最新VASP操作介绍-两次课
VASP使用总结

VASP使用总结VASP计算的理论及实践总结一、赝势的选取二、收敛测试1、VASP测试截断能和K 点2、MS测试三、结构弛豫四、VASP的使用流程(计算性质)1、VASP的四个输入文件的设置2、输出文件的查看及指令3、计算单电能(1) 测试截断能(2) 测试K点4、进行结构优化5、计算弹性常数6、一些常用指令一、赝势的选取VASP赝势库中分为:PP和PAW两种势,PP又分为SP(标准)和USPP(超软)。
交换关联函数分为:LDA(局域密度近似)和GGA(广义梯度近似)。
GGA 又分为PW91和PBE。
在VASP中,其中pot ,pot-gga是属于超软势(使用较少)。
Paw, paw-pbe ,和paw-gga是属于PAW。
采用较多的是PAW-pbe 和PAW-gga。
此外vasp 中的赝势分为几种,包扩标准赝势(没有下标的)、还有硬(harder)赝势(_h)、软(softer)赝势(_s), 所谓的硬(难以赝化),就是指该元素原子的截断动能比较大,假想的势能与实际比较接近,计算得到的结果准确,但比较耗时,难以收敛。
软(容易赝化),表示该元素原子的截断动能比较小,赝势模型比较粗糙,但相对简单,可以使计算很快收敛(比如VASP开发的超软赝势)。
即硬的赝势精度高,但计算耗时。
软的精度低,容易收敛,但节省计算时间。
另一种情况:如Gd_3,这是把f电子放入核内处理,对于Gd来说,f电子恰好半满。
所以把f电子作为价电子处理的赝势还是蛮好的(类似还有Lu,全满)。
(相对其他的4f元素来说,至于把f电子作为芯内处理,是以前对4f元素的通用做法。
计算结果挺好)常用的做法是:用两种赝势测试一下对自己所关心的问题的影响情况。
在影响不大的情况下,选用不含4f电子的赝势(即后缀是3),一来减少计算量,二来避免DFT对4f电子的处理。
【1.赝势的选择:vasp的赝势文件放在目录~/vasp/potentials 下,可以看到该目录又包含五个子目录pot pot_GGA potpaw potpaw_GGA potpaw_PBE ,其中每一个子目录对应一种赝势形式。
VASP简介ppt课件

☺可以在一行设置多个关键词(即参数)的值,但是每个关键值之间用分 号(;)隔开。如ISMEAR= 0; SIGMA= 0.2。 ☺当想不用INCAR中某个关键词的值时,在该行前面加上井号(#)注释掉, 如#ISMEAR=0; SIGMA = 0.2
5
POSCAR输入文件:描述体系结构
例:SiC体系的POSCAR文件
TITEL = US Si
LULTRA = T use ultrasoft PP ?
IUNSCR = 1 unscreen: 0-lin 1-nonlin 2-no
RPACOR = 1.580 partial core radius
POMASS = 28.085; ZVAL = 4.000 mass and valenz
子动力学模拟的软件包。 • 基于(有限温度下的,对电子气而言)局域密度近似,自由
能作为电子气密度的泛函 • 在每个MD时间步长内精确求解电子气的瞬时基态
2
基本任务
• 晶体的电子结构(如态密度、能带、电荷密度)计算 • 晶体的磁学性质计算 • 优化晶体的结构参数 • 内部自由度弛豫 • 结构弛豫 • 表面体系的基本性质的计算
标题或注释行,无特别意义 K点的数目 以字母R开头表示k点是按倒格子坐标系 前三个数是k点的坐标,最后一个数是相应k 点的权重(下面共5个k点)
如果是以卡笛尔坐标系来写k点坐 标,则第三行以字母C开头。
9
POTCAR输入文件: 赝势文件
Si 的一种势函数的部分内容
US Si 4.00000000000000000 parameters from PSCTR are: VRHFIN =Si: s2p2 LEXCH = CA EATOM = 115.7612 eV, 8.5082 Ry GGA = -1.4125 -1.4408 .0293 -.9884 eV
vasp学习
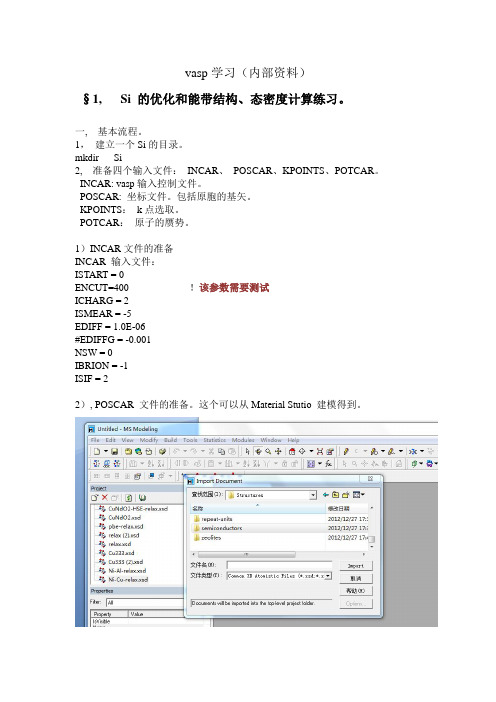
vasp学习(内部资料)§1, Si 的优化和能带结构、态密度计算练习。
一, 基本流程。
1,建立一个Si的目录。
mkdir Si2, 准备四个输入文件:INCAR、POSCAR、KPOINTS、POTCAR。
INCAR: vasp输入控制文件。
POSCAR: 坐标文件。
包括原胞的基矢。
KPOINTS:k点选取。
POTCAR:原子的赝势。
1)INCAR文件的准备INCAR 输入文件:ISTART = 0ENCUT=400 !该参数需要测试ICHARG = 2ISMEAR = -5EDIFF = 1.0E-06#EDIFFG = -0.001NSW = 0IBRION = -1ISIF = 22), POSCAR 文件的准备。
这个可以从Material Stutio 建模得到。
点击File, 选择import Document, 在structure里找到Semiconductor,选择Si.msi。
即找到Si的结构。
输出原子坐标,在MS中点击Build-->symmetry-->primitive cell (对于已经是原胞的情况,这一步省略)在点击Build-->symmety->make P1.再点击file-->export,选择输出文件为Si.cif。
(必须选择cif格式)输出以后,用文本文档的方式打开Si.cif。
在Si.cif中看到这样几行,Si1 Si 0.00000 0.00000 0.00000 0.00000 Uiso 1.00Si2 Si 0.25000 0.25000 0.25000 0.00000 Uiso 1.00这就是Si的原胞中原子的坐标。
根据POSCAR需要进行编辑。
(在vi中可以用列模式进行编辑。
“Ctr+v”进入列模式,键盘上下左右箭头进行区域选择,“ctr+p”粘贴选中的内容,“d”删除选中的内容。
)原胞的基矢可以通过在MS中点击原胞的白色边框,在MS 的左侧的Properties 一栏中,将出现原胞的详细信息。
VASP经典学习教程,有用

V ASP 学习教程太原理工大学量子化学课题组2012/5/25 太原目录第一章Linux命令 (1)1.1 常用命令 (1)1.1.1 浏览目录 (1)1.1.2 浏览文件 (1)1.1.3 目录操作 (1)1.1.4 文件操作 (1)1.1.5 系统信息 (1)第二章SSH软件使用 (2)2.1 软件界面 (2)2.2 SSH transfer的应用 (3)2.2.1 文件传输 (3)2.2.2 简单应用 (3)第三章V ASP的四个输入文件 (3)3.1 INCAR (3)3.2 KPOINTS (4)3.3 POSCAR (4)3.4 POTCAR (5)第四章实例 (5)4.1 模型的构建 (5)4.2 V ASP计算 (8)4.2.1 参数测试 (8)4.2.2 晶胞优化(Cu) (13)4.2.3 Cu(100)表面的能量 (14)4.2.4 吸附分子CO、H、CHO的结构优化 (15)4.2.5 CO吸附于Cu100表面H位 (17)4.2.6 H吸附于Cu100表面H位 (18)4.2.7 CHO吸附于Cu100表面B位 (19)4.2.8 CO和H共吸附于Cu100表面 (20)4.2.9 过渡态计算 (21)第一章Linux命令1.1 常用命令1.1.1 浏览目录cd: 进入某个目录。
如:cd /home/songluzhi/vasp/CH4 cd .. 上一层目录;cd / 根目录;ls: 显示目录下的文件。
注:输入目录名时,可只输入前3个字母,按Tab键补全。
1.1.2 浏览文件cat:显示文件内容。
如:cat INCAR如果文件较大,可用:cat INCAR | more (可以按上下键查看) 合并文件:cat A B > C (A和B的内容合并,A在前,B在后) 1.1.3 目录操作mkdir:建立目录;rmdir:删除目录。
如:mkdir T-CH3-Rh1111.1.4 文件操作rm:删除文件;vi:编辑文件;cp:拷贝文件mv:移动文件;pwd:显示当前路径。
VASP经典学习教程-有用

VASP 学习教程太原理工大学量子化学课题组2012/5/25目录第一章Linux命令 (1)1.1 常用命令 (1)1.1.1 浏览目录 (1)1.1.2 浏览文件 (1)1.1.3 目录操作 (1)1.1.4 文件操作 (1)1.1.5 系统信息 (1)第二章SSH软件使用 (2)2.1 软件界面 (2)2.2 SSH transfer的应用 (3)2.2.1 文件传输 (3)2.2.2 简单应用 (3)第三章VASP的四个输入文件 (3)3.1 INCAR (3)3.2 KPOINTS (4)3.3 POSCAR (4)3.4 POTCAR (5)第四章实例 (5)4.1 模型的构建 (5)4.2 VASP计算 (8)4.2.1 参数测试 (8)4.2.2 晶胞优化(Cu) (13)4.2.3 Cu(100)表面的能量 (2)4.2.4 吸附分子CO、H、CHO的结构优化 (2)4.2.5 CO吸附于Cu100表面H位 (4)4.2.6 H吸附于Cu100表面H位 (5)4.2.7 CHO吸附于Cu100表面B位 (6)4.2.8 CO和H共吸附于Cu100表面 (7)4.2.9 过渡态计算 (8)第一章Linux命令1.1 常用命令1.1.1 浏览目录cd: 进入某个目录。
如:cd /home/songluzhi/vasp/CH4 cd .. 上一层目录;cd / 根目录;ls: 显示目录下的文件。
注:输入目录名时,可只输入前3个字母,按Tab键补全。
1.1.2 浏览文件cat:显示文件内容。
如:cat INCAR如果文件较大,可用:cat INCAR | more (可以按上下键查看) 合并文件:cat A B > C (A和B的内容合并,A在前,B在后) 1.1.3 目录操作mkdir:建立目录;rmdir:删除目录。
如:mkdir T-CH3-Rh1111.1.4 文件操作rm:删除文件;vi:编辑文件;cp:拷贝文件mv:移动文件;pwd:显示当前路径。
Vasp入门+实例

(1). 生成4个输入文件: POSCAR POTCAR INCAR KPOINTS
(2). 优化晶格参数,求出体Mg的晶格参数
• 计算表面性质
• 应用
Building surfaces (1)asymmetric setup
(2)symmetric setup
unit cell coordinates are optimized
Fixed layers (bulk)
vacuum
示例1: 用VASP求1*1Mg(0001)的表面性质
NPAR=4 NSW=1 IBRION = 2 ISIF=2 ISYM = 1
(2). 优化晶格参数,求出能量最低所对应的晶格参数
运行VASP程序, 查看SUMMARY.fcc输出文件:
(3). 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量
(i) 找到平衡晶格常数后, 把该值写入到POSCAR文件中,并增加K点数 作一个离子步自洽计算(NSW = 0, IBRION = -1) . (ii) 从DOSCAR输出文件中读出态密度和费米能级,费米 费米能级也可从OUTCAR中读出.
VASP提供 各种POTCAR
c/a
K-Points 0 Monkhorst Pack 21 21 21 0 0 0
� 1� 3 � a1 � a ( i � j) 2 2 � 1� 3 � a2 � a ( i � j) 2 2 � � a3 � ck
NPAR=4 NSW=1 IBRION = 2 ISIF=2 ISYM = 1
(3). 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量 (4). 修改KPOINTS和INCAR输入文件,固定电荷密度,做非自洽 计算,得到输出文件EIGENVAL (5). 提取数据,画图
VASP介绍3

K-Points VASP提供 0 各种POTCAR Monkhorst Pack 21 21 21 0 0 0
NPAR=4 NSW=1 IBRION = 2 ISIF=2 ISYM = 1
(2). 优化晶格参数,求出能量最低所对应的晶格参数
运行VASP程序, 查看SUMMARY.fcc输出文件:
(3). 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量
(4). 修改KPOINTS和INCAR输入文件,固定电荷密度,做非自洽 计算,得到输出文件EIGENVAL (5). 提取数据,画图
(1). 生成4个输入文件: POSCAR POTCAR INCAR KPOINTS
Diamond Si 5.5 0.0 0.5 0.5 0.5 0.0 0.5 0.5 0.5 0.0 2 Direct 0.0 0.0 0.0 0.25 0.25 0.25 System =diamond Si ISTART = 0 ENCUT = 150.0 NELM= 200 EDIFF = 1E-04 EDIFFG = -0.02
(1). 生成4个输入文件: POSCAR POTCAR INCAR KPOINTS (2). 优化晶格参数,求出能量最低所对应的晶格参数
(3). 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量
(4). 修改KPOINTS和INCAR输入文件,固定电荷密度,做非自洽 计算,得到输出文件EIGENVAL (5). 提取数据,画图
c/a
1 3 a1 a( i j) 2 2 1 3 a2 a ( i j) 2 2 a3 ck
(2). 优化晶格参数,求出能量最低所对应的晶格参数
hcp结构晶体含有一个内部自由度, 晶格参数优化过程要比立方 结构费时
VASP单机操作流程(以Ni111表面为例)

八,能带计算(bands):
建立bands文件夹,输入命令:mkdir bands
将KPOINTS INCAR POSCAR POTCAR CHG CHGCAR WAVECAR复制到bands文件夹中,
计算完成后,输入命令:cat Etot.dat查看结果。如同截断能的优化一样,根据要求选择合适的K点。(在此我选择17)
四,优化晶格常数:
在优化晶格常数时,是对比例系数的优化,不能使用绝对值。一般在试验值的左右各取一些值,根据要求选择最合适的晶格常数。计算过程和上两步类似。
Cp En . sh POS . sh(为方便使用格式)
打开终端,编辑K点文件KPOINTS(在此使用自动生成的方法)。
Cat >KPOINTS<<!
>A
>0
>7 7 1
>0 0 0
>!
保存退出。
二,优化截断能:
优化过程的脚本为:
#!/bash/sh
for i in 200 250 300 350 400 450 500 550 600
do
cat >INCAR<<!
(其它内容省略)
(保存退出)
建立relax文件夹:mkdir relax
将CHG CHGCAR WAVECAR INCAR POSCAR KPOINTS POTCAR复制到该文件夹:
Cp CHG CHGCAR WAVECAR INCAR POSCAR KPOINTS POTCAR relax
则在relax文件夹下运行命令;
输入命令:vi INCAR
【Selected】VASP使用总结.docx

VASP计算的理论及实践总结一、赝势的选取二、收敛测试1、VASP测试截断能和A点2、MS测试三、结构弛豫四、VASP的使用流程(计算性质)1、VASP的四个输入文件的设置2、输出文件的查看及指令3、计算单电能(1)测试截断能(2)测试A点4、进行结构优化5、计算弹性常数6、一些常用指令一、赝势的选取VASP赝势库中分为:PP和PAW两种势,PP又分为SP(标准)和USPP (超软)。
交换关联函数分为:LDA(局域密度近似)和GGA(广义梯度近似)。
GGA 又分为PW91和PBE。
在VASP中,其中pot,pot-gga是属于超软势(使用较少)。
Paw,paw-pbe,和paw-gga是属于PAW。
采用较多的是PAW-pbe和PAW-gga。
此外vasp中的赝势分为几种,包扩标准赝势(没有下标的)、还有硬(harder)赝势(_h)、软(softer)赝势(_s),所谓的硬(难以赝化),就是指该元素原子的截断动能比较大,假想的势能与实际比较接近,计算得到的结果准确,但比较耗时,难以收敛。
软(容易赝化),表示该元素原子的截断动能比较小,赝势模型比较粗糙,但相对简单,可以使计算很快收敛(比如VASP开发的超软赝势)。
即硬的赝势精度高,但计算耗时。
软的精度低,容易收敛,但节省计算时间。
另一种情况:如Gd_3,这是把f电子放入核内处理,对于Gd来说,f电子恰好半满。
所以把f电子作为价电子处理的赝势还是蛮好的(类似还有Lu,全满)。
(相对其他的4f元素来说,至于把f电子作为芯内处理,是以前对4f元素的通用做法。
计算结果挺好)常用的做法是:用两种赝势测试一下对自己所关心的问题的影响情况。
在影响不大的情况下,选用不含4f电子的赝势(即后缀是3),一来减少计算量,二来避免DFT对4f电子的处理。
【1.赝势的选择:vasp的赝势文件放在目录~/vasp/potentials下,可以看到该目录又包含五个子目录potpot_GGApotpawpotpaw_GGApotpaw_PBE,其中每一个子目录对应一种赝势形式。
[实用参考]VASP经典学习教程
![[实用参考]VASP经典学习教程](https://img.taocdn.com/s3/m/0755a30710661ed9ad51f3a8.png)
V ASP学习教程太原理工大学量子化学课题组20PP/5/25太原目录第一章LinuG命令 (1)1.1 常用命令 (1)1.1.1 浏览目录 (1)1.1.2 浏览文件 (1)1.1.3 目录操作 (1)1.1.4 文件操作 (1)1.1.5 系统信息 (1)第二章SSH软件使用 (2)2.1 软件界面 (2)2.2 SSH transfer的应用 (3)2.2.1 文件传输 (3)2.2.2 简单应用 (3)第三章V ASP的四个输入文件 (3)3.1 INCAR (3)3.2 KPOINTS (4)3.3 POSCAR (4)3.4 POTCAR (5)第四章实例 (5)4.1 模型的构建 (5)4.2 V ASP计算 (8)4.2.1 参数测试 (8)4.2.2 晶胞优化(Cu) (13)4.2.3 Cu(100)表面的能量 (2)4.2.4 吸附分子CO、H、CHO的结构优化 (2)4.2.5 CO吸附于Cu100表面H位 (4)4.2.6 H吸附于Cu100表面H位 (5)4.2.7 CHO吸附于Cu100表面B位 (6)4.2.8 CO和H共吸附于Cu100表面 (7)4.2.9 过渡态计算 (8)第一章LinuG命令1.1常用命令1.1.1浏览目录cd:进入某个目录。
如:cd/home/songluzhi/vasp/CH4cd..上一层目录;cd/根目录;ls:显示目录下的文件。
注:输入目录名时,可只输入前3个字母,按Tab键补全。
1.1.2浏览文件cat:显示文件内容。
如:catINCAR如果文件较大,可用:catINCAR|more(可以按上下键查看) 合并文件:catAB>C(A和B的内容合并,A在前,B在后) 1.1.3目录操作mkdir:建立目录;rmdir:删除目录。
如:mkdirT-CH3-Rh1111.1.4文件操作rm:删除文件;vi:编辑文件;cp:拷贝文件mv:移动文件;pwd:显示当前路径。
最新VASP操作介绍-两次课PPT课件

3. K网格大小的选择:
对于一维至三维体系的计算,需涉及k点数目的选择,对 于K点的确定,它与布里渊区的形状以及对称性有关。VASP的 K点输入方法有多种,其中最常用的是直接给定K-mesh的大小, 然后程序根据布里渊区的形状以及对称性自动生成各K点的坐 标和权重。
对于K-mesh的确定方法,通常通过考察总能量/能量差的收敛 程度来确定,能量的收敛标准是1meV/atom。
基于平面波表示的Kohn—Sham方程:
G ' 2 m 2 |k G |2G G ' V i( o G n G ') V H ( ;) c i , k G i c i , k G
上式中动能项是对角化的,通过求解上式方括号中的哈密顿矩 阵来求解KS方程,该矩阵的大小由截至能(cutoff energy)来决定。
动力学模拟); DOSCAR : 态密度信息。
POSCAR文件内容说明:
Silicon bulk (Title) 2.9 (Scaling factor or lattice constant) 0.0 1.0 1.0 (第一个平移矢量的方向) 1.0 0.0 1.0 (第二个平移矢量的方向) 1.0 1.0 0.0 (第三个平移矢量的方向)
多数情况下,对半导体或绝缘体较小的K-mesh能量就可以 收敛,对于导体,一般需要较大的K-mesh。
VASP经典学习教程-有用

V ASP 学习教程太原理工大学量子化学课题组2012/5/25 太原目录第一章 Linux命令............................................ 错误!未定义书签。
常用命令................................................ 错误!未定义书签。
浏览目录............................................ 错误!未定义书签。
浏览文件............................................ 错误!未定义书签。
目录操作............................................ 错误!未定义书签。
文件操作............................................ 错误!未定义书签。
系统信息............................................ 错误!未定义书签。
第二章 SSH软件使用.......................................... 错误!未定义书签。
软件界面................................................ 错误!未定义书签。
SSH transfer的应用..................................... 错误!未定义书签。
文件传输............................................ 错误!未定义书签。
简单应用............................................ 错误!未定义书签。
第三章 VASP的四个输入文件................................... 错误!未定义书签。
VASP简介ppt课件
13
(2). 优化晶格参数,求出能量最低所对应的晶格参数
(3) 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量
(i) 找到平衡晶格常数后, 把该值写入到POSCAR文件中,并增加K点数 作一个离子步自洽计算(NSW = 0, IBRION = -1) .
(ii) 从DOSCAR输出文件中读出态密度和费米能级, 费米能级也可从 OUTCAR中读出.
☺可以在一行设置多个关键词(即参数)的值,但是每个关键值之间用分 号(;)隔开。如ISMEAR= 0; SIGMA= 0.2。 ☺当想不用INCAR中某个关键词的值时,在该行前面加上井号(#)注释掉, 如#ISMEAR=0; SIGMA = 0.2
5
POSCAR输入文件:描述体系结构
例:SiC体系的POSCAR文件
TITEL = US Si
LULTRA = T use ultrasoft PP ?
IUNSCR = 1 unscreen: 0-lin 1-nonlin 2-no
RPACOR = 1.580 partial core radius
POMASS = 28.085; ZVAL = 4.000 mass and valenz
标题或注释行,无特别意义 K点的数目 以字母R开头表示k点是按倒格子坐标系 前三个数是k点的坐标,最后一个数是相应k 点的权重(下面共5个k点)
如果是以卡笛尔坐标系来写k点坐 标,则第三行以字母C开头。
9
POTCAR输入文件: 赝势文件
Si 的一种势函数的部分内容
US Si 4.00000000000000000 parameters from PSCTR are: VRHFIN =Si: s2p2 LEXCH = CA EATOM = 115.7612 eV, 8.5082 Ry GGA = -1.4125 -1.4408 .0293 -.9884 eV
VASP使用总结

VASP计算的理论及实践总结一、赝势的选取二、收敛测试1、VASP测试截断能和K 点2、MS测试三、结构弛豫四、VASP的使用流程(计算性质)1、VASP的四个输入文件的设置2、输出文件的查看及指令3、计算单电能(1) 测试截断能(2) 测试K点4、进行结构优化5、计算弹性常数6、一些常用指令一、赝势的选取VASP赝势库中分为:PP和PAW两种势,PP又分为SP(标准)和USPP(超软)。
交换关联函数分为:LDA(局域密度近似)和GGA(广义梯度近似)。
GGA 又分为PW91和PBE。
在VASP中,其中pot ,pot-gga是属于超软势(使用较少)。
Paw, paw-pbe ,和paw-gga是属于PAW。
采用较多的是PAW-pbe 和PAW-gga。
此外vasp 中的赝势分为几种,包扩标准赝势(没有下标的)、还有硬(harder)赝势(_h)、软(softer)赝势(_s), 所谓的硬(难以赝化),就是指该元素原子的截断动能比较大,假想的势能与实际比较接近,计算得到的结果准确,但比较耗时,难以收敛。
软(容易赝化),表示该元素原子的截断动能比较小,赝势模型比较粗糙,但相对简单,可以使计算很快收敛(比如VASP开发的超软赝势)。
即硬的赝势精度高,但计算耗时。
软的精度低,容易收敛,但节省计算时间。
另一种情况:如Gd_3,这是把f电子放入核内处理,对于Gd来说,f电子恰好半满。
所以把f电子作为价电子处理的赝势还是蛮好的(类似还有Lu,全满)。
(相对其他的4f元素来说,至于把f电子作为芯内处理,是以前对4f元素的通用做法。
计算结果挺好)常用的做法是:用两种赝势测试一下对自己所关心的问题的影响情况。
在影响不大的情况下,选用不含4f电子的赝势(即后缀是3),一来减少计算量,二来避免DFT对4f电子的处理。
【1.赝势的选择:vasp的赝势文件放在目录~/vasp/potentials 下,可以看到该目录又包含五个子目录pot pot_GGA potpaw potpaw_GGA potpaw_PBE ,其中每一个子目录对应一种赝势形式。
vasp基本原理(入门)课件

N
2
n(r) i(r)
电子密度分布
i 1
Kohn-Sham方程是一个自洽方程组。先提供初始电子密度分布
n(r) , 它一般可由原子的nat(r) 叠加而成。依次求出经典Coulomb 势、交换关联势、有效势。再求解KS方程。再由KS波函数构造新
的电子密度分布。比较输入与输出的电子密度分布。如已自洽,
便计算总能,输出所有结果v。asp基本原理(入门)
解Kohn-Sham方程的流程图
.
n(r)=Σnat(r)
原子计算
求解φ、Vxc、Veff
计算总能Etot
求解Kohn-Sham方程 得到ψi
nin与nout混合
由ψi构造nout(r)
No
Yes
比较nin与 nout(r)
No 精度控制
Yes
输出结果: Etot、 ψi、 n(r) Vxc、Veff、En(k)、N(E)
第四章 密度泛函理论(DFT)
4.1 引言 4.2 DFT的优点 4.3 Hohenberg-Kohn定理 4.4 能量泛函公式
4.6 Kohn-Sham方程 4.7 总能Etot表达式 4.8 DFT的意义 4.9 小 结
vasp基本原理(入门)
4.1 引言
1。概述 • DFT = Density Functional Theory (1964):
i ( r ) i i ( r )
V eff (r ) (r ) V xc (r )
εi=Kohn-Sham本征值 称有效势
( r ) v ( r )
n ( r ') rr'
dr
'
v (r )
vH
VASP参数设置详解

VASP参数设置详解软件主要功能:采用周期性边界条件(或超原胞模型)处理原子、分子、团簇、纳米线(或管)、薄膜、晶体、准晶和无定性材料,以及表面体系和固体l 计算材料的结构参数(键长、键角、晶格常数、原子位置等)和构型l 计算材料的状态方程和力学性质(体弹性模量和弹性常数)l 计算材料的电子结构(能级、电荷密度分布、能带、电子态密度和ELF)l 计算材料的光学性质l 计算材料的磁学性质l 计算材料的晶格动力学性质(声子谱等)l 表面体系的模拟(重构、表面态和STM模拟)l 从头分子动力学模拟l 计算材料的激发态(GW准粒子修正)计算主要的四个参数文件:INCAR ,POSCAR,POTCAR ,KPOINTS,下面简要介绍,详细权威的请参照手册INCAR文件:该文件控制VASP进行何种性质的计算,并设置了计算方法中一些重要的参数,这些参数主要包括以下几类:l 对所计算的体系进行注释:SYSTEMl 定义如何输入或构造初始的电荷密度和波函数:ISTART,ICHARG,INIWAVl 定义电子的优化–平面波切断动能和缀加电荷时的切断值:ENCUT,ENAUG–电子部分优化的方法:ALGO,IALGO,LDIAG–电荷密度混合的方法:IMIX,AMIX,AMIN,BMIX,AMIX_MAG,BMIX_MAG,WC,INIMIX,MIXPRE,MAXMIX –自洽迭代步数和收敛标准:NELM,NELMIN,NELMDL,EDIFFl 定义离子或原子的优化–原子位置优化的方法、移动的步长和步数:IBRION,NFREE,POTIM,NSW–分子动力学相关参数:SMASS,TEBEG,TEEND,POMASS,NBLOCK,KBLOCK,PSTRESS–离子弛豫收敛标准:EDIFFGl 定义态密度积分的方法和参数– smearing方法和参数:ISMEAR,SIGMA–计算态密度时能量范围和点数:EMIN,EMAX,NEDOS–计算分波态密度的参数:RWIGS,LORBITl 其它–计算精度控制:PREC–磁性计算:ISPIN,MAGMOM,NUPDOWN–交换关联函数:GGA,VOSKOWN–计算ELF和总的局域势:LELF,LVTOT–结构优化参数:ISIF–等等。
[整理版]vasp攻略
![[整理版]vasp攻略](https://img.taocdn.com/s3/m/629019a9f021dd36a32d7375a417866fb84ac09a.png)
打包压缩命令:tar zcvf 文件名.tar.gz 源文件名采用link方式避免重复的文件浪费内存:ln –s 源文件命名Eg:ln –s ../optic/MME ./EuO.mme交互式绘图工具gnuplot: 命令行打:gnuplot进入格式:plot “文件名”退出:quit求磁矩:getmag如果由于节点掉线在提交任务后秒退,不输出outcar可以指定节点提交任务LJRS -l nodes=c0104:ppn=4chmod +x 名称——使脚本可以执行构造potcar,以A和B元素为例:如果是以Z为拓展名的文件:zcat A/POTCAR.Z B/POTCAR.Z > POTCAR如果是解压后的potcar文件:cat A/POTCAR B/POTCAR > POTCARINCAR中的RWIGS通过POTCAR文件获得单位晶胞体积:grep “vol”OUTCAR自动计算加应力情况下的最优化情况:C/a :vaspcaopt softmode-e -=*(画曲线)如果要看某种材料是FM还是AFM,需要以相同结构计算一次,看能量哪个低。
将POSCAR/CONTCAR/CHGCAR装换成xcrysden(进入xcrysden文件执行./xcrysden)可读取的形式Eg:(BFO)v2xsf CHGCAR -1 83 -2 26 -3 8 -dv2xsf POSCAR -1 83 -2 26 -3 8 -dv2xsf CONTCAR -1 83 -2 26 -3 8 -d晶胞放大时,k点需要等比例的缩小画出曲线图:ISMEAR如果是半导体/绝缘体取-5,如果是金属取1,SIGMA=0.2(一般不改变)如果求DOS,则ISMEAR=-5.如果求band,则ISMEAR=1.1、Relax ISTART=0,ICHARG=2, ISIF=3,NSW=200,EDIFFG=-1*10-3,IBRION=22、Scf NSW=0(关闭结构优化) 用Relax后的CONTCAR替换POSCAR3、DOS 添加scf后的CHGCAR,ISTART=1,ICHARG=11,k放大一倍DOSCAR 第六行:Emax Emin Emin与Emax之间点的数目Ef第七行:能量总的态密度(spin up)总的态密度(spin down)态密度积分(up)态密度积分(down)后面按原子分:能量s轨道态密度(spin up)s轨道态密度(spin down)p轨道态密度(spin up)p轨道态密度(spin down)d轨道态密度(spin up)d轨道态密度(spindown)f轨道态密度(spin up)f轨道态密度(spin dow n)Split_dos 对dos按原子区分4、band 与DOS所需文件一致,KPOINTS需要使用line模式手动输入。
太原理工大学--VASP--讲解

V ASP 学习教程太原理工大学量子化学课题组2012/5/25 太原目录第一章LINUX命令 (1)1.1 常用命令 (1)1.1.1 浏览目录 (1)1.1.2 浏览文件 (1)1.1.3 目录操作 (1)1.1.4 文件操作 (1)1.1.5 系统信息 (1)第二章SSH软件使用 (2)2.1 软件界面 (2)2.2 SSH transfer的应用 (3)2.2.1 文件传输 (3)2.2.2 简单应用 (3)第三章VASP的四个输入文件 (3)3.1 INCAR (3)3.2 KPOINTS (4)3.3 POSCAR (4)3.4 POTCAR (5)第四章实例 (5)4.1 模型的构建 (5)4.2 VASP计算 (8)4.2.1 参数测试 (8)4.2.2 晶胞优化(Cu) (13)4.2.3 Cu(100)表面的能量 (2)4.2.4 吸附分子CO、H、CHO的结构优化 (2)4.2.5 CO吸附于Cu100表面H位 (4)4.2.6 H吸附于Cu100表面H位 (5)4.2.7 CHO吸附于Cu100表面B位 (6)4.2.8 CO和H共吸附于Cu100表面 (7)4.2.9 过渡态计算 (8)第一章Linux命令1.1 常用命令1.1.1 浏览目录cd: 进入某个目录。
如:cd /home/songluzhi/vasp/CH4 cd .. 上一层目录;cd / 跟目录;ls: 显示目录下的文件。
注:输入目录名时,可只输入前3个字母,按Tab键补全。
1.1.2 浏览文件cat:显示文件内容。
如:cat INCAR如果文件较大,可用:cat INCAR | more (可以按上下键查看) 合并文件:cat A B > C (A和B的内容合并,A在前,B在后) 1.1.3 目录操作mkdir:建立目录;rmdir:删除目录。
如:mkdir T-CH3-Rh1111.1.4 文件操作rm:删除文件;vi:编辑文件;cp:拷贝文件mv:移动文件;pwd:显示当前路径。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1) 能量计算
J. Phys. Chem. C, 2008, 112, 191
2) 电子结构(能带结构、DOS、电荷密度分布)
能带结构
DOS
电荷密度分布
J. Phys. Chem. B, 2005, 109, 19270
3) 构型优化(含过渡态)和反应途径
J. Phys. Chem. B, 2006, 110, 15454
这样,电子波函数可以写为平面波的加和:
i ( k e G )r i (r ) c i , k G G
根据密度泛函理论,波函数通过求解Kohn—Sham方程来确定:
2 2 [ Vion (r ) VH (r ) V XC (r )] i (r ) i i (r ) 2m i:Kohn—Sham本征值
Vion:电子与核之间的作用势 VH和VXC:电子的Hartree势和交换-相关势
n ( r ') 3 2 VH (r ) e d r ' | r r '|
E XC [n(r )] V XC (r ) n(r )
基于平面波表示的Kohn—Sham方程:
4) 频率计算和HREELS能谱模拟
J. Phys. Chem. C, 2007, 111, 7437
5) STM图像模拟
Surf. Sci., 2007, 601, 3488
6) UPS能谱图像模拟
Surf. Sci., 2007, 601, 3488
7) 材料光学性质计算
8) 其它性质计算,包括功函、力学性质等
4) 严格意义上,通过考察体系总能量/能量差值对真空 区大小的收敛情况来确定合理的平移矢量长度。
Total energy
Length of vector
3. K网格大小的选择:
对于一维至三维体系的计算,需涉及k点数目的选择,对 于K点的确定,它与布里渊区的形状以及对称性有关。VASP的 K点输入方法有多种,其中最常用的是直接给定K-mesh的大小, 然后程序根据布里渊区的形状以及对称性自动生成各K点的坐 标和权重。 对于K-mesh的确定方法,通常通过考察总能量/能量差的收敛 程度来确定,能量的收敛标准是1meV/atom。 多数情况下,对半导体或绝缘体较小的K-mesh能量就可以
2 2 V (G G ' ) V (G G ' ) V | k G | G ion H XC (G G ' ) ci , k G i ci , k G G' G ' 2m
化学家习惯的原子轨道的概念相联系,即其结果与化学家
所感兴趣的成键和轨道作用图象很难联系出来,这就为我 们计算结果的分析带来了困难; 2) 考察某些物理量时,例如原子电荷,涉及到积分范围的选 取,这造成所得物理量的绝对值意义不大; 3) 有些方法,例如杂化密度泛函方法不易于采用平面波基组 方法实现。
3. VASP程序基本知识
FLAPW 相比,并且计算速度比 FLAPW 快很多。 已广泛应用于材料科学领域。
主要介绍内容
1. VESTA软件模型建立 2. VASP基本原理简介
பைடு நூலகம்
3. VASP软件基本知识
4. 常用关键词使用说明 5. 实例解析,实际操作?
2. VASP程序基本原理
VASP是基于赝势平面波基组的密度泛函程序,其前身
2. 重复平板模型(或层晶模型):
VASP程序采用重复平板模型来模拟零维至三维体系
零维分子体系
Dv: Vacuum thickness (~10 A)
二维固体表面
说明: 重复平板模型中的平移矢量长度必须合理选择,以保证: 1) 对于分子体系,必须保证相邻重复单元中最近邻原子之 间的距离必须至少7~10埃以上; 2) 对于一维体系,相邻两条链最近邻原子之间的距离必须 至少7~10埃以上; 3) 对二维体系,上下两个平板最近邻原子之间的距离必须 至少7~10埃以上;
是CASTEP 1989版本,其基本原理如下: 根据Bloch定理,对于周期体系,其电子波函数可以写 为单胞部分和类波部分的乘积:
ik r i (r ) e f i (r )
其中,单胞部分的波函数可以用一组在倒易空间的平面 波来表示:
i G e r f i (r ) c i ,G G
相同的精度;
3) 很方便地采用快速傅立叶变换 (FFT) 技术,使能量、力 等的计算在实空间和倒易空间快速转换,这样计算尽可
能在方便的空间中进行;
4) 计算的收敛性和精确性比较容易控制,因为通过截断能 的选择可以方便控制平面波基组的大小。
平面波基组方法的不足之处:
1) 所求得的波函数很难寻找出一个直观的物理或化学图象与
VASP软件介绍
说明:本PPT主要内容参考网络资源,其用于教
学是合适的。
主要参考:计算材料学:杨振华。
VASP计算软件包简介
VASP,其全称是Vienna Ab-initio Simulation Package。
VASP是一种使用赝势和平面波基组进行从头量子力学分 子动力学计算和第一性原理计算的软件包。 VASP主要用于具有周期性的晶体或表面的计算,可以采 用大单胞,也可以用于处理小的分子体系。
1. 与同类的软件相比,它比较早地实现了超软赝
势,计算量相对于一般的模守恒赝势方法大为减少。 2. 其对计算领域最大贡献无疑是在Blöchl的基础上
发展的投影缀加平面波(PAW)方法。这使得VASP
不仅计算速度快,而且精度是abinit和pwscf没法比
的。 VASP 的精度,比如磁性计算,很多可以跟
上式中动能项是对角化的,通过求解上式方括号中的哈密顿矩
阵来求解KS方程,该矩阵的大小由截至能(cutoff energy)来决定。
尝试电子密度和尝试波函数
程序流程:
写出交换相关势表达式
构造哈密顿量
子空间对角化,优化迭代
自由能的表达式E
新电子密度,与尝试电子密度比较 是 否
输出结果,写波函数
与原子轨道基组相比,平面波基组有如下优点: 1) 无需考虑BSSE校正; 2) 平面波基函数的具体形式不依赖于核的坐标,这样,一 方 面 , 价 电 子 对 离 子 的 作 用 力 可 以 直 接 用 H el lm a n Feymann 定理得到解析的表达式,计算显得非常方便, 另一方面也使能量的计算在不同的原子构象下具有基本