymerase.Subsequently,45cycles of95◦C for15s and60◦C for1min were performed in9600emula-tion mode.J.A.Garson et al./Journal of Virological Methods126(2005)207–2132092.5.Digene hybrid capture assayThe Digene Hybrid Capture®II HBV DNA Test was used in accordance with the manufacturer’s instructions(Di-gene Corp.,Gaithersburg,MD,USA).The standard test for-mat was employed giving a dynamic range of1.4×105to 1.7×109HBV copies/ml.Briefly,denatured HBV DNA in the test sample was hybridised with an HBV-specific RNA probe mix and the resulting RNA:DNA hybrids captured onto wells of a microtitre plate coated with hybrid-specific anti-bodies.The captured hybrids were detected and quantified by means of an alkaline phosphatase labelled antihybrid anti-body in conjunction with a chemiluminescent substrate.Cal-ibration of the Digene assay is based on a series of manufac-turer’s HBV DNA plasmid standards quoted as1.42×105, 2.83×107,5.66×108and1.7×109copies/ml.2.6.HBsAg and HBeAg assaysHepatitis B surface antigen(HBsAg)was detected by en-zyme immunoassay(Abbott Murex HBsAg Version3,ref-erence GE34/36)used according to the manufacturer’s in-structions.HBsAg was quantitated using a reverse passive haemagglutination test(Serodia®HBs antigen test kit,Fu-jirebio Inc.,Japan).HBV e antigen(HBeAg)was measured with an‘in house’assay(Ferns and Tedder,1985)using commercially available reagents(Murex Biotech Ltd.,UK, HBeAg/anti-HBe GE19reagents).3.Results3.1.Calibration of‘in house’standard against the WHO HBV International Standard97/746Two hundred andfifty millilitres of an HBeAg positive serum from an HBV-infected blood donor were aliquoted and frozen at−70◦C for use as an‘in house’standard.The titre of this material was found to be2×108IU/ml when calibrated against the WHO HBV International Standard97/746.The amplification efficiency of the‘in house’standard was the same as that of the WHO standard as judged by the identical slopes of their superimposed calibration curves(Fig.1).A typical amplification plot generated by the ABI Prism7000 software from calibration standards used to construct the stan-dard curve is shown in Fig.2.HBV amplification efficiency was not affected by the presence of∼250copies of the in-ternal control mCMV DNA in each reaction(data not pre-sented).NB:all of the data presented in this paper were de-rived from assays in which the mCMV internal control was utilised.3.2.Lower limit of detection and dynamic rangeThe lower limit of detection of the assay was assessed by testing multiple replicates of dilutions of the WHO HBVIn-Fig.1.Calibration of the‘in house’standard against the WHO HBV In-ternational Standard97/746.The‘in house’standard dilutions(diamond symbols)shown here are101,102,103,104,105,106and107fold,and the WHO standard dilutions(cross symbols)are101,2×101,102,2×102, 2×103and2×104fold.ternational Standard97/746.Dilutions equal to100,50,25, 10,and5IU/ml were tested in batches of8replicates on three separate assay runs,giving a total of24replicates at each dilution(Table1).Using Probit analysis(Arcus QuickStat Biomedical v1.0software),the number of positive results at each dilution was used to calculate the HBV DNA concentra-tion giving a95%probability of detection.The95%detec-tion limit was determined to be24.4IU/ml.Samples of HBV genotypes A–G exhibited equivalent amplification/detection efficiency,as predicted by the GenBank database analysis used for primer and probe design.HBV genotype H was not available for testing but would be expected on sequence grounds to be amplified and detected with the same efficiency as the other seven genotypes.The dynamic range of the as-say was found to be approximately9log10(10–1010IU/ml). Diluting samples of known titre in either serum or plasma produced identical results(data not presented).Table1Real-time PCR assay data used for Probit analysisHBV concentration a(IU/ml)Numberpositive/testedPercentagepositive 10024/24100.0 5024/24100.0 2523/2495.8 1017/2470.8515/2462.500/240.0a Based on dilutions of the WHO HBV International Standard97/746in normal human plasma.210J.A.Garson et al./Journal of Virological Methods 126(2005)207–213Fig.2.Typical amplification plot generated by ABI Prism 7000software.Reading from left to right the curves represent the following calibration standards:108,107,106,105,104,103,102and 10IU/ml.3.3.SpecificityForty-eight normal human sera were tested in the assay and all gave negative results.Thirty-one of 32plasma samples from patients with chronic hepatitis C infection also gave negative results and the remaining sample was subsequently shown to be doubly infected with HBV and HCV .Samples of normal human genomic DNA and DNA extracted from HIV infected human lymphocytes were also shown to be non-reactive in the assay as was human cytomegalovirus DNA.In a series of 100assays carried out over a two-year period,no evidence of cross contamination was observed between adjacent wells containing the top calibration HBV standard (108IU/ml)and normal human serum.3.4.ReproducibilityInter-assay coefficient of variation (CV%based on HBV DNA concentration in IU/ml),calculated from seven con-secutive assays run over a 2-month period,was 11.1%for a low-titre sample (200IU/ml)and 8.5%for a high-titre sample (107IU/ml).The intra-assay coefficient of variation was 7.0%on seven repeats of 105IU/ml and 7.1%on seven repeats of 103IU/ml.The threshold cycle for the mCMV internal con-trol signal was typically between 31and 33cycles for all samples.Rarely (less than 0.3%of specimens)test samples exhibited a delayed mCMV threshold cycle indicating either inhibition of the PCR reaction or inefficiency of nucleic acid extraction,for example,due to a blocked well in the QIAampvacuum plate.In such cases,repeat quantitation of the sample at a 1:10dilution was generally successful.3.5.Correlation with Digene hybrid capture assay Thirty-two samples from patients with chronic HBV in-fection which had had HBV DNA concentrations quantified by Digene hybrid capture assay were reanalysed using the real-time PCR assay.Correlation between the two assays was excellent (R 2=0.97)over the shared 4log 10dynamic range (Fig.3).For the purpose of this comparison,IU/ml determined by the real-time PCR assay were converted into copies/ml using a multiplication factor of 5.This conver-sion was required because quantitative data generated by the Digene assay are expressed in copies/ml.The conver-sion factor was based on the international collaborative study of Saldanha et al.(2001)in which the WHO International Unit was shown to be equivalent to approximately five HBV genome copies.3.6.Performance on clinical samplesIn a consecutive series of 256HBV DNA positive plas-mas,the geometric mean HBV DNA titre of the 181HBeAg containing samples was 5×106IU/ml.The geometric mean titre of the 75anti-HBe positive samples was significantly lower (P <0.001)at 2×104IU/ml.The real-time PCR assay was successfully used to monitor the response to antiviral therapy in 38patients with HBeAgJ.A.Garson et al./Journal of Virological Methods 126(2005)207–213211Fig.3.Correlation between HBV DNA titres in 32HBsAg positive samples measured by Digene hybrid capture assay and by the novel real-time PCR assay described in thisstudy.Fig.4.Monitoring of HBV DNA titre (solid line)and HBsAg titre (dashed line)in a patient with chronic hepatitis B infection over a three-year period.The duration of antiviral therapy with Lamivudine is indicated by the stippled box.Seroconversion from HBeAg to anti-HBe is shown at the top of the figure together with serum ALT data for each time point (normal range for ALT,10–35IU/ml).positive chronic HBV infection.A typical case is illustrated in Fig.4.This patient exhibited a dramatic (>4log 10)decline in circulating HBV DNA concentration soon after commencing Lamivudine followed by a more gradual decline over the sub-sequent 15months.The decline in HBV DNA concentration was accompanied by normalisation of serum alanine amino transferase (ALT)levels.Following cessation of Lamivudine therapy,HBV DNA concentration was shown to increase by approximately 3log 10.The changing levels of circulat-ing HBV DNA in this patient were reflected by concordant changes in HBsAg titre and accompanied by seroconversion from HBeAg to anti-HBe positivity.3.7.Performance on quality assurance panelResults obtained by testing a coded panel of HBV pro-ficiency samples supplied by Quality Control for Molecular Diagnostics (QCMD )are shown in Fig.5.Once again HBV DNA concentrations measured in IU/ml by the real-time PCR assay were converted into copies/ml using a multiplication factor of 5(Saldanha et al.,2001).There was a good corre-212J.A.Garson et al./Journal of Virological Methods126(2005)207–213Fig.5.Results obtained by blind testing the QCMD hepatitis B proficiency panel.NB:a negative result(i.e.below the lower limit of detection)is rep-resented by1.00E+00in thisfigure.lation between the data generated by the real-time PCR as-say and the titres provided by QCMD on decoding the panel (R2=0.9985).4.DiscussionA variety of commercial assays for HBV DNA quanti-tation are now available and routinely used in diagnostic laboratories worldwide.Although generally reliable and convenient,many of these assays offer only a relatively limited dynamic range so that clinical samples frequently have to be diluted and retested in order to avoid exceeding the upper limit of quantitation(Hendricks et al.,1995;Poljak et al.,2001;Kessler et al.,2000).The novel real-time PCR as-say described here exhibits an extraordinarily wide dynamic range(∼9log10)and efficiently overcomes this limitation. The advantage of this assay characteristic is well illustrated by Fig.4in which HBV DNA concentrations vary over a range of∼8log10within a single patient.Fig.4also illus-trates the importance of extreme assay sensitivity especially when monitoring patients on antiviral therapy;18of the21 plasma samples analysed in this case would have been below the level of detection by Digene hybrid capture assay.The lower limit of detection of the real-time PCR assay described in this study(Table1)matches that of the most sensitive tests yet described(Weiss et al.,2004;Sum et al.,2004).Pre-extraction centrifugation and/or use of a larger plasma/serum volume could further increase the sensitivity of the assay if required(Gobbers et al.,2001;Burgener et al.,2003).The use of‘safety’primers(Heermann et al.,1996)based on the primer set described here could also be considered.Such primers would reduce the small theoretical risk of inefficient amplification in case of the emergence of HBV strains with sequence variation corresponding to the3 end of the current primers.Good reproducibility is an essential requirement of nu-cleic acid quantitation tests and the very low inter-assay and intra-assay variability(CV%,7–11%)of this real-time PCR compares favourably with other protocols(Stelzl et al.,2004). Most previously described real-time PCR assays have not in-cluded an internal control in each sample from the lysis step onwards.This is important because such a strategy controls each sample for the efficiency of nucleic acid purification as well as revealing the presence of PCR inhibitors.The present assay,by employing whole virions(mCMV)rather than plas-mid DNA(Weiss et al.,2004)as internal control,goes even further by controlling for the efficiency of the viral lysis step itself.mCMV is readily available,non-hazardous to humans and easy to culture and harvest.It has proven useful for this purpose in other assays(Tedder et al.,2002a)and is now uni-versally employed as an internal control for all DNA virus ‘in house’PCR protocols in our laboratory.Statistically significant differences in HBV DNA titres be-tween HBeAg positive and anti-HBe positive samples were observed in this study and similar differences have been re-ported by others using real-time PCR and other assays(Ho et al.,2003;Jardi et al.,2001;Tedder et al.,2002b).To be able accurately to compare HBV DNA results generated by differ-ent laboratories using different tests,it is essential to have a well validated,internationally accepted HBV standard which can be used for assay calibration(Saldanha et al.,2001).We therefore used the World Health Organisation International HBV Standard97/746as the primary calibration standard and test results are therefore expressed in International Units per millilitre.By using a multiplication factor of5to con-vert IU/ml into copies/ml(Saldanha et al.,2001)an excellent correlation was observed between the results of the real-time PCR and the results of the Digene hybrid capture assay.Sim-ilarly,using the same correction factor,the results generated by the real-time PCR assay were in good agreement with the values defined by the QCMD proficiency panel.By using robotic technology for automated nucleic acid extraction and by exploiting real-timefluorescence for de-tection of amplicons,the length of time required to generate results has been greatly reduced in comparison with conven-tional PCR-based assays.High throughput is possible as all stages are conducted in a standard96-well microtitre plate format.The total assay duration for86patient samples is approximately4.5h of which only1h involves‘hands-on’activity.The assay is suitable for both plasma and serum samples,and reagent,royalty and consumables costs are sig-nificantly lower than for commercial assays.In conclusion,the novel real-time PCR assay described in this paper offers several important advantages for HBV DNA quantitation.These include:(i)an extremely wide dynamic range and a95%detection limit of24.4IU/ml,(ii)very high intra-assay and inter-assay reproducibility,(iii)primers and probes designed to accurately quantify all eight genotypes of HBV,(iv)calibration based on the WHO International Stan-dard97/746,(v)an excellent performance with the QCMD proficiency panel and a high correlation with Digene hybridJ.A.Garson et al./Journal of Virological Methods126(2005)207–213213capture assay over4log10,(vi)the inclusion of mCMV to control for the efficiency of all stages of the assay including viral lysis,(vii)a rapid high-throughput system with mini-mal‘hands-on’time due to robotic nucleic acid extraction and real-time amplicon detection and(viii)significantfinancial savings in comparison with commercial assays.We consider that this assay represents an ideal system in a busy diagnostic laboratory for monitoring the virological response in chron-ically infected patients undergoing antiviral therapy and for studies designed to explore the relationship between HBV viral load and stage of disease.AcknowledgmentsThis study was funded by a translational research grant from the UCL Hospitals NHS Foundation Trust.We thank S.Ijaz(Health Protection Agency)for providing genotyped HBV samples and S.Garson for assistance with preparation of the manuscript.J.Neyts(Katholieke Universiteit,Leuven, Belgium)kindly provided the C1217cell line and mCMV strain Smith,and blood donor samples were made available by the National Blood Service.ReferencesAltschul,S.F.,Gish,W.,Miller,W.,Myers,E.W.,Lipman,D.J.,1990.Basic local alignment search tool.J.Mol.Biol.215,403–410. Berger,A.,Preiser,W.,Doerr,H.W.,2001.The role of viral load de-termination for the management of human immunodeficiency virus, hepatitis B virus and hepatitis C virus infection.J.Clin.Virol.20, 23–30.Burgener,M.,Candrian,U.,Gilgen,M.,parative evaluation of four large-volume RNA extraction kits in the isolation of viral RNA from water samples.J.Virol.Methods108,165–170.Chu,C.J.,Hussain,M.,Lok,A.S.,2002.Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection.Hepa-tology36,1408–1415.Corden,S.,Ballard,A.L.,Ijaz,S.,Barbara,J.A.,Gilbert,N.,Gilson,R.J., Boxall,E.H.,Tedder,R.S.,2003.HBV DNA levels and transmission of hepatitis B by health care workers.J.Clin.Virol.27,52–58. Ferns,R.B.,Tedder,R.S.,1985.Detection of both hepatitis B e antigen and antibody in a single assay using monoclonal reagents.J.Virol.Methods11,231–239.Gobbers,E.,Oosterlaken,T.A.,van Bussel,M.J.,Melsert,R.,Kroes,A.C., Claas,E.C.,2001.Efficient extraction of virus DNA by NucliSens Extractor allows sensitive detection of hepatitis B virus by PCR.J.Clin.Microbiol.39,4339–4343.Heermann,K.H.,Seitz,H.,Thomssen,R.,1996.Capture and RT-PCR of hepatitis C virus RNA with safety primers.J.Virol.Methods59, 33–43.Hendricks, D.A.,Stowe, B.J.,Hoo, B.S.,Kolberg,J.,Irvine, B.D., Neuwald,P.D.,Urdea,M.S.,Perrillo,R.P.,1995.Quantitation of HBV DNA in human serum using a branched DNA(bDNA)signal ampli-fication assay.Am.J.Clin.Pathol.104,537–546.Ho,S.K.,Yam,W.C.,Leung,E.T.,Wong,L.P.,Leung,J.K.,Lai,K.N., Chan,T.M.,2003.Rapid quantification of hepatitis B virus DNA byreal-time PCR usingfluorescent hybridization probes.J.Med.Micro-biol.52,397–402.Jardi,R.,Rodriguez,F.,Buti,M.,Costa,X.,Cotrina,M.,Valdes,A., Galimany,R.,Esteban,R.,Guardia,J.,2001.Quantitative detection of hepatitis B virus DNA in serum by a new rapid real-timefluorescence PCR assay.J.Viral Hepat.8,465–471.Kessler,H.H.,Stelzl,E.,Daghofer,E.,Santner,B.I.,Marth,E.,Lackner,H.,Stauber,R.E.,2000.Semiautomated quantification of hepatitisB virus DNA in a routine diagnostic b.Immunol.7,853–855.Lavanchy,D.,2004.Hepatitis B virus epidemiology,disease burden,treat-ment,and current and emerging prevention and control measures.J.Viral Hepat.11,97–107.Mommeja-Marin,H.,Mondou, E.,Blum,M.R.,Rousseau, F.,2003.Serum HBV DNA as a marker of efficacy during therapy for chronic HBV infection:analysis and review of the literature.Hepatology37, 1309–1319.Norder,H.,Hammas,B.,Lee,S.D.,Bile,K.,Courouce,A.M.,Mushah-war,I.K.,Magnius,L.O.,1993.Genetic relatedness of hepatitis B vi-ral strains of diverse geographical origin and natural variations in the primary structure of the surface antigen.J.Gen.Virol.74,1341–1348. Poljak,M.,Marin,I.J.,Seme,K.,Brinovec,V.,Maticic,M.,Meglic-V olkar,J.,Lesnicar,G.,Vince,A.,2001.Second-generation Hybrid capture test and Amplicor monitor test generate highly correlated hep-atitis B virus DNA levels.J.Virol.Methods97,165–169. Saldanha,J.,Gerlich,W.,Lelie,N.,Dawson,P.,Heermann,K.,Heath,A.,2001.An international collaborative study to establish a WorldHealth Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques.V ox Sang.80,63–71. Selgrade,M.K.,Nedrud,J.G.,Collier,A.M.,Gardner,D.E.,1981.Effects of cell source,mouse strain,and immunosuppressive treatment on production of virulent and attenuated murine cytomegalovirus.Infect.Immun.33,840–847.Simmonds,P.,Zhang,L.Q.,Watson,H.G.,Rebus,S.,Ferguson,E.D., Balfe,P.,Leadbetter,G.H.,Yap,P.L.,Peutherer,J.F.,Ludlam,C.A., 1990.Hepatitis C quantification and sequencing in blood products, haemophiliacs,and drug ncet336,1469–1472.Stelzl,E.,Muller,Z.,Marth,E.,Kessler,H.H.,2004.Rapid quantification of hepatitis B virus DNA by automated sample preparation and real-time PCR.J.Clin.Microbiol.42,2445–2449.Sum,S.S.,Wong,D.K.,Yuen,M.F.,Yuan,H.J.,Yu,J.,Lai,C.L.,Ho,D.,Zhang,L.,2004.Real-time PCR assay using molecular beaconfor quantitation of hepatitis B virus DNA.J.Clin.Microbiol.42, 3438–3440.Tedder,R.S.,Ayliffe,U.,Preiser,W.,Brink,N.S.,Grant,P.R.,Peggs,K.S., Mackinnon,S.,Kreig-Schneider,F.,Kirk,S.,Garson,J.A.,2002a.Development and evaluation of an internally controlled semiautomated PCR assay for quantification of cell-free cytomegalovirus.J.Med.Virol.66,518–523.Tedder,R.S.,Ijaz,S.,Gilbert,N.,Barbara,J.A.,Corden,S.A.,Gilson, R.J.,Boxall,E.H.,2002b.Evidence for a dynamic host–parasite rela-tionship in e-negative hepatitis B carriers.J.Med.Virol.68,505–512. Thompson,J.D.,Gibson,T.J.,Plewniak, F.,Jeanmougin, F.,Higgins,D.G.,1997.The CLUSTAL X windows interface:flexible strategiesfor multiple sequence alignment aided by quality analysis tools.Nu-cleic Acids Res.25,4876–4882.Weiss,J.,Wu,H.,Farrenkopf,B.,Schultz,T.,Song,G.,Shah,S.,Siegel, J.,2004.Real time TaqMan PCR detection and quantitation of HBV genotypes A–G with the use of an internal quantitation standard.J.Clin.Virol.30,86–93.Zaaijer,H.L.,ter Borg,F.,Cuypers,H.T.,Hermus,M.C.,Lelie,P.N., parison of methods for detection of hepatitis B virus DNA.J.Clin.Microbiol.32,2088–2091.。