GeneMarker处理SSR原始数据详细教程
Genemarker中文说明书

Gene Marker®The Biologist Friendly SoftwareGeneMarker®是唯一为基因研究者设计的“生物学家友好”基因分型软件。
软件界面具有引导功能并且布局直观,使用非常简便,同时兼顾精准、快速等优点,并集合大量应用,是一个功能强大的科研工具。
软件特点总结如下:特点:操作简单精准读取片段及等位基因快速界面引导自定义设置低成本临床研究:脆性X综合征三染色体/非整倍性研究MLPAMS-MLPA囊胞性纤维症微卫星不稳定基础研究:AFLPT-RFLP微卫星标记(STR/SSR)SNP标记(SnapShot/SNPlex)聚类分析亲缘关系鉴定GeneMarker可同时分析多达1000个四色或五色荧光平板胶或毛细管电泳泳道的数据,并兼容全部主流毛细管和平板胶电泳系统产生的文件,如ABI的文件(*.FSA、*.AB1、*.ABI、*.HID)、SCF的文件、MegaBACE™ 的文件(*.RSD、*.ESD)、Beckman-Coulter 的文件,以及与我们JelMarker®软件结合使用分析LI-COR、DNA Analyzers和柯达成像系统产生的平板胶图像(TIFF、BIP、JPEG 、TXT格式)。
GeneMarker是卓越的遗传分析和片段分析软件,可完美替代其他同类软件,如LI-CO (RSAGA软件)、ABI(GeneMapper®、Genotyper®、和GeneScan®)和MegaBACE® 的分析软件。
用户操作GeneMarker 的“Run Wizard ”简化用3个简单的步骤指引用户,使分析变准确,用户可选择嵌入式的模板或自定板。
GeneMarker 的模式识别技术可以自器或试剂造成的错误,如饱和峰、噪声重叠、钉子峰及影子峰。
目前,GeneMarker 的各项功能均于选择模板或自定义创建模板后自动分显示及报告。
基因转录数据分析软件使用过程简介
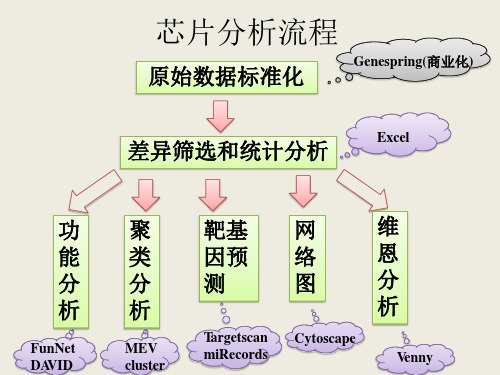
此处可以选择用 图表形式展示
23
GO分析下拉菜单: 选择默认选项: BP/CC/MF
Pathway分析下拉菜单: 选择KEGG_PATHWAY
24
4 数据导出
Options选项展开: 可以对阈值进行设置
点击此处导 出分析数据
此处两个值都设置为1, 可以查看全部数据的富 集分析情况,然后点击 Rerun Using Options
25
• 聚类分析 MEV软件 Cluster软件
MEV软件操作流程
1 打开MeV,导入数据。
选择源文件, 文件格式见 下一页
点击最左上角的第 一个数据,load
28
导入文件格式.txt 基因或miRNA名称, 可以是probe ID,gene symbol,miRNA等 相应样本的标准化 信号值
29
2 数据中位化(一般需对gene进行中位化)
Gene中位化
30
3 进行聚类分析
选择层次聚类
选择欧式全连接
31
4 数据参数设置(1)
点击此处进行 参数设置
参数设置选 项分别设为 -n,0,n
32
数据参数设置(2)
在界面上右击后,对 该两处设置,去除黄 色圈中标志。
分别勾选掉此处
33
5 保存图像
• t检验(使用的函数是ttest)
VLOOKUP函数匹配多列
多行匹配: $A1:绝对引用被查找的那一列 工作薄名!$A $2: $EW$17736:绝对引用查找区域的行和列 COLUMN():返回第几列 FALSE:精确匹配
功能分析
• FunNet:/
• DAVID: ttp:///home.jsp
FunNet分析流程
SSR标记实验步骤

SSR标记实验步骤简介:SSR简单序列重复标记(Simple sequence repeat, 简称SSR标记),也叫微卫星序列重复,是由一类由几个核苷酸(1-5个)为重复单位组成的长达几十个核苷酸的重复序列,长度较短,广泛分布在染色体上。
由于重复单位的次数的不同或重复程度的不完全相同,造成了SSR长度的高度变异性,由此而产生SSR标记或SSLP标记。
虽然SSR在基因组上的位置不尽相同,但是其两端序列多是保守的单拷贝序列,因此可以用微卫星区域特定顺序设计成对引物,通过PCR技术,经聚丙烯酰胺凝胶电泳,即可显示SSR位点在不同个体间的多态性。
优点:(1)标记数量丰富,具有较多的等位变异,广泛分布于各条染色体上;(2)是共显性标记,呈孟德尔遗传;(3)技术重复性好,易于操作,结果可靠。
缺点:开发此类标记需要预先得知标记两端的序列信息,而且引物合成费用较高。
操作程序:取叶片→磨样→提取DNA→PCR扩增→电泳检测→染色→读带标记1、DNA提取按照Doyle和Dickson(1987)CTAB法(`Cetyl triethyl ammonium bromide)并略加改进的程序进行,具体步骤如下:①成熟期取叶片加液氮研磨成粉末状,转入1.5ml离心管中,-20℃冰箱保存;②加入600ul 65℃的2×CTAB混匀并置于65℃水浴中保温30~60分钟;③取出,冷却,上下摇匀后,加入600微升24:1的氯仿异戊醇;④12000转/分钟离心10分钟,取上清液于另一个1.5ml的离心管中;⑤加异丙醇,12000转/分钟离心10分钟,倒掉上清液;⑥干后用100%的洒精清洗,干后加适量TE2、PCR扩增模板DNA的浓度大约25ng/ul,扩增反应体系为20ul。
具体如下:Sterile ddH2O 11ulPCR Buffer 3uldNTP-mix 0.5ulPrimer1 1ulPrimer2 1ulTaq polymerase 0.5ul(2U/μL)DNA 3ulPCR扩增程序为:(2h30min)①预变性:94℃,5分钟;②变性:94℃,40秒;③退火:55℃40秒;④延伸:72℃,1分钟;⑤循环:从2到4共38个循环;⑥72℃下最后延伸5分钟;4℃5min,扩增产物置于4℃的冰箱保存。
原始的转录组测序结果处理流程

原始的转录组测序结果处理流程
原始的转录组测序结果处理流程包括以下步骤:
1. 质量控制:使用软件如FastQC对原始测序数据进行质量评估,包括检查测序质量、序列长度分布、GC含量等。
2. 去除低质量序列:根据质量评估结果,使用软件如Trimmomatic 或Cutadapt去除低质量的测序序列、接头序列和低质量碱基。
3. 序列比对:使用软件如Bowtie、STAR或HISAT2将已处理的测序序列比对到参考基因组或转录组序列上。
4. 拼接转录本:使用软件如StringTie或Cufflinks对比对结果进行转录本拼接,得到基因和转录本的注释信息。
5. 差异表达分析:使用软件如DESeq2、edgeR或limma对不同样本之间的基因表达水平进行差异分析,找出差异表达的基因。
6. 功能注释与富集分析:对差异表达基因进行GO、KEGG等功能注释和富集分析,了解差异表达基因的生物学功能和通路。
7. 可变剪接分析(可选):使用软件如rMATS或MAJIQ对转录组数据中的可变剪接事件进行分析,探索不同样本之间的剪接差异。
8. 数据可视化:使用软件如R、Python或基因组浏览器将分析结果进行可视化展示,如热图、曲线图、柱状图等。
9. 结果解读:根据分析结果,对差异表达基因和功能富集结果进行解读,探索转录组的生物学意义和可能的调控机制。
总结起来,原始的转录组测序结果处理流程包括质量控制、序列去除、比对、拼接、差异分析、功能注释与富集分析、可变剪接分析、数据可视化和结果解读等步骤。
生物信息学中的基因组数据处理教程

生物信息学中的基因组数据处理教程随着基因组测序技术的快速发展,生物学研究进入了一个数据驱动的时代。
基因组数据的处理和分析对于理解生物体的功能和进化具有重要意义。
生物信息学中的基因组数据处理涉及到多个步骤,包括基本的数据预处理、序列比对、变异检测和功能注释等。
本教程将向您介绍这些基本步骤以及使用常见的工具和软件进行基因组数据处理的方法。
1. 基本的数据预处理在进行任何类型的基因组数据分析之前,首先需要对原始数据进行预处理。
这包括数据质量控制和去除低质量的序列。
其中,数据质量控制涉及到过滤掉带有低质量碱基或含有接头序列的reads。
常用的工具包括FastQC和Trim Galore。
Trim Galore可以去除接头序列并进行质量控制,还可以指定过滤条件和截断参数来提高数据质量。
2. 序列比对序列比对是将测序reads与参考基因组进行比对的过程。
比对可以帮助我们确定reads的起始和终止位置,并对其进行定量分析。
常用的比对工具有Bowtie、BWA和HISAT。
这些工具提供了快速、高效的比对算法,可以根据用户的需求进行参数配置和定制化操作。
3. 变异检测变异检测是基因组数据处理中的重要步骤之一,可以帮助我们发现个体之间的遗传差异或氨基酸突变。
常用的变异检测工具有GATK、SAMtools和FreeBayes。
这些工具可以检测单核苷酸多态性、插入/缺失突变和结构变异等不同类型的变异。
4. 功能注释功能注释是对基因组变异进行生物学解释的过程。
该过程包括鉴定变异位点的功能影响、基于数据库进行注释,并推断可能的生物功能。
常用的功能注释工具有ANNOVAR、Variant Effect Predictor (VEP)和SnpEff。
这些工具提供了丰富的注释信息和分析功能,可以帮助我们理解变异的生物学意义。
5. 数据可视化与解释基因组数据处理的最后一步是将处理后的数据进行可视化和解释。
通过绘制柱状图、散点图和热图等图表,我们可以更好地理解数据结果并从中发现潜在规律。
SSR技术原理,方法及步骤课件.doc

SSR技术1. SSR 简介说明:简单重复序列(Simple Sequence Repeat,SSR),简单重复序(SSR)也称微卫星DNA ,其串联重复的核心序列为1-6 bp,其中最常见是双核苷酸重复,即(CA) n 和(TG) n 每个微卫星DNA 的核心序列结构相同,重复单位数目10-60 个,其高度多态性主要来源于串联数目的不同。
SSR标记的基本原理:根据微卫星序列两端互补序列设计引物,通过PCR 反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR 的方法扩增出不同长度的PCR 产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。
在真核生物中,存在许多2-5bp 简单重复序列,称为“微卫星DNA”其两端的序列高度保守,可设计双引物进行PCR 扩增,揭示其多态性。
SSR具有以下一些优点:(l) 一般检测到的是一个单一的多等位基因位点;(2)微卫星呈共显性遗传,故可鉴别杂合子和纯合子;(3)所需DNA 量少。
显然,在采用SSR 技术分析微卫星DNA 多态性时必须知道重复序列两端的DNA 序列的信息。
如不能直接从DNA 数据库查寻则首先必须对其进行测序。
SSR的分类:根据SSR 核心序列排列方式的不同,可分为 3 种类型:1)完全型(perfect) ,指核心序列以不间断的重复方式首尾相连构成的DNA 。
如:ATATATATATATATATATATATATATATATATAT ;2)不完全型(imperfect) ,指在SSR 的核心序列之间有 3 个以下的非重复碱基,但两端的连续重复核心序列重复数大于 3 。
如:ATATATATGGATATATATATCGATATATATATATATATGGATATATATAT ;3)复合型(compound) ,指2 个或 2 个以上的串联核心序列由 3 个或3 个以上的连续的非重复碱基分隔开,但这种连续性的核心序列重复数不少于 5 。
SSR技术原理-方法及步骤

SSR技术1. SSR简介说明:简单重复序列(Simple Sequence Repeat,SSR),简单重复序(SSR)也称微卫星DNA,其串联重复的核心序列为1-6 bp,其中最常见是双核苷酸重复,即(CA) n和(TG) n每个微卫星DNA的核心序列结构相同,重复单位数目10-60个,其高度多态性主要来源于串联数目的不同。
SSR标记的基本原理:根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR 产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。
在真核生物中,存在许多2-5bp简单重复序列,称为“微卫星DNA”其两端的序列高度保守,可设计双引物进行PCR扩增,揭示其多态性。
SSR具有以下一些优点:(l)一般检测到的是一个单一的多等位基因位点;(2)微卫星呈共显性遗传,故可鉴别杂合子和纯合子;(3)所需DNA量少。
显然,在采用SSR技术分析微卫星DNA多态性时必须知道重复序列两端的DNA序列的信息。
如不能直接从DNA数据库查寻则首先必须对其进行测序。
SSR的分类:根据SSR核心序列排列方式的不同,可分为3种类型:1)完全型(perfect),指核心序列以不间断的重复方式首尾相连构成的DNA。
如:ATATATATATATATATATATATATATATATATAT;2)不完全型(imperfect),指在SSR的核心序列之间有3个以下的非重复碱基,但两端的连续重复核心序列重复数大于3 。
如:ATATATATGGATATATATATCGATATATATATATATATGGATATATATAT;3)复合型(compound) ,指2个或2个以上的串联核心序列由3个或3个以上的连续的非重复碱基分隔开,但这种连续性的核心序列重复数不少于5 。
如:ATATATATATATATGGGATATATATATATA 。
SSR技术原理-方法及步骤

SSR技术1. SSR简介说明:简单重复序列(Simple Sequence Repeat,SSR),简单重复序(SSR)也称微卫星DNA,其串联重复的核心序列为1-6 bp,其中最常见是双核苷酸重复,即(CA) n和(TG) n每个微卫星DNA的核心序列结构相同,重复单位数目10-60个,其高度多态性主要来源于串联数目的不同。
SSR标记的基本原理:根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR 产物,将扩增产物进行凝胶电泳,根据别离片段的大小决定基因型并计算等位基因频率。
在真核生物中,存在许多2-5bp简单重复序列,称为“微卫星DNA”其两端的序列高度保守,可设计双引物进行PCR扩增,揭示其多态性。
SSR具有以下一些优点:(l)一般检测到的是一个单一的多等位基因位点;(2)微卫星呈共显性遗传,故可鉴别杂合子和纯合子;(3)所需DNA量少。
显然,在采用SSR技术分析微卫星DNA多态性时必须知道重复序列两端的DNA序列的信息。
如不能直接从DNA数据库查寻则首先必须对其进行测序。
SSR的分类:根据SSR核心序列排列方式的不同,可分为3种类型:1〕完全型(perfect),指核心序列以不间断的重复方式首尾相连构成的DNA。
如:ATATATATATATATATATATATATATATATATAT;2〕不完全型(imperfect),指在SSR的核心序列之间有3个以下的非重复碱基,但两端的连续重复核心序列重复数大于3 。
如:ATATATATGGATATATATATCGATATATATATATATATGGATATATATAT;3〕复合型(compound) ,指2个或2个以上的串联核心序列由3个或3个以上的连续的非重复碱基分隔开,但这种连续性的核心序列重复数不少于5 。
如:ATATATATATATATGGGATATATATATATA 。
微卫星序列分析GeneMapper 使用教程(中文版)

ABI PRISM® GeneMapper™ Software Version 3.0微卫星分析使用教程(中文版)SSR条带读取设置向导© Copyright 2002, Applied Biosystems. All rights reserved.For Research Use Only. Not for use in diagnostic procedures.Information in this document is subject to change without notice. Applied Biosystems assumes no responsibility for any errors thatmay appear in this document. This document is believed to be complete and accurate at the time of publication. In no event shallApplied Biosystems be liable for incidental, special, multiple, or consequential damages in connection with or arising from the useof this document.Notice to Purchaser: License DisclaimerPurchase of this software product alone does not imply any license under any process, instrument or other apparatus,system, composition, reagent or kit rights under patent claims owned or otherwise controlled by Applera Corporation,either expressly, or by estoppel.TRADEMARKS:ABI PRISM and its design, Applied Biosystems, GeneScan, Genotyper, SNaPshot, LIZ, and VIC are registered trademarks ofApplera Corporation or its subsidiaries in the U.S. and certain other countries.AB (Design), ABI, Applera, GeneMapper, NED, PET, ROX, and 6-FAM are trademarks of Applera Corporation or its subsidiariesin the U.S. and certain other countries.Macintosh is a registered trademark of Apple Computer, Inc.All other trademarks are the sole property of their respective owners.Part Number 4335525 Rev. B09/2002目录:第一章总论关于微卫星分析使用教程------------------------1-1数据提供---------------------------------------------1-2操作流程---------------------------------------------1-3第二章创建Custom Kits,Panels,and Markers 操作流程--------------------------------------------2-1创建Kits,Panels,and Markers----------------2-2第三章运用Panels,Bins进行数据分析操作流程--------------------------------------------3-1导入Panels------------------------------------------3-2导入和设定相关数据------------------------------3-3创建Bits---------------------------------------------3-4附录:Panel,Bin结构认识总论本章内容主要包括:关于微卫星分析使用教程-----------------------1-1数据提供--------------------------------------------1-2操作流程--------------------------------------------1-3关于微卫星分析使用教程概述:微卫星分析方法可以让您高效快速的运用ABIPRISM®GeneMapper™ Software Version 3.0.对微卫星进行分析目标:通过学习微卫星分析方法,您可以●建立GeneMapper 分析项目●导入样本文件进行数据分析●利用自动bin功能创建bin如何使用此教程:当您按照后面几个章节中的流程操作时,切记:●按照顺序进行每一步操作●不要导入其他无关样本和bins所有的操作过程都按照菜单操作方式编写,如:选择Tools > GeneMapper Manager当然您也可以点击工具菜单上的快捷按钮教程中的术语:教程中使用以下术语表格1-1 术语和定义表格1-1 术语和定义数据提供概述:GeneMapper Software Version 3.0包括以下数据Panel 信息:主要提供:●LMS-HD5 Version 2.5 and LMS-MD10 Version 2.5 kits●GeneScan™ Installation Standard DS-33 (6-FAM™, VIC®,NED™, and PET™ dyes)●GeneScan™ Installation Standard DS-30 (6-FAM™, HEX™,NED™, and ROX™ dyes)●This tutorial注意:更多信息请阅读您说使用的Kit所对应Panel信息,若要需要创建Custom Kit,请阅读第二章Bin 信息:Bin 信息对应GeneScan Installation Standard DS-33, 采用GeneScan™ 500LIZ_3730 标准,只用于Applied Biosystems3730/3730xl DNA Analyzer默认设置:提供下列默认设置表1-2 默认设置带型标准设置:为微卫星数据分析提供下列带型标准:●GeneScan™ 400HD size standard●GeneScan™ 500 size standard●GeneScan™ 500 (-250) size standard●GeneScan™ 500 (-250) LIZ® size standard●GeneScan™ 500 LIZ® size standard注意:可以设定用户带型标准,阅读ABI PRISM® GeneMapper™Genotyping Software User’s Manual (PN 4335526). 样本文件:本教程提供四种微卫星样本文件,它们均采用5个客户PETmarker ,利用LMSMD-10 panel 9在3100型仪器上产生操作流程:分析微卫星数据:以下表格为运用GeneMapper 软件分析微卫星数据的流程概述,请链接相关章节,阅读详细流程。
genalex ssr格式

genalex ssr格式【原创实用版】目录1.GenaLex SSR 格式的概述2.GenaLex SSR 格式的特点3.GenaLex SSR 格式的应用领域4.GenaLex SSR 格式的优缺点分析正文1.GenaLex SSR 格式的概述GenaLex SSR 格式是一种基因序列数据存储和交换的格式,由GenBank、EBI 和 DDBJ 三个主要的核酸序列数据库共同制定。
该格式旨在为科学家提供一个通用、简单且易于理解的方式来描述和分享基因序列数据。
2.GenaLex SSR 格式的特点GenaLex SSR 格式具有以下特点:(1) 可读性强:采用纯文本格式,使得数据易于阅读和理解。
(2) 结构简单:数据以记录(record)为单位进行组织,每个记录包含一个序列及其相关注释信息。
(3) 通用性:适用于各种类型的核酸序列数据,包括基因组、转录组和蛋白质组等。
(4) 可扩展性:支持对新数据类型的添加,以满足不断发展的科研需求。
3.GenaLex SSR 格式的应用领域GenaLex SSR 格式在多个领域都有广泛应用,包括:(1) 基因组学:用于存储和分享基因组序列数据,有助于基因组学研究的进展。
(2) 转录组学:转录组数据通常以 GenaLex SSR 格式存储,便于研究人员分析基因表达。
(3) 蛋白质组学:用于表示蛋白质序列及其相关信息,有助于研究蛋白质结构和功能。
(4) 生物信息学:GenaLex SSR 格式为生物信息学研究提供了丰富的数据资源,便于研究人员开展序列比对、进化分析等研究。
4.GenaLex SSR 格式的优缺点分析优点:(1) 易于阅读和理解:采用纯文本格式,方便科学家直接阅读和理解序列数据。
(2) 数据通用性强:适用于多种类型的核酸序列数据,便于不同领域的研究人员共享和交流数据。
(3) 支持数据扩展:能够适应新数据类型的添加,有利于科研领域的发展。
缺点:(1) 存储空间需求大:由于采用纯文本格式,序列数据的存储空间需求相对较大。
Genemapper微卫星分析中文操作指南

GeneMapper 软件微卫星分析中文简易操作手册微卫星分析流程:软件术语介绍:一.设定微卫星分析方法:1.登录GeneMapper软件:1.1选择开始 -- All Programs -- Applied Biosystems -- GeneMapper--GeneMapper ;或双击桌面快捷方式打开软件;1.2登录GeneMapper软件:用户名默认为gm,在Passwprd中输入密码,点击OK进入GeneMapper软件2创建分析所需kit、panel和marker等参数:2.1 点击软件上方工具栏Tools,选择Panel Manager;2.2 在Panel Manager对话框中,选中1 Panel Manager,点击2 新建 Kit;于New Kit对话框中依次输入Kit名称,Kit类型,并点击OK保存设置;2.3 在Panel Manager对话框中,选中1 STR KIT,点击2 新建 Panel;3中输入Panel名称;2.4 选中1 STR panel,点击2新建Marker,在3位置依次输入Marker Name(Marker名称) ,Dye Color (荧光标记种类) ,Min Size,Max Size (片段最小值和最大值) ,Marker Repeat (重复次数) ;2.5重复2.4步骤,完成所有Marker设置,点击OK保存;3. 创建新文件,导入数据:3 .1 点击1 新建文件夹,在2 Project Type中文件夹类型选择Microsatellite ,点击OK;3.2 导入数据:点击1 Add Samples To Project 添加数据,在弹出的对话框中选择待分析数据,通过3 Add To List 添加到右边的面板中,点击4 Add完成数据添加;4. 设置分析参数:4.1 完成数据添加后,在1 位置,Analysis Method中选择New Analysis Method新建分析方法;在弹出的对话框中选择分析类型2 Microsatellite,点击OK确认;4.2 设置分析参数:在Analysis Method Editor中,点击1 General,填写2 Name(分析方法名称);之后点击3 Allele,在将4 Bin Set选择none;其它参数为软件默认,不需要修改;点击OK确认;4.3 选定好之前设定的分析方法,在后续的栏目中设定Panel 和Size Standard(分子量内标);4.4 完成分析参数设定后,选中Analysis Method,Panel和Size Standard栏目,点击1File,选择2Fill Down;此时,Analysis Method,Panel和SizeStandard栏目会使用之前选定的分析参数;也可以使用鼠标分别选择;4.5 保存分析文件:点击File,选择Save Project,输入文件夹名称:5. 执行初始分析:5.1 点击分析按钮1,分析数据:5.2 在1 Genotypes中查看初步分析结果,或通过2 Display Plots选项查看样品分型图:5.3 在样品分型图界面中,Plot Setting选择 Microsatellite Default;通过软件上方的快捷按钮编辑数据显示方式:6 创建Bin Set:6.1 打开Tools—Panel Manager,选中1 STR Kit,点击2 新建Bin Set,在弹出的New Bin Set 对话框3中输入Bin Set名称:6.2 添加参考数据:6.2.1 确保1 Bin Set已选择新建的Bin Set,选中2 STR Panel,此时会显示出之前新建的每个Marker,点击3 Add Reference Data 添加参考数据;6.2.3 在Add Microsatellite Reference Data对话框中,选择1 参考数据,通过2Add To List 添加至右面的面板中,点击3 Add,完成添加:6.3 生成Bin Set:完成参考数据添加后,点击1 Auto Bin,之后使用软件默认参数,点击2 OK,自动生成Bin Set:6.4 编辑Bin Set:分别选中每个Marker 1 查看其对应的Bin,选中2 Bin,右键进行Bin的编辑或者删除,也可通过软件3处的快捷图标进行Bin的编辑;完成后点击OK生成Bin Set;二.分析并检查结果:1.编辑分析方法:点击软件上方快捷图标1 Analysis Method Editor,将Allele中的2 Bin Set修改为之前新建的Bin Set,其他参数保持默认,点击OK;2.点击分析按钮再次分析数据,通过Genotypes查看分型结果,或选中数据通过Display Plots查看样品分型图;选中需要查看的数据,否则无法查看Samples Plot:查看样品分型图:三.打印并导出数据(可选):1.在View中选择不同的界面(Samples,Genotypes等),再通过File—Export/Print 导出或者打印相应的数据:2.在Analysis 中选择不同的界面(Display Plots,Report Manager等),再通过File—Export/Print 导出或者打印相应的数据:。
使用GeneMarker软件分析SnaPshot和SBE

使用GeneMarker软件分析SnaPshot和SBE引言单核苷酸多态性(SNP)在人类基因组中每100至300bp就有一个位点,构成了人类90%的遗传变异(1)。
功能SNP或非同义SNP (nsSNPs)发生在基因的编码区,调控SNP(rSNPs)发生在基因的调控子改变蛋白质功能或基因的表达(2,3),基因内或基因间的SNP 可能不会改变蛋白质功能,但是可以用于研究进化生物学(4),复杂疾病、药物反应、环境因子的关联性分析,或QTL定位(5)。
有一种检测SNP基因型的方法是单核苷酸引物延伸法(SBE),无荧光标记的引物的3’末端直接挨着SNP 位点,通过Taq聚合酶扩增一个碱基即为SNP位点,荧光标记的ddNTP与SNP多态位点互补并终止反应。
最终扩增片段增加了一个碱基,但是由于荧光染料对这些小片段电泳迁移率的影响,电泳观察到的片段要比预计的大。
SNP 可以被单色或双色荧光的峰及引物的长度进行鉴定。
SBE技术可以快速的鉴定数量较少的SNP,使用MegaBACE SnuPe基因分型试剂盒及ABI的SnaPshot分型系统广泛应用在在动植物研究中(4,5)。
为了更好地发挥SBE技术的潜力,需要一个强大的基因分型和数据分析系统。
GeneMarker基因分析软件可快速高效的分析和报告出扩增数据。
GeneMarker是“生物学家友好”软件,包含强大的片段读取算法,高效的等位基因读取使片段读取精度小于1bp,对SNuPE 和SnaPshot产生的数据,进行小片段迁移率变化调整,一起显示两种颜色的SNP峰图。
该软件能够分析来自各种毛细管电泳系统的数据,包括ABI、MegaBACE和Beckman。
GeneMarker与我们的JelMarker 软件结合可以进行平板胶数据的分析。
ProcedureGeneMarker软件软件的如下设置可分析出SBE最好的结果,从原始数据到分析报告导出的过程分为三步:片段大小读取、创建SBE panel、利用panel分析数据。
POPGENE32下载及SSR共显性数据分析相关教程

POPGENE32下载及SSR共显性数据分析相关教程生物专题2010-05-31 11:13:19 阅读673 评论4 字号:大中小订阅有关共显性标记数据判读问题,现在这再做个解释:在中有一个双倍体SSR数据的计算实例:/* Test Data Set II: Diploid Data */Number of populations = 4Number of loci = 21Locus name :AAT-1 AAT-2 AAT-3 ACO ADH DIA-1 DIA-3 EST-2 GDH G6P HAIDH MDH-1 MDH-2 MDH-3 MDH-4 PEP-1 PEP-2 PGI-2 PGM SPG-2AA AA AA AA BB AB AA AB BB AA AA AA AA AA AA AA AA AA AB AA AAAA AA AA AA AA AA AA BB AA AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AB BB AC AA AB BB AA AA AA AA AA AA AA AA AA AB AA AAAA AA AA AA BC AC AA AB AB AB AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA BB CC AA BB AB AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA AB AC AA BB AA AA AA AA AA AA AA AC AA AA AA AB AAAA AA AA AB AB AC AA AB AB AA AA AA AA AA AA AB AA AA AA AA AAAB AA AA AA BC AC AA AB AB AA AA AA AA AB AA AB AA AA AA AA AAAA AA AA AA BB AA AA AA AA AA AA AA AA AA AA AA AA AA AC AA AAAA AA AA AA AA BC AA AB AA AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AB BC BC AA BB AB AA AA AA AA AB AA AA AA AA AA AC AAAA AA AA AB AC AB AA BB BB AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA AB AC AA BB AA AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA AA BC AB AB AA AA AA AA AA AA AA AA AA AA AC AA AAAA AA AA AB AA AC AA AB AB AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AA AA AB AA AB AB AC AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA AA AB AA AB AA AB AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA AC AA AAAA AA AA AA AA BB AA AB AA AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AA AB BC AA AA AB AC AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA BB BB AA AA AA AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AA AC AC AA BB AB AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AB BB BC AA BB AA AC AA AA AA AA AA AA AA AA AC AD AAAA AA AA AA AB BC AA AB AB AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA BB BC AA BB AA AA AA AA AA AA AA AB AA AA AA AC AAAA AA AA AA AA BC AA AB AB AA AA AA AA AA AA BC AA AA AA AA AAAA AA AA AA AB BC AA AB AB AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AA BD BC AA AB AB AA AA AA AA AA AA AA AA AA AC AE AAAA AA AA AB CC BB AA BB AA AA AA AA AA AA AA AA AA AA AA AB AAAA AA AA AB CC BB AA AA AB AA AA AA AA AA AA AA AA AA AA AB AA AA AA AA AA BB BC AA AB AB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AB BC AA AA BB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AB AC AB AA BB AA AA AA AA AA AA AA AA AA AA AA AB AA AA AA AA AB BC BB AA BB BB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AB AB AB AA BB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AB CC AB AA AA AB AA AA AA AA AA AA AB AA AA AC AA AA AA AA AA AB AB AB AA BB AA AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA BB AB AA AA AA AA AA AA AA AA AA BB AA AA AA AA AA AA AA AA AA AC BC AA BB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA CC BC AA AB AB AA AA AA AA AA AA AB AA AA AA AC AA AA AA AA AA BB AC AA BB AA AA AA AA AA AA AA AA AA AA CC AA AA AA AA AB AA BE BB AA BB AB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA AB BC AA AB AA AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA BB AA CC AA AB AB AA AA AA AA AA AA BC AA AA AA AA AA AA AA AA AB AC BC AA BB BB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AB AA BB AA AC AB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AB BB AC AA BB AA AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA AC AC AA AA AB AA AA AA AB AA AA AB AA AA AA AA AA AA AA AA AA AA AB AA AB AB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA BC BC AA BB AB AA AA AA AA AA AA AB AA AA AA AC AA AA AB AA AA AB CC AA BB AA AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA AA BC AA AB BB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AC AB AA AA AA AA AA AA AA AA BC AA AA AA AA AA AA AA AA AA BC BC AA AB AB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA AA AC AA AA BB AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AB AB BB AA AB AA AA AA AA AA AA AA AB AA AA AC AA AA AA AA AA AB BC AB AA AB AB AA AA AA AA AA AA BB AA AA AA AB AA AA AA AA AB BC AB AA AB AB AA AA AA AA AA AA BB AA AA AA AB AAAA AA AA AA BB AB AA AA AA AA AA AA AA AA AA BB AA AA AA AA AA AA AA AA AA AC BC AA BB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA AA CC BC AA AB AB AA AA AA AA AA AA AB AA AA AA AC AA AA AA AA AA BB AC AA BB AA AA AA AA AA AA AA AA AA AA CC AA AA AA AA AB AA BE BB AA BB AB AA AA AA AA AB AA AB AA AA AA AA AA AA AA AA AA AB BC AA AB AA AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA BB AA CC AA AB AB AA AA AA AA AA AA BC AA AA AA AA AA AA AA AA AB AC BC AA BB BB AA AA AA AA AA AA AA AA AA AA AA AA AA AA AB AB AA BB AA AC AB AA AB AA AA AC AA AB AA AA AA AA AA AA AA AA AB BB AC AA BB AA AA AA AA AA AA AA AB AA AA AA AA AA AA AA AA AA AC AC AA AA AB AA AA AA AB AA AA AB AA AA AA AA AA AC AA AA AA AA AB AA AB AB AA AA AA AA AA AA AB AA AA AA AA AAAA AB AA AA AB CC AA BB AA AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AA AA BC AA AB BB AA AA AA AA AA AA AA AA AA AA AA AAAA AA AA AA AA AC AB AA AA AA AA AA AA AA AA BC AA AA AA AA AAAA AA AA AA BC BC AA AB AB AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AA AA AC AA AA BB AA AA AA AA AA AA AB AA AA AA AA AAAA AA AA AB AB BB AA AB AA AA AA AA AA AA AA AB AA AA AC AA AAAA AA AA AB BC AB AA AB AB AA AA AA AA AA AA BB AA AA AA AB AAAA AA AA AB BC AB AA AB AB AA AA AA AA AA AA BB AA AA AA AB AA其中我们能看出这里分析了某个物种4个群体的遗传多样性,标记的个数为21,locus name下的为21对SSR,我们来看引物AAT-1,群体型数据中左侧第一纵列为该引物的扩增情况,依次类推,我们再看引物ACO,可观察到有3种等位基因:AA/BB/CC/;我们看前4个个体的扩增情况应该为0100(BB)、1000(AA)、0100(BB)、0110(BC)(其中0为空白,1为有带扩出),依次类推,我们就能将扩增情况与等位基因一一对应起来。
SSR分析的原理及操作技术

下降;酶量过少影响反应产量。 ▪ 10×buffer缓冲液(不含Mg2+ ):维持PCR pH的稳定。
PCR循环条件
▪ 高温变性:双链DNA模板加热变性成单链;
▪ 低温退火:在低温下引物与单链DNA互补配对; 退火温度针对不同的引物差别较大,退火温度过低,极易形成非特
SSR分析的原理及操作技 术
DNA分子标记技术类型
▪ 以Southern杂交为基础的分子标记技术: 限制性内切酶片段长度多态性标记(RFLP)
▪ 以PCR为基础的分子标记技术:随机引物PCR标记和特异引物PCR标记 随机扩增多态性DNA(RAPD) 扩增的限制性内切酶片段长度多态性(AFLP) 相关序列扩增多态性(SRAP) 简单重复序列(SSR)或简单序列长度多态性(SSLP)
▪ 以mRNA为基础的分子标记技术: 差异显示(DD) 逆转录PCR(RT-PCR)
▪ 以单核苷酸多态性为基础的分子标记技术: 单核苷酸多态性(SNP)
SSR简介
▪ 在生物的基因组中,特别是高等生物的基因组中含有大量的重复序列, 根据重复序列在基因组中的分布形式可分为:
串联重复序列(Tandemly repeated sequences) 分散重复序列(Interspersed repeated sequences)
▪ 微卫星在植物基因组中的含量非常丰富,均匀分布于整个植物基 因组中,但不同植物中微卫星出现的频率变化非常大,但 (AT)n最多。
SSR标记的基本原理
▪ 尽管微卫星DNA分布于整个基因组中的不同位置,但 某一特定的微卫星的两端侧翼序列通常都是保守性较强
基于EST和Genome序列设计SSR引物的步骤

EST-SSR 引物开发步骤1、首先计算机安装PC 版perl 程序,然后从GenBank/dbEST (http://www.ncbi.nlm.nih/entrez)中以FASTA 格式下载该种中所有的EST 序列,共29830 条。
2、利用Blastclust(v.2.2.10;http://www. /web/newsltr/spring04/blastlab.htm)软件,对这些EST 序列进行冗余性查找,利用CD_HIT(http:///cd-hit/)快速批量去冗余序列。
3、利用est_timmer(http://pgrc.ipk-gatersleben.de/misa/)去除EST 序列中过短的序列(<100bp)和过长的序列(>700bp)以及mRNA 的“帽子”和“尾巴”(A或T)。
4、利用misa.pl(http://pgrc.ipk-gatersleben.de/misa/)识别和定位SSR,其中配置文件misa.ini 用来设置识别SSR 标记的标准。
5、利用primer3 模块批量设计SSR 引物,引物设计的主要参数是,引物长度20~26bp;退火温度Tm 值48℃~65℃;(G+C)含量30%~70%;PCR 扩增产物长度大于150bp。
6、利用MacV ector7.0 软件对所设计的引物进行验证,引物由上海生工生物技术有限公司合成。
Genome-SSR 的引物开发步骤1、通过Genbank数据库,检索相近种的全基因组序列。
保存成FAST格式。
2、利用SSRHunter1.3搜索基因组序列中的简单重复序列信息位点。
3、选取信息位点核苷酸重复基序为2~6个的序列为靶标序列,利用软件Primer Premier 5.0设计SSR引物,引物设计的主要参数是,引物长度20~26bp;退火温度Tm 值48℃~65℃;(G+C)含量30%~70%;PCR 扩增产物长度大于150bp。
SSR引物富集与设计方法

一、研究材料与方法1.样品的采集与保存液氮冷冻后-70℃冷冻保存,或加入无水乙醇-70℃冷冻保存。
2.主要试剂DNAeasy基因组提取试剂盒(Qiagen公司),限制性内切酶MboⅠ(Takara公司),生物素吸附剂Vectrex Avidin D(Vector Laboratories),binding buffer(150 mM NaCl/100 mM Tris,pH=7.5),PMD19-T载体(Takara公司),大肠杆菌(Escherichia coli)DH5α,琼脂糖(Gene Tech公司),胰蛋白胨(OXOID),酵母提取物(OXOID),NaCL(国药集团化学试剂有限公司),Tris(上海鼎国生物技术有限公司),冰乙酸(上海化学试剂有限公司),EDTA(北京鼎国生物技术有限责任公司),溴酚蓝、二甲苯氰(广州威佳有限科技公司),甘油(国药集团化学试剂有限公司),酚(国药集团化学试剂有限公司),异戊醇(国药集团化学试剂有限公司),SybgreenⅠ(北京鼎国生物技术有限责任公司)。
3.主要耗材1.5 ml离心管(江苏省海门三和通东玻璃仪器厂),96孔PCR板(Axygen公司),枪头(江苏省蓝天仪器厂)。
4.主要实验仪器移液器(Effendorf),台式离心机(Effendorf),电热恒温水槽DK-8D型(上海一恒科技有限公司),PCR仪(Biorad),电泳仪DYY-6C型(北京六一仪器厂),凝胶成像仪。
5.主要试剂和缓冲液的制备DNA消化缓冲液:100mmol/L Tris-Cl,50mmol/L EDTA(pH 8.0),150 mmol/L NaCl,0.1% SDS 50×TAE电泳贮存液/L(缓冲液为10×):242 g Tris碱,57.1 ml冰乙酸,100 ml 0.5 mol/LEDTA凝胶加样缓冲液(Loading buffer)(10×):0.25%溴酚蓝(或二甲苯氰),50%甘油。