实验二: 双序列比对实验
05_双序列比对

简单的序列比对
• 将两条序列左端对齐,放在两行中。 • 如果某列中两条序列的字符相同,则用竖线(|)将
它们连起来。 • 这是最佳比对结果吗?
是否有更好的比对结果?
• 如果我们仅仅将它们相对移动两个位置, 就可得到更好的比对结果:
结论
• We can not simply put both sequences one alongside the other, but we must compare them in all possible shiftings looking for the best alignment.
双序列比对多序列比对序列比对dna序列比对蛋白质序列比对序列比对软件氨基酸序列比对序列全局比对图片比对照片比对
复习
1. 数据库检索和数据库搜索的概念 2. Entrez数据库检索系统 3. SRS 数据库检索系统
双序列比对 (Pairwise Sequence Alignment)
1. 为什么要进行序列比对? 2. 序列比对相关术语 3. 最佳比对 4. 利用计算机进行序列比对 5. 序列的点阵作图比对 6. 双序列比对工具
potential similarities
30-40
30 Longer, weaker local alignments
< 30
4.1.3 PAM 和 BLOSUM 的差别
• PAM 矩阵是从蛋白质序列的全局比对 结果推导出来的;
• BLOSUM 矩阵则是从蛋白质序列局部 比对推导出来的。
Rat versus mouse RBP
“午夜区”( midnight zone )
• 当相似性低于10%时,就进入了“午 夜区”( midnight zone ),这时无法 检测结构的相似性。
实验2 序列比对

实验二:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析。
实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlign用dotplot方法能够直观地认识两条序列比对,但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍由DNASTAR公司开发的一个比较全面的生物信息学软件包--Lasergene,它包含了7个模块,其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 2.3),选择完毕点击Done回到程序页面。
实验二 双序列比对分析

实验三双序列比对分析一.实验目的Tay-Sachs是一种常染色体隐性遗传疾病,它的起因是第15号染色体的等位基因HEXA突变。
人类的HEXA基因在GenBank中的编号为“NM_000520”,小鼠的HEXA 基因在GenBank中的编号为“AK080777”,它们是核苷酸序列,以这两条序列为例,学习双序列比对分析。
1.学习和掌握在MATLAB平台上应用Bioinformatics工具包有关核苷酸和蛋白质双序列比对的命令和功能。
2.学习和掌握在MATLAB平台上应用Bioinformatics工具包访问GenBank,并提取核苷酸和蛋白质序列数据的方法。
3.学习和掌握在MATLAB平台上应用Bioinformatics工具包制作核苷酸或蛋白质两条序列比对的点阵图的方法。
4.学习和掌握在MATLAB平台上应用Bioinformatics工具包进行核苷酸或蛋白质双序列的局部比对和全局比对的方法。
二.实验内容1.在MATLAB平台上应用Bioinformatics工具包访问GenBank,提取核苷酸序列并转换为蛋白质序列。
①用“web”命令在MATLAB平台上打开NCBI网页。
web('/')web('/books/bv.fcgi?call=bv.View..ShowSection&rid=gnd')②用“getgenbank”功能从GenBank中读序列信息到MARLABhumanHEXA = getgenbank('NM_000520')mouseHEXA = getgenbank('AK080777')在MATLAB的workshop打开humanHEXA 和mouseHEXA查看其内容。
③从GenBank中提取2条核苷酸序列后,首先要做的是用全局比对来寻找两条序列中的相似序列。
因为进行蛋白质序列的比对更能体现其生物学本质,所以常常进行蛋白质序列的比对。
实验二_数据库相似性搜索与序列比对

实验二_数据库相似性搜索与序列比对实验二数据库相似性搜索与序列比对实验原理:数据库相似性搜索以两两序列比对为基础,将感兴趣的基因序列与序列数据库中的每个序列进行比较,鉴别出相似的序列。
搜索结果显示出与最佳匹配序列的对位排列及匹配记分。
序列数据库搜索对发现基因的功能非常有效。
fasta和blast是两个著名的用于数据库相似性搜索的软件包。
其中blast(basiclocala1ignmentsearchtool)基于局部比对的搜索工具,是一种启发式搜索算法服务软件,包括blastp,blastn,blastx,tblastn 和tblastx程序。
实验目的和要求:学习数据库相似性检索和序列比对的程序的使用,能够理解程序给出的结果,从中获取有关功能和结构的信息。
(1)要求学生使用所学的数据库检索方法检索数据库中的特定基因(2)掌握数据库相似性搜索工具blast的基本比对方法,参数设置及结果分析(3)掌握核酸和蛋白质序列的比对方法、参数设置和结果分析实验材料:未知核酸序列;未知氨基酸序列;SOD基因工具软件:(1)数据库检索工具Entrez一、利用blast中的special类下的aligntwosequences(bl2seq)比较人与老鼠的sod基因蛋白质序列的相似性程度(1)人类aab27818是通过NCBI 1的ntrez和小鼠3gtt_E的SOD基因氨基酸序列或登录号(SOD分为SOD1或SOD2等,检索时注意选择完全相同的SOD基因)搜索蛋白质数据库获得的。
(2)进入NCBI的blast网页,然后选择specializedlast下的align two sequences(bl2seq)程序来比较这两个序列(3)选择blastp子程序,将序列或登录号分别粘贴到序列框中(4)其他选项采用默认的设置,运行程序(5)分析结果,并回答以下问题NCBI的Entrez搜索中使用了哪些关键词?humanandsodmouseandsod人和小鼠SOD基因蛋白质序列的注册号是多少?人aab27818 1和鼠标3gtt_e两序列比对得到的一致性百分比和相似性百分比分别为多少?识别127/153(83%)阳性135/153(88%)两序列比对结果中哪些区域出现了gap?差距0/153(0%)二、利用specielizedblast的conserveddomain进行蛋白质保守结构域分析(1)进入ncbi的blast网页(2)选择specialize last to enter下的保守域超链接(3)在cazy数据库查找一个糖苷水解酶glycosidehydrolases(gh+学号),获得其蛋白质序列或蛋白质序列的genbank登录号aek59386.1(4)在保守域页面的输入框中输入糖苷水解酶的登录号或蛋白质(5),选择默认参数,点击提交进行提交分析(6)阅读得到的结果,点击各hit的超链接了解找到的结构域的功能(7)将结构域图形和表格记录在实验报告中三、利用blast在数据库中搜索不同物种的同源基因(1)利用文献检索工具检索clostridiumthermocellum嗜热梭菌与其纤维素降解功能相关的基因,例如糖苷水解酶glycosidehydrolases(gh+学号)或多糖裂解酶polysaccharidelyases(pls)或碳水化合物酯酶carbohydrateesterases(ces)等(2)利用ncbi的entrez检索该基因获得其核酸序列ab125373或者使用(2)中的蛋白质注册号通过NCBI数据库中的相关信息链接到核酸数据库,以获得基因的核酸注册号或序列(3)利用blastn进行数据库相似性搜索搜索其他微生物中的同源基因(4)分析blast结果,并回答以下问题检索获得基因名称是?chi19-1该基因的登录号是多少?ab125373进行blastn搜索的数据库选项为?nr请列出其他3-5种具有该基因及其同源基因的微生物的注册号?ap009493.1。
双序列比对算法

双序列比对算法
/// 双序列比对用于研究两个序列定义的DNA有多少相似之处,或者蛋白质序列有多少相似之处。
/// 这种比较在DNA鉴定和遗传暗示步骤中是非常重要的,在生物信息学应用中,两个序列之间比较也特别重要,特别是在研究顺序的进化关系和鉴定功能。
///
/// 双序列比对算法主要用于非完全比对,因为完全比对可以利用穷举法,把序列中的元素两两比对,以期发现所需的最近分数最高的所需的比对对。
/// 双序列比对算法可以利用动态规划算法,其中首先定义一个矩阵,表示序列i和序列j的最佳比对,在这个矩阵中,每个条目用于表示以矩阵中元素为末尾的两个序列段的相似度/距离,元素i和元素j越来越相似,这个度量值越大,距离越小。
///
/// 动态规划法大概有两个步骤:第一个步骤是填充一个矩阵,第二个步骤是从矩阵中搜索出最有可能产生最高得分的比对对。
双序列比对的具体步骤

双序列比对的具体步骤双序列比对,这个名字听起来挺复杂,其实就像在一场舞会上找搭档。
想象一下,一对舞者在舞池里翩翩起舞,他们的动作得协调,节奏得一致,才能让人眼前一亮。
双序列比对就是在生物信息学中的“舞会”,我们要把两条DNA、RNA或者蛋白质序列“比对”起来,看它们之间的相似性和差异性。
今天就来聊聊这个过程,有点轻松有点幽默,希望大家能跟着我的节奏走。
你得有两条序列,像是两个舞者。
可能一条是来自于某种植物,另一条是来自于一只可爱的海豚。
哦,别小看这些序列,它们的长度可能就像一条大长河,从头到尾可真不少。
你得准备好工具,比如生物信息学里的软件工具。
这个工具就像是舞会的DJ,帮你调节音乐,确保舞者在合适的节拍下起舞。
比如BLAST、Clustal Omega、MAFFT之类的,你选一个就行。
别担心,它们都很“给力”,能帮你快速找到相似部分。
接下来就是预处理。
这一步就像是给舞者穿上合适的舞鞋,得把序列中那些多余的空格、特殊字符都处理掉。
想象一下,鞋子磨脚,舞者可跳不好呀。
这时,你还可以考虑一下序列的质量。
如果有些序列质量差,像是舞步不稳的舞者,建议把它们剔除掉,留下那些稳稳的“舞者”。
然后,真正的比对开始了!你将这两条序列输入工具,就像把舞者推入舞池。
软件开始工作了,开始寻找匹配的部分。
这个过程就像在找节拍,找到相似的地方,打出“合拍”的舞步。
如果两个序列在某些位置上有相同的碱基或者氨基酸,哇塞,那就是完美的配合!你会看到一条条的对齐线,心里不禁暗暗叫好。
不过,双序列比对可不是只有完美的配合,有时也会出现差异。
就像舞会上的舞者,可能有些人跳得慢,有些人快,动作也各有不同。
有的地方可能有缺失,有的地方则是多了几步。
这时候,你得好好分析这些差异,看看它们背后的意义。
是因为物种间的进化差异,还是因为某些环境因素造成的?哎呀,真是个好奇的事儿,像是在追溯舞者的历史。
你得把比对的结果给整理出来。
这个结果就是你的“舞会记录”,记录了舞者们的精彩表演。
序列比对(双序列比对)

二:实验内容及操作步骤
1. 进入
/blast/bl 2seq/wblast2.cgi
2. 下载核酸或蛋白质序列分别放在 sequence1和sequence2中 3. 核酸使用blastn程序,蛋白使用blastp程序 4. 设置不同罚分值进行比对,比较结果
二:实验内容及操作步骤
1wblast2.cgi
2. 下载核酸或蛋白质序列分别放在 sequence1和sequence2中 3. 核酸使用blastn程序,蛋白使用blastp程序 4. 设置不同罚分值进行比对,比较结果 5. 自己选取 个序列来比对。 自己选取2个序列来比对 个序列来比对。
cgcggttccg acggcgggga gggggcgagg tggggccgtg gcggcgagcc caccgacgga ggaggcggtg cagatgacgg agccgctcac caaggaggac ctcgtggcct acctcgtctc cgggtgcaag cccaaggaga actggagaat tggcacagaa catgagaagt tcggttttga agttgataca ttgcgtccta taaagtacga tcagatccgt gacatcctga atggacttgc tgagaggttt gattgggaca agatagttga agaaaataac gttatcggtc tcaagcaggg aaaacaaagc atttcactag aacctggcgg tcagtttgaa cttagtggtg ctcctcttga
运用:
请查询(或搜索)Os11g37990的 请查询(或搜索) 的 DNA、mRNA(cDNA)和蛋白质序列。 、 和蛋白质序列。 和蛋白质序列
双序列对比的原理

双序列对比的原理双序列对比是指对比两个序列之间的结构、相似性、差异性或演化关系的一种方法。
它广泛应用于生物学研究中,可以帮助研究人员理解不同物种的关系、进化过程以及功能区域等方面的信息。
双序列对比的原理基于两个序列的比对和分析。
在双序列对比中,两个序列通常是DNA、RNA或蛋白质序列。
在进行对比之前,需要先经过序列比对的步骤,即将两个序列按照一定的规则对齐,使得相同的部分对齐在一起并显示出来,不同的部分则可以通过间隔、替代或插入的方式标示出来。
比对的目标是找到两个序列之间的共同特征,以便更好地分析和解释它们之间的关系。
在进行双序列对比时,常用的比对算法包括Smith-Waterman算法和Needleman-Wunsch算法。
这些算法通过动态规划的方法,计算出两个序列之间的最优比对方案。
最优比对方案是指在符合一定规则的前提下,使得比对结果得分最高或最接近实际情况的方案。
通过比对算法计算得到的比对结果,可以用于查找相似性或共有区域,也可以用于推测序列之间的演化关系。
双序列对比还可以通过比对结果进行比对分析。
比对分析可以通过不同的方法和步骤来实现。
常见的比对分析方法包括序列标注、格局分析、功能预测和进化树构建等。
序列标注是指对比对结果中的不同部分进行注释和分析,比如标示出替代、插入和缺失的位置,这对于比对结果的解读非常重要。
格局分析是指对比对结果中重复出现的序列单元进行统计和分析,以便预测可能的功能区域。
功能预测是指根据比对结果推测序列的功能和结构特征,比如预测蛋白质序列中的位点、结构域和功能基序。
进化树构建是指基于比对结果推测不同序列之间的演化关系,从而揭示物种的进化历程和亲缘关系。
双序列对比的核心原理是在比对的基础上进行分析和解读。
通过比对两个序列,可以获得它们之间的结构和相似性信息,并进一步推测它们之间的差异性和演化关系。
双序列对比可以应用于多个领域,例如进化生物学、比较基因组学、疾病研究等。
双序列对比的原理有哪些

双序列对比的原理有哪些
双序列对比是一种常用的分析方法,用于比较两个序列之间的相似性和差异性。
其原理主要有以下几个方面:
1. 序列比对:双序列对比的第一步是将两个序列进行比对。
比对方法可以是全局比对、局部比对或者半全局比对。
全局比对是将整个序列进行比对,局部比对是比对序列的一部分,半全局比对则是在全局比对的基础上,允许序列的一部分有插入或删除。
2. 得分计算:比对之后,需要根据比对的结果计算得分。
得分的计算方法可以是简单计数,也可以是根据序列的相似性进行加权计算。
一般来说,相同的碱基或氨基酸会得到较高的得分,而不同的碱基或氨基酸会得到较低的得分。
3. 相似性分析:得到比对的得分之后,可以通过计算相似性指数来衡量两个序列的相似性。
常用的相似性指数有百分比相似性、相似度矩阵等。
相似性分析可以帮助研究人员理解两个序列之间的关系,比如判断它们是否属于同一个家族或亲缘关系。
4. 差异性分析:通过比对和得分计算,可以找出两个序列之间的差异。
差异性分析可以用于寻找突变、插入、缺失等变异类型,从而帮助研究人员研究序列的功能和进化。
总的来说,双序列对比的原理是通过比对、得分计算和相似性分析来研究两个序列之间的相似性和差异性,从而帮助理解序列的功能和进化。
两条序列比对与多序列比对
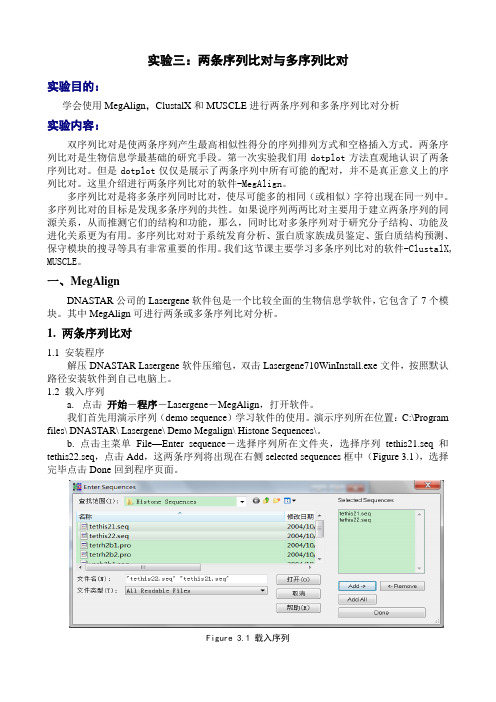
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
实习二:两条序列比对及蛋白质三维结构预测

实习二:两条序列比对及蛋白质三维结构预测实习二:两条序列比对及蛋白质三维结构预测学号姓名专业年级实验时间提交报告时间实验目的:1. 学会使用EMBOSS软件包的NEEDLE和WATER进行两条序列比对2. 通过PRSS理解两条序列比对得分的显著性检验3. 会使用SWISS-MODEL进行蛋白质三维结构预测实验内容:依据实验教材学会使用EMBOSS软件包在比对资源,能够使用全局局部的比对方法比对蛋白和核酸的序列;PRSS比对两条序列的p-value;及通过SWISS-MODEL 系统的全自动及提供模板的方式预测蛋白的三维结构。
作业:1. Align two nucleotide sequences of your interest with Needle and Water at EBI respectively. Interpret the alignments and explain the differences between the Needle and Water alignment results. Do the same procedure with two protein sequences and analyze the results.首先进入EMBOSS Needle页面,选择Nucleotide alignment;上传上次保存的脱水素dehydrin的山茶Camellia和大豆Glycine 的mRNA的FASTA格式文件;再设定比对参数,使用默认的评分矩阵即匹配5分,错配-4分同理进行EMBOSS Needle页面的protein alignment;上传山茶和大豆的蛋白序列的FASTA格式文件;评分系统选择BLOSUM62,使用默认的分值。
其次EMBOSS Water页面,用同上核酸和蛋白的比对方法分别进行山茶Camellia和大豆Glycine的mRNA和蛋白的比对;1、mRNA的全局与局部比对:共同部分# Aligned_sequences: 2# 1: JN400597.1# 2: NM_001253177.2# Matrix: EDNAFULL# Gap_penalty: 10.0# Extend_penalty: 0.5#全局比对结果:# Length: 1210# Identity: 611/1210 (50.5%)# Similarity: 611/1210 (50.5%)# Gaps: 437/1210 (36.1%)# Score: 1671.5#局部比对结果# Length: 1099# Identity: 613/1099 (55.8%)# Similarity: 613/1099 (55.8%)# Gaps: 314/1099 (28.6%)# Score: 1678.5由此发现局部和全局比对的长度和得分均不相同,局部比对的长度小于全局比对的序列长度,且得分高于全局比对,因此经过局部比对得到的序列相似性较高,空位较少。
双序列比对-

• 局部相似性比对的生物学基础是蛋白质功能位点往往是由 较短的序列片段组成的,这些部位的序列具有相当大的保 守性,尽管在序列的其它部位可能有插入、删除或突变。 此时,局部相似性比对往往比整体比对具有更高的灵敏度, 其结果更具生物学意义。
局部比对搜索基本工 具BLAST
• BLAST是目前常用的数据库搜索程序,它是 Basic Local Alignment Search Tool的缩 写,意为“基本局部相似性比对搜索工具”
•
相似性和同源性关系
• 序列的相似性和序列的同源性有一定的关系,一 般来说序列间的相似性越高的话,它们是同源序 列的可能性就更高,所以经常可以通过序列的相 似性来推测序列是否同源。
• 正因为存在这样的关系,很多时候对序列的 相似性和同源性就没有做很明显的区分,造成经 常等价混用两个名词。所以有出现A序列和B序列 的同源性为80%一说。
• 替换
在比对结果是在同一位置上出现两个不同的氨基 酸
• 插入,删除
发生于残基添加或消除时,比对中由一无效字符表 示,加在一条或另一条序列中。
• 间隙
反映了进化上发生的变化,在应用上,间隙使得 两个蛋白质能进行全长比对。
比对:全局和局部
• 序列比对的数学模型大体可以分为两类,一类从全长序列 出发,考虑序列的整体相似性,即全局比对;第二类考虑 序列部分区域的相似性,即局部比对。
图形示意结果
结果页面(二)
目标序列描述部分
带有genbank的链接,点击可以进入 相应的genbank序列
匹配情况,分值,e值
结果页面(三)
详细的比对上的序列的排列情况
一个具体的例子(blastp)
假设以下为一未知蛋白序列
>query_seq MSDNGPQSNQRSAPRITFGGPTDSTDNNQNGGRNGARPKQRRPQGLPNNTAS WFTALTQHGKEELRFPRGQGVPINTNSGPDDQIGYYRRATRRVRGGDGKMKEL SPRWYFYYLGTGPEASLPYGANKEGIVWVATEGALNTPKDHIGTRNPNNNAATV LQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRGNSRNSTPGSSRGNSPARM ASGGGETALALLLLDRLNQLESKVSGKGQQQQGQTVTKKSAAEASKKPRQKRT ATKQYNVTQAFGRRGPEQTQGNFGDQDLIRQGTDYKHWPQIAQFAPSASAFFG MSRIGMEVTPSGTWLTYHGAIKLDDKDPQFKDNVILLNKHIDAYKTFPPTEPKKDK KKKTDEAQPLPQRQKKQPTVTLLPAADMDDFSRQLQNSMSGASADSTQA
2双序列比对

哈尔滨医科大学 生物信息学院
李霞教授
第一节
引言
同源(homology)- 具有共同的祖先
直向同源(Orthologous ) 共生同源(paralogous )
相似(similarity)
同源序列一般是相似的,相似序列不 一定是同源的
2
3
4
通过点矩阵进行序列比较
5
6
7
Normalize Total Mutation Rate to 1%
This defines an evolutionary period: the period during which the 1% of all sequences are mutated (accepted of course)
1
2 1 2 2 1 0 1 2 2 2 1 2 1 2 2
1
1 1 2 2 2 1 0 2 1 1 2 2 2 1 2
1
2 2 1 1 2 2 2 0 1 2 2 2 1 2 2
1
2 1 1 2 2 2 1 1 0 2 2 1 1 1 1
2
1 2 2 2 2 2 1 2 2 0 1 1 2 2 2
但这并不意味100次PAM后,每个氨基酸都发生变化,因为其中一些位置可能 会经过多次突变,甚至可能会变回到原来的氨基酸。
PAM矩阵的制作步骤 •构建序列相似(大于85%)的比对 •计算氨基酸 j 的相对突变率mj(j被其它氨基酸替换的次数) •针对每个氨基酸对 i 和 j , 计算 j 被 i 替换次数 •替换次数除以相对突变率(mj)
41
取最小值!
计算过程:
42
计算过程:
•按行计算
双序列比对案例

双序列比对案例
那我就来讲个双序列比对的超有趣案例。
想象一下,你有两条神秘的密码纸条(这就好比是我们的两个序列啦)。
一条纸条上写着“ABCDEFG”,另一条写着“ABCFEG”。
咱们现在要做双序列比对,就像是玩一个超级找茬兼连连看的游戏。
首先呢,我们一眼就看到开头的“AB”是一样的,这就像是两个老朋友一见面就热情拥抱,很容易就对上了。
然后呢,第一条纸条的“C”和第二条纸条的“C”也完美匹配,这就好比是两个小伙伴戴着相同的帽子,一下子就被认出来了。
可是,第一条纸条的“D”在第二条纸条里找不到对应的小伙伴,这就有点尴尬啦,就像一个人去参加聚会结果发现没有和自己同好的人。
再往后看,“E”在两条纸条里又对上了,就像失散的双胞胎又重逢了一样。
接着第一条纸条的“F”和第二条纸条的“F”也匹配上了。
但是第一条纸条的“G”和第二条纸条的“E”“G”顺序有点乱,这里就像是两个人跳舞跳错了步骤。
在这个简单的例子里,我们通过这样的比对,就能知道这两条“密码纸条”(序列)哪些部分是相似的,哪些部分是不一样的。
这在生物学里可有用啦,比如说比较不同生物的基因序列,看看哪些基因是保守的(就像那些容易匹配上的部分),哪些基因发生了变异(就像那些对不上的部分)。
这就像是在探索生物进化这个超级大迷宫里的小线索,是不是超级酷呢?。
双序列比对

双序列比对序列比对的理论基础是进化学说,如果两个序列之间具有足够的相似性,就推测二者可能有共同的进化祖先,经过序列内残基的替换、残基或序列片段的缺失、以及序列重组等遗传变异过程分别演化而来。
序列相似和序列同源是不同的概念,序列之间的相似程度是可以量化的参数,而序列是否同源需要有进化事实的验证。
在残基-残基比对中,可以明显看到序列中某些氨基酸残基比其它位置上的残基更保守,这些信息揭示了这些保守位点上的残基对蛋白质的结构和功能是至关重要的,例如它们可能是酶的活性位点残基,形成二硫键的半胱氨酸残基,与配体结合部位的残基,与金属离子结合的残基,形成特定结构motif的残基等等。
但并不是所有保守的残基都一定是结构功能重要的,可能它们只是由于历史的原因被保留下来,而不是由于进化压力而保留下来。
因此,如果两个序列有显著的保守性,要确定二者具有共同的进化历史,进而认为二者有近似的结构和功能还需要更多实验和信息的支持。
通过大量实验和序列比对的分析,一般认为蛋白质的结构和功能比序列具有更大的保守性,因此粗略的说,如果序列之间的相似性超过30%,它们就很可能是同源的。
在进行序列两两比对时,有两方面问题直接影响相似性分值:取代矩阵和空位罚分。
粗糙的比对方法仅仅用相同/不同来描述两个残基的关系,显然这种方法无法描述残基取代对结构和功能的不同影响效果,缬氨酸对异亮氨酸的取代与谷氨酸对异亮氨酸的取代应该给予不同的打分。
因此如果用一个取代矩阵来描述氨基酸残基两两取代的分值会大大提高比对的敏感性和生物学意义。
虽然针对不同的研究目标和对象应该构建适宜的取代矩阵,但国际上常用的取代矩阵有PAM和BLOSUM等,它们来源于不同的构建方法和不同的参数选择,包括PAM250、BLOSUM62、BLOSUM90、BLOSUM30等。
对于不同的对象可以采用不同的取代矩阵以获得更多信息,例如对同源性较高的序列可以采用BLOSUM90矩阵,而对同源性较低的序列可采用BLOSUM30矩阵。
实验二.双序列比对

实验二.双序列比对一.实验目的:练习使用动态规划算法进行双序列比对;理解打分矩阵和参数对双序列比对结果的影响;理解动态规划算法的原理。
二.实验要求:动态规划算法是序列比对最基本的算法,可以确保找到最优比对。
分为全局比对和局部比对算法。
通过本实验的练习,更好的理解动态规划算法。
三.实验内容提要:对如下的两条序列进行双序列比对分析:> Drosophila Sex-lethal proteinASNT NLIVNYL PQDMTDRELYALFRAIGPINTCRIMRDYKTGYS YGYAFVD FTSEMD SQRAIKVLNG> Mouse Huc RBDMDSKT NLIVNYL PQNMTQDEFKSLFGSIGDIESCKLVRDKITGQS LGYGFVN YSDPN DADKAINTLNGL这些蛋白质包含一个RNA 识别模体。
该模体包含两个高度保守的两个功能区RNP1 和RNP2。
通过ebi 网站的在线工具完成练习(/Tools/psa/emboss_needle/)1.RNP1 和RNP2 是否得到比对?选择至少三个(差别大的)空位罚分和延伸值来进行比对。
2a.算法是否找到RNP1 和RNP2 的正确比对?2b.当空位开启罚分高时,结果发生什么变化?2c.当空位延伸罚分高时,结果发生什么变化?2d.为什么k 个连续的空位罚分要小于k 个间隔的空位罚分?使用PAM250 矩阵重复上述过程。
3. 比对结果是否发生变化?继续进行这两条序列的局部比对,通过ebi 网站的在线工具完成练习,网址:(/Tools/psa/emboss_water/)4a. RNP1 和RNP2 是否在局部比对中得到比对?4b. 局部比对的生物学意义是什么?4c. 为什么在这种比对中我们选择局部比对而不是全局比对?采用不同的打分参数和其它打分矩阵。
5.比对结果发生了什么变化?四.实验结果:1.全局比对1.1EBLOSUM62矩阵a.空位罚分10,延伸值0.5:b.空位罚分10,延伸值10c.空位罚分5,延伸值为10:1.2使用PAM250打分矩阵:a.空位罚分10,延伸值0.5:b.空位罚分10,延伸值10c.空位罚分5,延伸值为10:2.局部比对2.1采用Blosum62打分矩阵:a.空位罚分10,延伸值0.5:b.空位罚分10,延伸值10c.空位罚分5,延伸值为10:2.2采用PAM250打分矩阵:a.空位罚分10,延伸值0.5:b.空位罚分10,延伸值10c.空位罚分5,延伸值为10:五.回答问题:1.RNP1 和RNP2 得到比对。
双序列比对

耿红卫cauglacier@Bio informatics24 Pairwise Sequence Alignment1Homology, Similarity, Identity2Accepted Point Mutations and PAM matrix 3Log-Odds Score Matrix 4BLOSUM scoring matrix1Homology, Similarity, Identity 同源性、相似性、一致性4Homology 同源性1Two sequences are homologous if they share a common evolutionary ancestry2There are no degrees of homology; sequences are either homologous or not3Homologous proteins almost always share a significantly related three-dimensional structure4When two sequences are homologous, their amino acid or nucleotide sequences usually share significant identity5Homology, Similarity, Identity同源性,相似性,一致性1Homology is a qualitative inference (sequences arehomologous or not), identity and similarity are quantities that describe the relatedness of sequences2Two molecules may be homologous without sharingstatistically significant amino acid (or nucleotide) identity6Two types of homolog物种间的同源ortholog同一物种内由于基因复制、分离导致的同源paralog7Ortholog直系(直向,垂直)同源1Orthologs are homologous sequences in different species that arose from a common ancestral gene during speciation.在形成物种的过程中,来自共同祖先的基因分布到不同的物种,这些同源基因称为直系同源2从进化的角度来看,这类基因通常具有相同的功能,但并非绝对3是物种形成的结果,基因的历史反映了物种的历史8Paralog旁系(横向,并行)同源1Paralog describes homologous genes within a single species that diverged by a mechanism such as gene duplication旁系同源是指在同一个物种内,由于基因重复而产生分支,形成的一系列同源基因2是基因复制的结果,旁系同源基因在同一物种中平行演化9Globin: 球蛋白10An example for homologsHemoglobin 血红蛋白myoglobin 肌红蛋白1112Myoglobin1314Identitygi_119389635_pdb_2H35_B gi_119389637_pdb_2H35_D gi_230638_pdb_2MM1_gi_413915760_pdb_2H35_A gi_413915761_pdb_2H35_Cgi_119389635_pdb_2H35_B 100254444gi_119389637_pdb_2H35_D 254444gi_230638_pdb_2MM1_2626gi_413915760_pdb_2H35_A 100gi_413915761_pdb_2H35_C1516A group of myoglobin orthologs18 2Accepted Point Mutations and PAM matrix可接受点突变和PAM矩阵20Dr. Margaret Oakley Dayhoff, credited as the founder of Bio‐Informatics, ca. 198021有关双序列比对的意义实质1、排列两条序列,使之达到最大程度的相同的过程2、在比对氨基酸的情况下,就是考量最大程度的保守性目的1、衡量两个分子相似和同源的可能性2、如果序列相同的程度显著,那么可能同源,但不存在一定程度上的同源性,要么同源,要么不同源如何判断是否同源?(序列应该相似到什么程度?)必须以生物统计学为基础,用合适的算法进行判断是否同源的最终标准来自结构研究和进化分析22Dayhoff 模型是什么?任何两个序列之间都呈现不同程度的保守性Dayhoff 模型给任意两个蛋白质打分,衡量它们或远或近的相关性Dayhoff模型是如何给蛋白打分的?23Problem considered by Dayhoff and colleaguesHow to assign scores to aligned amino acid residues24Definition of Accepted point mutation (PAM)Accepted point mutation (PAM) is a replacement of one amino acid in a protein by another residue that has been accepted by natural selection 可接受点突变是指蛋白质的一个氨基酸被另外一个氨基酸替代,并且该氨基酸又被自然选择接受25What’s PAM?蛋白质的进化就是氨基酸的替代、缺失或者插入但无论蛋白质怎样变化,该蛋白质必须被自然界接受那些被自然选择接受的替代就叫做可接受点突变(PAM)26When PAM occurred?1A gene undergoes a DNA mutation such that it encodes a different amino acid基因发生突变,从而编码一个不同的蛋白2The entire species adopts that change as the predominant form of the protein整个物种接受了这种改变,并将这种蛋白作为主要形式27一种氨基酸能够被另一种氨基酸替代,而替代后形成的新蛋白质又被自然选择接受了哪种替代能够被自然界接受?显然,保守型取代最有可能被接受比如丝氨酸取代苏氨酸29丝氨酸和苏氨酸30To determine all possible changes1572 changes in 71 groups of closely related proteinswere examined得出了任意一对氨基酸相互替换的次数Dayhoff 对于“可接受”突变的选择是基于对氨基酸替代的经验观察31Dayhoff’s protein superfamiliesProtein 每亿年的PAMProtein 每亿年的PAM免疫球蛋白k 链C 区37胰蛋白酶 5.9k 酪蛋白33胰岛素 4.4上皮生长因子26细胞色素c 2.2血清白蛋白19谷氨酸脱氢酶0.9血红蛋白α 链12组蛋白H30.14肌球蛋白8.9组蛋白H40.10神经生长因子8.5泛素蛋白32Numbers of accepted point mutations in 1572 cases of amino acid substitutions (×10)33Dayhoff: 部分可接受点突变(PAM)的数目(×10)共有1572个氨基酸替换PAMs原氨基酸A Ala R ArgN AsnD Asp C CysQGlnE GluG Gly替代氨基酸A R 30N 10917D 1540532C 331000Q 9312050760E 2660948310422G 579101561621030112H211032264310243231034Dayhoff :氨基酸的相对突变能力The relative mutability of amino acids(氨基酸发生突变的次数/氨基酸出现的总次数×100%)Asn 134His 66Ser 120Arg 65Asp 106Lys 56Glu102Pro 56丙氨酸Ala 100Gly 49Thr 97Tyr 41Ile96Phe 41Met94Leu 40Gln93Cys 20Val74Trp1835Frequency with which each amino acid is foundGly 8.9%Arg 4.1%Ala 8.7%Asn 4.0%Leu 8.5%Phe 4.0%Lys 8.1%Gln 3.8%Ser 7.0%Ile 3.7%Val 6.5%His 3.4%Thr 5.8%Cys 3.3%Pro 5.1%Tyr 3.0%Glu 5.0%Met 1.5%Asp4.7%Trp1.0%总和为100%,如果每种氨基酸出现的频率相等,则均为5%36氨基酸代码表丙氨酸alanine Ala A 亮氨酸leucine Leu L 精氨酸arginine Arg R 赖氨酸lysine Lys K 天冬酰胺asparagine Asn N 甲硫氨酸methionine Met M 天冬氨酸aspartic acid AspD笨丙氨酸phenylalanine PheF半胱氨酸cysteine Cys C 脯氨酸proline Pro P 谷氨酰胺glutanineGlnQ丝氨酸serine Ser S 谷氨酸glutamic acid Glu E 苏氨酸threonine Thr T 甘氨酸glicine Gly G 色氨酸tryptophan TrpW组氨酸histidine His H 酪氨酸tyrosine Tyr Y 异亮氨酸isoleucineIle I颉氨酸valineVal V37The PAM1 matrixA mutation probability matrixEach element of the matrix Mij shows the probability that an original amino acid j (see the columns) will be replaced by another amino acid I (see the rows) over a defined evolutionary interval 数字表示:原氨基酸(列)被其他氨基酸(行)在一个进化区间内被替代的可能性For the PAM1 matrix, the proteins have undergone 1% change (that is,1 accepted point mutation per 100 amino acid residues)38The PAM1 mutation probability matrix39The PAM1 matrix1The PAM1 matrix was based on the alignment of closely related protein sequences, all of which were at least 85% identical within a protein familyPAM1矩阵基于紧密相关的蛋白质序列的比对而得,这些蛋白质一致性至少达85%2We aim to construct matrices for proteins that share any degree of amino acid identity40The PAM1 matrix用可接受突变的数目,结合每种氨基酸出现的频率,就产生了突变概率矩阵Mutation probability matrix 矩阵中的数值表示:在一定的进化时期内,原氨基酸(列)被替代氨基酸(行)替代的概率PAM1矩阵进化的时间是一个PAM ,而不是以年为单位这里PAM 的定义为进化趋异(evolution divergence)的单位,表示两个蛋白质之间1%的氨基酸发生改变的时间PAM1 矩阵表示进化时间是一个PAM时氨基酸的替代概率41PAM1突变概率矩阵(部分)原氨基酸A Ala R Arg N Asn D Asp C Cys Q Gln E Glu G Gly H His I Ile 替代氨基酸A 9867291038172126R 199131011000103N 419822360466213D 604298590653641C 1100997300011Q 394509876271231E 1007560359865423G 2111211137993510H 181831201099120I223121200987242解读PAM1 Matrix1、最大值位于左上右下的对角线上,每列值的和是10000,对应于100%2、第一列9867表示在一个PAM 进化期内,丙氨酸有98.67%的可能被替换成丙氨酸(A),有0.01%的可能被替换为精氨酸(R),以此类推3、最容易发生突变的天冬酰胺Asn 有98.22%的可能不改变,最不容易突变的色氨酸Cys 有99.73的可能不改变4、氨基酸如果发生突变,很容易找出最可能变成哪个氨基酸,合理的替代得到奖励分,不合理的替代将会被罚分43Substitution Matrix of PAM1 Matrix 其他PAM 矩阵是PAM1乘以自身数次得到的PAM 矩阵的两种极端情况1、PAM=0时,成为单位矩阵,因为没有氨基酸发生替代2、PAM 趋于无穷时,每种替代氨基酸等概率出现,每行的数值趋近于一个固定值,该值就是氨基酸出现的概率3、其他PAM 由PAM1推测而来,如PAM25044PAM0 matrixA Ala R Arg N Asn D Asp C Cys Q Gln E Glu G Gly A 100%0%0%0%0%0%0%0%R 0%100%0%0%0%0%0%0%N 0%0%100%0%0%0%0%0%D 0%0%0%100%0%0%0%0%C 0%0%0%0%100%0%0%0%Q 0%0%0%0%0%100%0%0%E 0%0%0%0%0%0%100%0%G0%0%0%0%0%0%0%100%45The PAM250 mutation probability matrix46The PAM250 matrix1It is produced when the PAM1 matrix is multiplied against itself 250 times, and it is one of the common matrices used for BLASTsearches of databasesPAM250矩阵由PAM1矩阵自乘250次得到,是BLAST 的常用打分矩阵2The PAM250 matrix applies to an evolutionary distance where proteins share about 20% amino acid identityPAM250适用于一定的进化距离,即蛋白质大约有20%的一致性3相比于PAM1,氨基酸的整体突变概率变化幅度减小,但表达的意义同PAM14PAM-N 矩阵值表示两条相距N 个PAM 的序列氨基酸替换的概率3Log ‐Odds Score Matrix对数打分矩阵48构建打分矩阵的思想基础1一些氨基酸性质接近,一些氨基酸差异较大,因此替换不是随机发生的,替代不会剧烈影响蛋白质结构,否则该蛋白质可能被自然选择淘汰2最常用的方法是统计自然界各种氨基酸的相互替代率,如果两种氨基酸相互替代发生的比较频繁,那么这一对氨基酸在打分矩阵中的得分就高3调查相关度较高的蛋白,能够更明显地看出相互取代率49从突变概率矩阵到对数比值打分矩阵From a Mutation Probability Matrix to a Log-OddsScore Matrix研究PAM 的目的是导出一个打分系统,用以评价两条序列的相关性。
双序列比对算法研究

国防科学技术大学硕士学位论文双序列比对算法研究姓名:何万双申请学位级别:硕士专业:计算机科学与技术指导教师:骆志刚20060301鱼堕型堂塾查盔堂堑塞生堕竺垡笙奎第二章序列比对相关知识现代科学技术各学科领域的日益发展和交叉融和给生命科学的研究提供了巨大的推动力。
在我们将实验室产生的序列信息递交到GenBank等公共数据库之前,有一个非常重要的预备工作,就是将我们的序列与公共数据库中的序列进行比较。
本章引入了基本的生物学知识和基因组的概念,并着重介绍序列比对的相关内容。
§2.1基因组基本概念众所周知,生物由细胞组成,而细胞中含有一种与遗传有关的高分子化合物,称为脱氧核糖核酸(DNA).DNA的基本组成单位为腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶四种核苷酸,分别由A、G、C和T四个字母(称为碱基)表示,这些核苷酸按照一定的顺序和方向排歹Ⅱ,就形成了DNA的序列。
一般的DNA分子都有两条互补的分子链,两条链间通过A.T、G.C问的配对形成了双螺旋结构(如图2.1)。
图2.1DNA的双螺旋结构、碱基配对在解旋酶的作用下,DNA的两条链分离开,分别作为一个模板,又在聚合酶的作用下合成一条新链。
这个过程称为DNA的复制(如图2.2)。
国防科学技术大学研究生院学位论文图2-2DNA的复制核糖核酸(RNA)是与DNA相似的一种高分子化合物,它的组成中以尿嘧啶(用字母U表示)替代胸腺嘧啶,并只形成单链结构。
通过一定化学作用下的A.U、G-C问的配对,一段RNA序列可以获取DNA部分离散区域(称为编码区或基因)的序列信息,这个过程称为转录。
RNA序列中连续的三个字母可以确定唯一的氨基酸,而氨基酸的序列进一步确定了蛋白质的组成进而决定蛋白质的功能,这一过程称为翻译。
生命活动始终遵循从DNA序列到RNA序列,再从RNA序列到蛋白质这一规律,考虑到蛋白质是生命活动的基本单位,因此,DNA序列在某种程度上“主宰”了生命活动,这便是分子生物学中著名的中心法则【19l(如图2.3),它指出,生物活动都是通过由DNA序列转录成为RNA序列再由RNA序列翻译成为蛋白质这一过程进行的,即DNA序列蕴含了包括遗传在内的生物体所有活动的秘密.。
生物信息学-序列比对-实验报告

姓名
学号
专业年级
基础学院生物信息学教研室
题目
序列比对
日期
实验者
一、实验目的
掌握BLAST 2的使用和功能
了解点阵法进行双序列比对的优点
二、实验器材
电脑
三、方法与步骤
见下文
四、结果与讨论
1,例题中其它的最佳比对结果
2,用动态规划法找出两序列的所有最佳比对,要求写出详细过程。打分矩阵采用{(4,-3,-4},即匹配得4分,不匹配得-3分,空位得-4分。序列1:AAAG,序列2:ACG。
RBP4(NP_006735),前列腺素D2合成酶(NP_000945)
载脂蛋白D(NP_001638),前列腺素D2合成酶(NP_000945)
•双序列比对RBP4的直系同源蛋白:人(NP_006735),小鼠(NP_035385.1),大鼠(NP_037294.1)。
3,点阵法
序ห้องสมุดไป่ตู้1自身比对
1 AAGGTCAGGAACAAAGAAACAGCTGAATACCAAACAGGATATCTGTGGTAAGCGGTTCCT
61 GCCCCGGCTCAGGGCCAAGAACAGATGAGACAGCTGAGTGATGGGCCAAACAGGATATCT
121 GTGGTAAGCAGTTCCTGCCCCGGCTCGGGGCCAAGAACAGATGGTCCCCAGATGCGGTCC
序列2与序列3比对:两者为反向互补序列,可以发现可能的发夹状结构。
–比对全部采用默认参数。
–结果用截图。
–没有显著相似的注明“No significant similarity found”即可。
•2,双序列比对RBP4的直系同源蛋白:人(NP_006735),小鼠(NP_035385.1),大鼠(NP_037294.1),并对各自核酸进行两两比对。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
BLAST搜索的统计学显著性
假 阳 性 升 高 E 10 5 2 1 0.1 0.05 0.001 0.0001 P 0.99995 0.99326 0.86466 0.63212 0.09516 0.04877 0.0009995 0.0001
BLAST 应用实例1:H1N1
• 登录NCBI BLAST主页面
用检测序列蛋白质搜索由核酸序列 数据库按6条链翻译成的蛋白质序 列数据库 将核酸序列按6条链翻译成蛋白质 序列后搜索由核酸序列数据库按6 条链翻译成的蛋白质序列数据库
Tblastx
核酸
核酸
一个DNA链可以编码6个蛋白序列
5’ CAT CAA 5’ ATC AAC 5’ TCA ACT 5’ CATCAACTACAACTCCAAAGACACCCTTACACATCAACAAACCTACCCAC 3’ 3’ GTAGTTGATGTTGAGGTTTCTGTGGGAATGTGTAGTTGTTTGGATGGGTG 5’ 5’ GGG TAG 5’ TGG GTA 5’ GTG GGT
• P (s≥x) = 1 - exp( -Kste-λx )
• BLAST迒回比对பைடு நூலகம்分大于阈值S的期望值为:
• E = -Kste-λS • 随着S的增加,E值呈指数下降,比对随机发生的可 能性就接近于0 (阈值越高,序列相似就越可信) • 数据库的大小和探测序列的长度影响比对随机发生 的可能性 (序列越长,序列相似就越可信)
BLAST程序检测序列和数据库类型
程序名 Blastp Blastn 检测序列 蛋白质 核酸 数据库类型 蛋白质 核酸 方法
用检测序列蛋白质搜索蛋白质序列 数据库
用检测序列核酸搜索核酸序列数据 库
Blastx
Tblastn
核酸
蛋白质
蛋白质
核酸
将核酸序列按6条链翻译成蛋白质 序列后搜索蛋白质序列数据库
BLAST程序比较
Program Input
1 blastn blastp blastx tblastn DNA DNA
Database 1
protein 6 DNA
protein protein
6
protein 36 DNA
tblastx
DNA
DNA
BLAST搜索的统计学显著性
• 对于两个随机序列s和t,随机观察到比对得 分大于等于x的概率:
– 最常用,各著名的生物信息中心都提供本服务 – 有在线版和本地运行版
• /
– 一组程序
• blastp, blastn, blastx, tblastn, tblastx
BLAST算法
• 查找完全匹配或者高出匹配阈值的短小序 列片段对; • 将它们延伸得到较长的相似性匹配; • 用替换记分矩阵计算得分,直到达到最大 可能得分。
– PAM30 vs. PAM70 – BLOSUM45 vs. BLOSUM62 – PAM70 vs. BLOSUM62
• 2. BLAST搜索中参数Expect threshold的含义是什 么?该参数调整对搜索结果有什么影响? • 要求:
– 一周内完成 – 作业提交页面中的备注栏标明:学号后6位-姓名-实验2
双序列比对实验
Pairwise Sequence Alignment
实验目的
• 掌握BLAST对比搜索工具的工作原理 • 掌握BLAST的基本操作和参数选择 • 分析BLAST应用不同的打分矩阵所得到的 结果的异同
BLAST
• 基本局部比对搜索工具 (basic local alignment search tool, BLAST)
– 脂质运载蛋白: sp|P31025 – Blosum62 – PAM30
使用Blosum62矩阵搜索
使用PAM30矩阵搜索
作业
• 1. 用在线BLAST工具对载脂蛋白D (NP_001638) 和RBP4 (NP_006735)进行双序列比对,比较下面3 组比对结果的差异,截图并分析差异的原因。
– /
• 选择”protein blast”程序 • 探测序列框输入H1N1氨基酸序列,或直接 输入”gi|224983683” • 选择蛋白质数据库PDB • 点击BLAST按钮搜索结果
BLAST 应用实例2:脂质运载蛋白
• 改变打分矩阵对结果的影响