TXIB-FDA approval
FDA仿制药一致性评价指导指南(中英文版)

Guidance for Industry Controlled Correspondence Related to Generic DrugDevelopment行业指南:有关仿制药研发的书面咨询This guidance represents the current thinking of the Food and Drug Administration (FDA, or the Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff responsible for this guidance as listed on the title page.该指南代表了FDA对该主题目前的看法。
它并不会赋予任何人任何权利,也不会约束FDA或公众,如果有替代的方法能够满足法律法规的要求,可以使用替代的方法。
如果想探讨替代的方法,请联系该指南首页中FDA 负责执行该指南的工作人员。
I.INTRODUCTION 简介This guidance provides information regarding the process by which generic drug manufacturers and related industry can submit correspondence to FDA requesting information related to generic drug development. This guidance also describes the Agency’s process for providing communications related to such correspondence. FDA is issuing this guidance as part of its implementation of the Generic Drug User Fee Amendments of 2012 (Public Law 112-144, Title III), commonly referred to as GDUFA.该指南描述了仿制药生产商以及相关行业向FDA提交书面咨询,询问有关仿制药研发信息的过程,同时还描述了FDA针对这些书面咨询提供交流的过程。
FDA批准新药信息
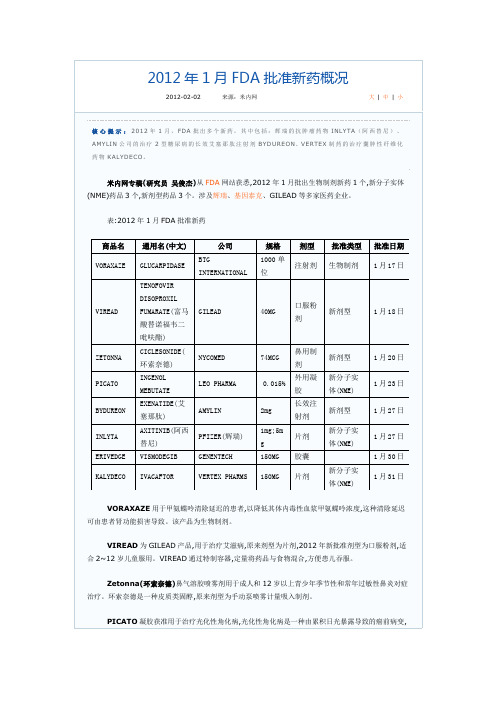
可能进展为鳞状细胞癌。
BYDUREON(艾塞那肽),普通注射剂的英文商品名为BYETTA,2012年FDA批准该药品注射液每周1次缓释剂型的上市,用于治疗2型糖尿病。
研究表明,治疗24周,艾塞那肽缓释剂型可较基线降低糖化血红蛋白1.6%,而每日2次的剂型降低0.9%。
该药最常见的不良反应是恶心、低血糖、呕吐、腹泻、紧张不安感、眩晕、头痛、消化不良、便秘和无力。
INLYTA(阿西替尼)辉瑞公司产品,用于治疗对其它药物没有应答的晚期肾癌(肾细胞癌)。
INLYTA是一种小分子酪氨酸激酶抑制剂,对多个靶点有效,包括VEGF受体1,2和3。
最常见不良反应包括腹泻,高血压,疲劳,食欲下降,恶心。
FDA表示,高血压患者在接受阿西替尼治疗之前应当控制好血压。
ERIVEDGE由基因泰克公司生产,用于治疗基底细胞癌(basal cell carcinoma,BCC),适用于无法开刀或使用化疗的癌症晚期患者,及癌细胞已经扩散到其他身体器官的病人。
患者每日服用一次。
ERIVEDGE药瓶上的标签警告,此药若用于孕妇将可能导致婴儿死亡,它最常见的副作用包括肌肉痉挛,脱发,消瘦,腹泻,疲劳,改变或丧失味觉,食欲下降,便秘,呕吐。
KALYDECO用于治疗囊肿性纤维化(CF)。
每12小时服用1片。
中度和重度肝功能不全患者剂量相应减少。
与中度或强CYP3A的抑制剂的药物合用时,减少剂量。
医学药学缩写符号

A :Aa :Ae :Ae l(m) :Ae s S :Ae∞:A(m) :Ami n:AN,max::AN,min:AN,t:ARE :AS Sμ:AS S,AU:ASS,max::A s s,m i n:A s s,t:AUC :AUCb:AUC(m) :AUCS S :AUMC :C :Ca :CA :Cb :Cbd :CD :C i nf :CL :CL:CLb :CLbD :CLb,H:CLcr :CLD :CLf :CLH :CL i nt :CL(m) :CLpD :CLR :C l u :C(m) :Cmax :C(m)s s :Cmin :Cn :CN,max::CN,mi n:Cpc:C s s :C0:(C 1)max:(C 1)m i n :C∞:(C∞) max:(C∞) m i n:Css,av:C s s,max::C s s,m i n:CT :CTW :Cupper::Clower :Cu :Cu I :Cv :D2 :DM :DM,max:E :EC5 0 :EH :Emax :fp :fs :F :Fbd:fO :fp :FEV 1 :FH :fm :Fm :FR :fu:fu :fub :fup :fur :fuT :γ:G FR :K :ka :KA :kD :Ke:K O:K l O:kf :K 1:Km :K i :Kp :RT :kt :m :MRT :Mu :n :P :Q:Q0:Qc :QD :Qf :QH : P : Rac : Rd : RF : Ro : Rt : S : SA :τ:τ1:Tmax :Tmax :t d:tp :t,p:T i nf :Tm :t 1/2 ,t0.5:t 1/2α:t 1/2β:T 1/2Ka:Tt :V :VB :Vc :Vd :V l :Vm:Vp :VPC :VR :VT :V s S :VTW :Vu :X :X A:Xa :Xc :Xn :Xt :Xu :X0:α:β:GVHD:TRANSNET:IFI: IFD:GM:HEPA:LAF:G-CSF:GM-CSF:M-CSF:Candida albicans:C.tropicalis:C.guilliermondii:C.krusei:C.parapsilosis:C.glabrata:C.kefyr:C.catenulata:C. dubliniensis:C.zeylanoides:C.viswanathii:C.utilis:C.rugosa:C.pulcherrima:C.norvegensis:C.norvegensis:C.lusitaniae:C.lipolytica:C.lipolytica:C.haemulonii:C.ciferrii:C. neoformans:cryptococcosis:Cryptococcus neoformans:Trichosporon:dermatophytosis:Pneumocystis carinii, Pc:体内药量(mg或μmo1)在吸收部位有待于吸收的药量(mg或μmo1)尿中累积原形药物排泄量(mg或μmo1)已消除的代谢物的量(mg或μmo1)在稳态下,一个给药间隔期间尿中累积原形药物排泄量(mg或μmo1)单次给药后,至无穷大时间内尿中累积原形药物排泄量(mg或μmo1)体内代谢物的量(mg或μmo1)达到预期药物效应的所需最小药量(mg或μmo1)固定给药剂量和给药问隔,第N次给药后体内的最大药量(mg或μmo1)固定给药剂量和给药问隔,第N次给药后体内的最小药量(mg或μmo1)在第N次给药后的t时间点的体内药量(mg或μmo1)单次给药质,有待从尿中排泄的药量(mg或μmo1)在恒速静脉滴注时,.稳态时的体内药量(mg或μmo1)稳态时,一个给药间隔期间的平均体内药量(mg或μmo1)固定剂量,固定给药间隔,稳态时一个给药期间的最大和最小体内药量(mg或μmo1)固定剂量,固定给药间隔,稳态时一个给药期间的最小体内药量(mg或μmo1)在固定剂量和给药间隔,稳态时一个给药剂量后t时间点的体内药量(mg或μmo1)血浆药物浓度一时间曲线下面积,它代表一次用药后的吸收总量,反映药物的吸收程度。
美国食品药品管理局(FDA)

美国食品药品治理局〔FDA〕〔一〕美国药政治理机构美国食品药品治理局〔FoodandDrugAdmistraton简称FDA〕,隶属于美国卫生教育福利部,负责全国药品、食品、生物制品、化装品、兽药、医疗器械以及诊断用品等的治理。
FDA下设药品局、食品局、兽药局、放射卫生局、生物制品局、医疗器械及诊断用品局和国家毒理研究中心、区域工作治理机构,即6个局〔有的刊物也称6个中心〕,一个中心和一个区域治理机构。
美国食品药品治理机构共有职工约7500人,FDA总部有1143人,其中药品局为350人。
药品局〔也称药品评价和研究中心〕负责人用药品审批工作,设有8个处和假设干科室。
1.药品治理处。
下设药品信息、信息系统设计、行政治理和预算、医学图书馆4个科室。
2.药品监督办公室。
下设有药品质量评价、药品标签监督、生产和产品质量、科研调查、法规等7个科室。
3.药品标准处。
设有常用药品评价、药品上市和广告2个科。
4.药品审评一处。
下设心血管——肾脏药、抗肿瘤药、营养药、医用造影外科和齿科药、肠胃药和凝血药5个科室。
5.药品审评二处。
下设抗感染药、代谢和内分泌药、抗病毒药3个科室。
6.流行病和生物统计处。
下设流行病及调查、生物统计2个科室。
7.研究处。
下设研究和测试、药物分析2个科室。
8.仿制药品处。
下设仿制药品、生物等效2个科室。
美国食品药品治理局设在华盛顿特区及马利兰州罗克威尔城,机构庞大,分支机构遍布全国各地。
为了加强药品质量治理,FDA将全国划分成6个大区,即太平洋区〔旧金山、西雅图、洛杉肌〕、西南区〔达拉斯、丹佛、堪萨斯〕、中西区〔芝加哥、明尼阿波利斯、底特律〕、东北区〔波士顿、纽约、布法罗〕、中大西洋区〔费城、辛辛那提、纽瓦克、巴尔的摩〕、东南区〔亚特兰大、纳什维尔、新奥尔良、奥兰多、波多利各的圣吉安〕。
每区设立一个大区所,大区所下又设假设干个地区所。
太平洋区的大区所所在地为旧金山,西南区的大区所所在地为达拉斯,中西区的大区所所在地为芝加哥,东北区的大区所所在地为波士顿,中大西洋区的大区所所在地为费城,东南区的大区所所在地为亚特兰大。
FDA警告信

「FDA警告信」这是美国本土API生产商的GMP水平?April 7, 2016WARNING LETTER NO.2016-NOL-02UNITED PARCEL SERVICEDelivery Signature RequestedChris Lemley, President & CEOApotheca Supply, Inc. dba Apothecares3220 Highway 31 SouthBuilding BDecatur, Alabama 35603-1731Dear Mr. Lemley:The U.S. Food and Drug Administration (FDA) inspected your pharmaceutical manufacturing (repackaging and relabeling) facility, Apotheca Supply, Inc. dba Apothecares, at 3220 Highway 31 South, Building B, Decatur, Alabama, from February10-12, 2015.We identified significant deviations from current good manufacturing practice (CGMP) for the manufacture of active pharmaceutical ingredients (API).These deviations cause your drugs to be adulterated within the meaning of Section 501(a) (2)(B) of the Federal Food, Drug, and Cosmetic Act(the Act), 21 United State Code (USC) 351(a)(2)(B), because the methods used in, or the facilities or controls used for, their manufacture, processing, packing, or holding do not conform to, or are not operated or administered in conformity with CGMP.We reviewed your February 23, 2015, response in detail; and, note it lacks sufficient corrective actions. We also acknowledge receipt of your subsequent correspondence.Our investigator observed specific deviations during the inspection, including, but not limited to, the following:调查员在检查时发现的特定的偏差 ,包括但不限于:1.Your quality unit failedto review and approve all quality related documents; and, the mainresponsibilities of your quality unit were not described in writing.你的质量部门未能审核和批准所有质量相关文件;质量部门的主要职责没有书面描述.During our inspection, we found your quality unit did not approve your written standard operating procedures (SOPs) for numerous critical processes, such as quality unit responsibilities, expiration date extension, material quarantine, product distribution, equipment cleaning, product return, complaint handling, product recalls, supplier qualification, raw material testing, and annual product reviews. We also found your quality unit did not review and approve quality-related documents, batch records, certificates of analysis, and the extension of API expiration dates. Specific examples include:在检查中,我们发现你们的质量部门不批准一些关键程序的SOP,例如质量部门职责, 失效日期延长, 物料隔离, 产品物流分布, 设备清洁, 产品返回, 投诉处理, 产品召回, 供应商授权, 原料检验, 和产品质量年度回顾. 我们也发现你们质量部门不审核和批准与质量相关的文件, 批记录, COA和延长后的API失效日期. 特定的例子包括:a. Your quality unit didnot routinely release or reject all API. We found that your fi rm’s office manager releases your drugs from quarantine to distribution, even though this person is neither identified as a member of your quality unit nor has documented CGMP training.你们质量部门不对API进行常规的放行和拒绝 . 我们发现你们公司办公室经理负责放行药品到流通部门, 尽管这个人既不属于质量部门,也没有文件显示接受过CGMP培训.b. Your quality unit did not routinely complete annual product quality reviews you're your repacking operations as required in your SOP Procedural Review. This deficiency is similar to deficiencies we found during our June 2009 and January 2010 inspections.你们质量部门没有按照你们的SOP《产品回顾》对你们再包装操作完成年度产品质量回顾。
药品注册英语术语

Glossary(术语):Regulatory Affairs(RA):药政事务drug authority :药政当局investigation and research before project approval :立项前的调研Market Authorization(MA):上市许可post-approval commitment study :上市后的承诺研究post-approval variation application :补充申请life cycle :生命周期Chemistry,Manufacturing,and Controls(CMC):药品的化学、生产和控制cross-functional teams :公司内部各部门look at the big picture :从大局考虑think strategically :进行战略性思考risks and benefits:风险和获益Food and Drug Administration(FDA):美国食品药品监督管理局European Medicines Agency(EMA):欧洲药品管理局International Multi-center Clinical Tria l(IMCT):国际多中心临床试验Bioequivalence study(BE study):生物等效性试验generic drug:仿制药Center for Drug Evaluation(CDE):SFDA下属的药品审评中心Quality by Design(QbD):质量源于设计CMC Pilot Program :FDA 在业内开展的关于 QbD 的试点研究early launch :早日上市design space :设计空间Business Development(BD):业务发展部门Imported Drug License(IDL):进口药品注册证Manufacturing License(ML):生产许可证Clinical Trial Permission(CTP):临床试验批件Active Pharmaceutical Ingredient(API):原料药Orange Book :橙皮书business value :商业价值the Pharmacopoeia o f the Pe o p le's Re pu b lic o f China(ChP):中国药典the United States Pharmacopoeia(USP):美国药典the European Pharmacopoeia(Ph.Eu r.或EP):欧洲药典List o f Essential Drugs(EDL):基本药物目录Reimbursement Drug List(RDL):医保目录)typing e r or :打印错误slip of the pen :笔误Drug Master File(DMF):药物主文件Certificate o f Analysis(Co A):检验报告Marketing(MKT):市场部market share :市场占有率sales volume :销量investigator b ro chure(IB):研究者手册protocol :临床试验方案priority:优先度package insert(PI):说明书labeling :包装标签Patie nt Information Leaflet(PIL):患者使用的说明书Summary of Product Characteristics(SmPC,SPC):产品特性摘要foil:铝箔carton:装药品的小盒shipping label :运输包装标签Medical :医学部provincial drug administration(PDA):省级药监局,包括省、自治区和直辖市药品监督管理部门Institute for Food and Drug Control:药检所National Institute for the Control of Pharmaceutical and Biological Produc ts(NICPBP):中国药品生物制品检定所,简称“中检所”supplementary dossier :补充资料approval letter :注册批件out o f specification(OOS):超出标准、不合格adverse effect(AE):不良事件trial waiver :减免临床试验Clinical :临床部门Commercial :商业部门new chemical entity(NCE):新化学实体ke y opinion leader(KOL):关键意见领袖off-label use:标签外使用patient pool :患者库deadline :最后期限global trial :全球性的临床试验,即国际多中心临床试验regional trial :区域性的临床试验。
2013-2014 年FDA批准的新药

2013年FDA批准的27个新药汇总作者:scifans来源:scifans2013-12-26 13:21:00分享到:53关键词:FDA新药今年FDA批准的新药数量上少于去年,但质量上却高于去年,其中10个有重磅潜力。
1. Nesina:苯甲酸阿格列汀片阿格列汀(alogliptin)是Takeda研发的新型DPP-4抑制剂,用于治疗II型糖尿病,已获得FDA批准的同类药物还有西他列汀(sitagliptin)、沙格列汀(saxagliptin)、利拉利汀(Linagliptin),另外维格列汀(vildagliptin)已在欧洲上市。
此外,FDA还一同批准了两个含阿格列汀的复方制剂,即Oseni(阿格列汀/吡格列酮)和Kazano (阿格列汀/二甲双胍)。
在14项涉及8500名II型糖尿病患者的临床试验中,相比于安慰剂,Nesina能额外降低糖化血红蛋白(HbA1c)0.4%-0.6%。
在4项涉及2500名II型糖尿病患者的临床试验中,相比于二甲双胍,Kazano 能额外降低HbA1c 0.5%。
在4项涉及1500名II型糖尿病患者的临床试验中,相比于吡格列酮,Kazano能额外降低HbA1c 0.4%-0.6%。
2. Kynamro:米泊美生钠注射液/industry/internation/535623.shtml米泊美生(mipomersen)是Genzyme研发的一种合成的硫代磷酸寡核苷酸,被FDA批准用于治疗纯合子型家族性高胆固醇血症(homozygous familial hypercholesterolemia,FoFH)。
作为反义核酸类药物,米泊美生通过与Apo B-100蛋白mRNA的编码区互补配对,抑制Apo B-100蛋白(LDL和VLDL的主要载脂蛋白)的翻译合成,降低FoFH患者的LDL-C、TC、Non-HDL-C水平。
在为期26周涉及56名FoFH的多国随机对照试验中,治疗组平均LDL-C、TC、Apo B、Non-HDL-C、TG水平分别降低25、21、27、25、18 mg/dL,平均HDL-C水平增加15,而安慰剂组各项指标变化均在5 mg/dL以内。
2020年10—11月FDA批准新药概况

2020年10—11月FDA批准新药概况2020年10月,FDA批出1个新分子实体和1个新生物药物,分别为治疗COVID-19药品Veklury(瑞德西韦)和治疗埃博拉病毒药物Inmazeb(atoltivimab+ maftivimab+odesivimab-ebgn)。
11月,FDA批出3个新分子实体和1个新生物药物,分别为治疗早衰综合征药品Zokinvy(lonafarnib)、治疗高草酸尿症药品Oxlumo (lumasiran)、治疗肥胖症药品Imcivree(setmelanotide)和治疗神经母细胞瘤药物Danyelza(naxitamab-gqgk)。
1 Veklury(瑞德西韦)Veklury有注射液和冻干粉两种剂型,被正式批准用于治疗年龄在12岁及以上、体重至少40 kg的需要住院治疗的COVID-19患者,成为美国首个也是唯一一个获批的新冠肺炎治疗药物。
瑞德西韦是核苷类RNA依赖的RNA聚合酶竞争性抑制剂,通过阻碍SARS-CoV-2的复制而起作用。
2 Inmazeb(atoltivimab+maftivimab+odesivimab-ebgn)Inmazeb为注射液,被批准用于治疗由扎伊尔型埃博拉病毒引起的成人和儿童患者感染,包括感染检测呈阳性母亲的新生儿。
Inmazeb靶向埃博拉病毒表面表达的糖蛋白,这种糖蛋白通过与细胞表面的受体相结合,导致病毒和宿主细胞膜的融合,使病毒进入细胞。
组成Inmazeb的3种中和抗体可同时与这种糖蛋白结合,从而阻断病毒的附着和进入细胞。
3 Zokinvy(lonafarnib)Zokinvy为口服胶囊,被批准用于减少哈金森-吉尔福德早衰综合征(Hutchinson-Gilford progeria syndrome, HGPS)患者的死亡风险,以及治疗患有特定早衰样核纤层蛋白病的1~2岁的患者。
Zokinvy是一种首创性的口服法尼基转移酶抑制剂,可阻断早衰蛋白的法尼基化,故而降低早衰蛋白在细胞核中的积累,使得早老症儿童生存获益。
FDA相关术语

FDA(FOOD AND DRUG ADMINISTRATION):(美国)食品药品管理局4*7NDA(NEW DRUG APPLICATION):新药申请LF=R8@rrmINDA(INVESTIGATIONAL NEW DRUG APPLICA TION):NDA前申报阶段~4}1yANDA(ABBREVIATED NEW DRUG APPLICA TION):简化新药申请>CEPA(EXPORT APPLICA TION):出口药申请(申请出口不被批准在美国销售的药品)Sz12 Q'gTREA TMENT IND:研究中的新药用于治疗(il'w`)NABBREVIATED(NEW)DRUG:简化申请的新药D4GYBDMF(DRUG MASTER FILE):药物主文件(持有者为谨慎起见而准备的保密资料,可以包括一个或多个人用药物在制备、加工、包装和贮存过程中所涉及的设备、生产过程或物品。
只有在DMF持有者或授权代表以授权书的形式授权给FDA,FDA在审查IND、NDA、ANDA时才能参考其内容)="b #HOLDER:DMF持有者@v-B5[VljdCFR(CODE OF FEDERAL REGULATION):(美国)联邦法规w*Rb*$-<pFPANEL:专家小组]h_,aDqBATCH PRODUCTION:批量生产;分批生产?orsD6BATCH PRODUCTION RECORDS:生产批号记录<v+onE}uDPOST-OR PRE- MARKET SURVEILLANCE:销售前或销售后监督1Lhfo/)_rCKINformED CONSENT:知情同意(患者对治疗或受试者对医疗试验了解后表示同意接受治疗或试验)-?:wz \PRESCRIPTION DRUG:处方药9Y~4[OTC DRUG(OVER—THE—COUNTER DRUG):非处方药46Z9*U.S.PUBLIC HEALTH SERVICE:美国卫生福利部Oxl^NIH(NATIONAL INSTITUTE OF HEALTH):(美国)全国卫生研究所eWNe3DCLINICAL TRIAL:临床试验fsH.Xj%^ANIMAL TRIAL:动物试验m7d+-)ACCELERA TED APPROV AL:加速批准r3($Dl9jmOSTANDARD DRUG:标准药物J/INVESTIGA TOR:调研人员7&dc\[9G0/PREPARING AND SUBMITTING:起草和申报TOwR^SUBMISSION:申报;递交+zOs4XIyBENIFIT(S):受益#VY<~b5O1NRISK(S):受害8KBv]xvlDRUG PRODUCT:药物产品SMKDRUG SUBSTANCE:原料药zpi,JQ}ESTABLISHED NAME:确定的名称1|&P*vGENERIC NAME:非专利名称7H&N*=W1oY8PROPRIETARY NAME:专有名称;p}7,INN(INTERNATIONAL NONPROPRIETARY NAME):国际非专有名称$UINARRATIVE SUMMARY记叙体概要JE`ADVERSE EFFECT:副作用Yp8LD>ulI9ADVERSE REACTION:不良反应bD~W_*IU{PROTOCOL:方案6zYZ6ARCHIVAL COPY:存档用副本YruREVIEW COPY:审查用副本h'ND'{pF,eOFFICIAL COMPENDIUM:法定药典(主要指USP、NF).%N2KNUSP(THE UNITED STATES PHARMACOPEIA):美国药典(现已和NF合并一起出版)U' H+ 0NF(NATIONAL formULARY):(美国)国家药品集j#yTM]%OFFICIAL=PHARMACOPEIAL= COMPENDIAL:药典的;法定的;官方的NQ=| :AGENCY:审理部门(指FDA))^*SPONSOR:主办者(指负责并着手临床研究者)izcIDENTITY:真伪;鉴别;特性)FYFFSTRENGTH:规格;规格含量(每一剂量单位所含有效成分的量)-vLABELED AMOUNT:标示量\'#~~REGULA TORY SPECIFICA TION:质量管理规格标准(NDA提供)4Jk{}REGULA TORY METHODOLOGY:质量管理方法(FDA用于考核原料药或药物产品是否符合批准了的质量管理规格标准的整套步骤)(Y^w!REGULA TORY METHODS V ALIDATION:管理用分析方法的验证(FDA对NDA提,5f}b供的方法进行验证)q+y@"w4PDietary supplement:食用补充品72[IODE (ORPHAN DRUG EXCLUSIVITY): 器官用药市场独占权y=k,XmSvL:RwNCE (NEW CHEMICAL ENTITY) : 新化合物Vu。
辉瑞启动非处方版立普妥临床试验计划

辉瑞启动非处方版立普妥临床试验计划辉瑞正计划推进其有关销售柜台出售版抗胆固醇药立普妥的申请,希望能打消监管机构的怀疑情绪,说服其相信消费者能在没有医嘱的情况下正确服用这种药物。
辉瑞已在最近启动了一项针对1200名病人进行的临床试验,以便考察服用非处方药版立普妥的消费者是否能对自己进行血液测试,从而查看这种药物是否正在改善其胆固醇水平,并基于查看的结果来作出是否服用的决定。
就目前而言,市场上销售的立普妥及其相关仿制药均为处方药。
通常情况下,医生会要求病人进行血液测试,从而对其胆固醇水平进行监控,并观察立普妥是否会对病人造成肌无力等副作用。
在这项临床试验中,辉瑞通过美国的35家药房招募愿意接受测试的病人,预计该试验将在年底以前完成,届时辉瑞将依据测试结果来决定是否向美国监管机构提出申请。
如果辉瑞提出申请并成功获批,则立普妥将成为第一种可通过柜台出售的非处方抑制素。
对于辉瑞来说,非处方药版立普妥将帮助该公司重新夺回一部分市场,从而有助于提振其业绩表现。
从立普妥的专利保护权在2011年底到期以来,价格较低的仿制药开始进入市场,从而导致辉瑞丧失了一些市场份额。
去年,辉瑞来自于立普妥的销售额为23亿美元,远低于2006年时创下的将近130亿美元的历史最高水平。
而根据高盛集团作出的预测,柜台出售版立普妥将为辉瑞带来超过10亿美元的年销售额。
辉瑞首席执行官晏瑞德表示,该公司正在投入大量资源来推进柜台出售版立普妥相关计划,并表示启动上述临床试验对这一计划来说是个“重大的里程碑”。
辉瑞并未透露这种药物的售价将是多少,仿制药的售价在每片50美分到5美元之间。
辉瑞称,柜台出售版立普妥将有助于填补一个“治疗缺口”,为那些面临心脏病发作风险但目前并未服用抑制素的患者提供帮助。
辉瑞还表示,这种药物能让更多人接受治疗,因此可以给整个医疗保健体系都带来节省资源的效果,从而有可能减轻医疗成本高昂的问题。
辉瑞旗下消费者医疗保健部门的全球研发高级副总裁马克戈尔贝特表示:“非处方药版是另一个进入点。
TKI类药物

过去十年,许多酪氨酸激酶抑制剂(TKI)药物已经在肿瘤领域应用,根据一篇最近的综述,看看截止到2013年8月份,由FDA 和EMA批准的所有TKI药物都有那些。
1、阿西替尼(axitinib,Inlyta)在2012年1月27日获FDA批准治疗对其它药物没有应答的晚期肾癌(肾细胞癌),由辉瑞(Pfizer)公司开发。
阿西替尼是多靶点酪氨酸激酶抑制剂,具体用法为5mg 空腹口服 2/日。
2、克唑替尼(crizotinib,XALKORI)用于治疗ALK阳性的局部晚期或转移的非小细胞肺癌,推荐剂量和方案是250 mg口服每天2次。
3、达沙替尼(Dasatinib,施达赛Sprycel)治疗对包括甲磺酸伊马替尼在内的治疗方案耐药或不能耐受的慢性髓细胞样白血病。
FDA也经正常程序批准达沙替尼治疗对其他疗法耐药或不能耐受的费城染色体阳性的急性淋巴细胞性白血病成人患者。
4、厄洛替尼(Erlotinib,特罗凯Tarceva)既往接受过至少一个化疗方案失败后的局部晚期或转移的非小细胞肺癌。
厄洛替尼单药用于非小细胞肺癌的推荐剂量为150mg/日,至少在进食前1小时或进食后2小时服用。
5、吉非替尼(Gefitinib,易瑞沙Iressa)适用于治疗既往接受过化学治疗或不适于化疗的局部晚期或转移性非小细胞肺癌。
推荐剂量为250mg(1片)每日1次,空腹或与食物同服。
6、伊马替尼(Imatinib,格列卫)用于治疗慢性粒细胞性白血病(CML),胃肠道间质瘤(胃肠道间质瘤)和其他一些疾病。
到2011年,该药已被FDA批准用于治疗10个不同的癌症。
7、拉帕替尼(Lapatinib;泰立沙Tykerb)联合卡培他滨治疗ErbB-2过度表达的,既往接受过包括蒽环类,紫杉醇,曲妥珠单抗(赫赛汀)治疗的晚期或转移性乳腺癌。
推荐剂量为1250mg,每日1次,第1~21天服用,与卡培他宾2000mg/d,第1~14天分2次服联用。
8、尼洛替尼(Nilotinib,达希纳Tasigna)适应症为对既往治疗(包括伊马替尼)耐药或不耐受的费城染色体阳性的慢性髓性白血病(Ph+ CML)慢性期或加速期成人患者。
2017年12月美国FDA公布的药品安全信息Ⅱ

ABSTRACT There exist great individual diferences in the eoncentration 0I vaneomy(‘in,SO It IS necessary
t()I11()nitor its c0n( el1trati0n in clini(·.An enzyme nmhiplied immunoassay teehnique (EMIT)and a two—di— n1ensional liquid chromatography(:oupled with ultraviolet deteetion method (2D-HP[ C—UV)were established
健 康 损 害 的 相 关 病 例 .FI)A 认 为 对 于 批 ;隹的 GBCAs收 益
仍 然 大 于 任 何 潜 在 风 险 然 而在 经 过 更 多的审 查 和 咨询 医 学成 像 咨询 委 员会
后 .FI)A 要 求 采 取 更 多 的 措 施 ,就 MRl使 用 GBcAs残 留 问题 向卫 生 保健 专业 人 员和 患者 发 出警示 ,这 些措 施 有助
就 有 肾 功 能 衰 竭 的 患 者 中 发 生 FI)A 也 收 到 肾 功 能 正 常
的患 者发 生 多 器官 系统 不 良事件 的报 告 ,这 些 不 良事伟 与
钆 残 留 的 因 果 关 系 未 建 立
背 景 : 这 是 对 2Il17年 5 月 22 日 MedW arch safety
vaneomyein. KEY W O RDS Vaneomyein;EMIT:2D——HPLC——UV;Coneentration
· 资 讯 概 览
2024药事管理法规教材

2024药事管理法规教材## English.### Chapter 1: Introduction to Pharmacy Law and Ethics.1. What is the definition of pharmacy law?Pharmacy law is the body of laws and regulations that govern the practice of pharmacy. It includes laws that govern the dispensing of drugs, the operation of pharmacies, and the conduct of pharmacists.2. What is the purpose of pharmacy law?The purpose of pharmacy law is to protect the public health and safety by ensuring that drugs are dispensedsafely and effectively.3. What are the sources of pharmacy law?Pharmacy law is derived from a variety of sources, including federal and state statutes, regulations, and court decisions.4. What are the ethical principles that guide pharmacists?Pharmacists are guided by a number of ethical principles, including the following:Beneficence: The pharmacist must act in the best interests of the patient.Non-maleficence: The pharmacist must do no harm to the patient.Autonomy: The pharmacist must respect the patient's right to make decisions about their own health care.Justice: The pharmacist must treat all patients fairly and equitably.### Chapter 2: The Pharmacy Practice Act.1. What is the Pharmacy Practice Act?The Pharmacy Practice Act is a state law that governs the practice of pharmacy. It typically includes provisions that define the scope of practice of pharmacists, set forth the requirements for licensure, and establish the grounds for disciplinary action.2. What are the key provisions of the Pharmacy Practice Act?The key provisions of the Pharmacy Practice Act typically include the following:Definition of pharmacy: The Act defines pharmacy as the practice of preparing, compounding, dispensing, and distributing drugs.Scope of practice: The Act sets forth the scope of practice of pharmacists, which typically includes thefollowing activities:Dispensing drugs.Providing drug information.Compounding drugs.Administering drugs.Consulting with patients on drug therapy.Licensure requirements: The Act sets forth the requirements for licensure as a pharmacist, which typically include the following:Graduation from an accredited pharmacy school.Passing a national licensure examination.Meeting continuing education requirements.Grounds for disciplinary action: The Act sets forththe grounds for disciplinary action against pharmacists, which typically include the following:Violating the Pharmacy Practice Act.Engaging in unprofessional conduct.Committing a felony.### Chapter 3: The Federal Food, Drug, and Cosmetic Act.1. What is the Federal Food, Drug, and Cosmetic Act(FD&C Act)?The FD&C Act is a federal law that governs the manufacture, distribution, and labeling of food, drugs, and cosmetics. It is administered by the Food and Drug Administration (FDA).2. What are the key provisions of the FD&C Act?The key provisions of the FD&C Act include the following:Definition of a drug: The Act defines a drug as any substance intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease.New drug approval process: The Act requires that all new drugs be approved by the FDA before they can be marketed. The FDA reviews the safety and effectiveness of new drugs before approving them.Labeling requirements: The Act requires that all drugs be labeled with certain information, such as the drug's name, strength, dosage, and directions for use.Prohibited acts: The Act prohibits the following acts, among others:Adulterating or misbranding drugs.Distributing unapproved drugs.Making false or misleading claims about drugs.### Chapter 4: The Controlled Substances Act.1. What is the Controlled Substances Act (CSA)?The CSA is a federal law that regulates the manufacture, distribution, and use of controlled substances. It is administered by the Drug Enforcement Administration (DEA).2. What are the key provisions of the CSA?The key provisions of the CSA include the following:Definition of a controlled substance: The Act definesa controlled substance as any drug or other substance thatis listed in one of the five schedules of the CSA.Scheduling of controlled substances: The CSA divides controlled substances into five schedules based on their potential for abuse and dependence. Schedule I drugs havethe highest potential for abuse and dependence, while Schedule V drugs have the lowest potential for abuse and dependence.Registration requirements: The Act requires that all manufacturers, distributors, and dispensers of controlled substances be registered with the DEA.Prescribing requirements: The Act requires that all prescriptions for controlled substances be written by a licensed physician.Prohibited acts: The Act prohibits the following acts, among others:Manufacturing, distributing, or dispensing controlled substances without a license.Prescribing controlled substances for non-legitimate medical purposes.Possessing controlled substances without aprescription.### Chapter 5: The Health Insurance Portability and Accountability Act.1. What is the Health Insurance Portability and Accountability Act (HIPAA)?HIPAA is a federal law that protects the privacy of health information. It is administered by the Department of Health and Human Services (HHS).2. What are the key provisions of HIPAA?The key provisions of HIPAA include the following:Privacy Rule: The Privacy Rule protects the privacy of health information by requiring covered entities to take steps to protect the confidentiality of health information.Security Rule: The Security Rule protects the security of health information by requiring covered entities toimplement security measures to protect health information from unauthorized access, use, or disclosure.Enforcement: HIPAA is enforced by HHS through a variety of mechanisms, including audits, investigations, and civil and criminal penalties.## 中文回答:### 第一章药事法律法规与职业道德概论。
利妥昔单抗质控标准

利妥昔单抗质控标准
利妥昔单抗(Rituximab)是一种常用于治疗B细胞相关疾病的单克隆抗体药物。
质控标准是用于确保药物质量和一致性的参考样品。
对于利妥昔单抗,通常有以下类型的质控标准:
1. 原料药(Active Pharmaceutical Ingredient, API)标准:这是用于制造利妥昔单抗的原材料的标准。
它包括对活性成分(利妥昔单抗)的纯度、杂质含量、理化性质等方面的要求。
2. 终端制剂(Finished Product)标准:这是利妥昔单抗最终制剂的质控标准。
它包括对药物的含量、纯度、杂质含量、微生物质量等方面的要求。
这些标准通常由药品制造商、药典组织或其他相关机构制定,并提供给药品制造商、药品监管机构和实验室进行质量控制和检测。
需要注意的是,具体的质控标准可能会因不同的国家或地区而有所不同。
因此,建议参考当地的药典(如美国药典USP、欧洲药典Ph. Eur.、中国药典等)或联系利妥昔单抗的制造商或供应商,以获取最准确和最新的质控标准信息。
1/ 1。
【高中生物】晚期肾细胞癌:新联合疗法有望获FDA批准上市

【高中生物】晚期肾细胞癌:新联合疗法有望获FDA批准上市一种治疗晚期肾细胞癌(RCC)的新联合疗法在早期临床试验中显示出了良好疗效,可能很快就会获得美国食品药品监督管理局(FDA)的上市批准。
1月17日,FDA同意优先审批lenvatinib(Lenvima)的补充新药申请,用于联合依维莫司(Afinitor)治疗无法手术切除、既往接受过一种血管内皮生长因子(VEGF)靶向疗法的晚期或转移性RCC患者。
此前,lenvatinib已获FDA突破性疗法认定,用于在临床试验中治疗晚期或转移性RCC患者。
Lenvatinib是由卫材公司发现和研制的一种多受体酪氨酸激酶抑制剂,可抑制血管内皮生长因子受体VEGFR1-3的激酶活性,同时还可抑制与致病性血管生成、肿瘤生长和癌症进展有关的其他受体酪氨酸激酶。
Lenvatinib已获FDA上市批准,商品名为Lenvima,用于治疗对放射性碘治疗耐药的局部复发性或转移性、进展性、分化型甲状腺癌患者。
“随着FDA接受lenvatinib的补充新药申请,我们向我们的目标??为无法手术切除的晚期或转移性RCC患者提供首个酪氨酸激酶和mTOR抑制剂联合疗法?又迈近了一步,”卫材药品研发系统的肿瘤药品研发部主席KenichiNomoto在卫材公司向媒体发布的新闻稿中说。
“我们期待在未来几个月里协助FDA审批lenvatinib联合依维莫司作为晚期RCC 患者的潜在治疗新选择。
”FDA同意优先审批lenvatinib的补充新药申请的依据是一项II期三臂临床试验最新取得的无进展生存期数据。
该试验对比了lenvatinib+依维莫司联合治疗、依维莫司单药治疗、lenvatinib单药治疗对RCC患者的疗效。
该试验共入组153名RCC患者,其中99%的患者都既往接受过一种VEGF靶向疗法。
研究人员报道称,相比依维莫司单药治疗,lenvatinib+依维莫司联合治疗延长了患者的无进展生存期(风险比0.40),并提高了患者的客观缓解率。
国内首个美国FDA制剂文号落户华海药业

国内首个美国FDA制剂文号落户华海药业
佚名
【期刊名称】《医药工程设计》
【年(卷),期】2007(28)4
【摘要】华海药业日前公布公告称,公司已经收到美国食品药品监督管理局(下称“FDA”)的通知,公司向FDA申报的奈韦拉平片的新药简略申请(ANDA)已获得批准,批准文号为ANDA078644。
这是国内获得的首个美国FDA制剂文号,标志着该产品具备了在美国市场销售的资格,预计公司原料药的供应出口量将会有大的提升。
【总页数】1页(P11-11)
【关键词】美国FDA;药业;制剂;国内;食品药品监督管理局;批准文号;市场销售;出口量
【正文语种】中文
【中图分类】TQ461
【相关文献】
1.华海药业:度洛西汀美国获批国内有望加速上市 [J],
2.华海药业固体制剂车间通过关国FDA认证 [J], 无
3.华海药业成国内首个通过FDA认证仿制药企 [J],
4.美国FDA批准首个SGLT2抑制剂降糖药上市 [J],
5.美国FDA批准首个长效C5补体抑制剂Ultomiris治疗PNH [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
FDA、TGA、PMDA、EDQM一些基础知识

FDA、TGA、PMDA、EDQM一些基础知识CFDA系将食品安全办的职责、食品药品监管局的职责、质检总局的生产环节食品安全监督管理职责、工商总局的流通环节食品安全监督管理职责整合组建而成,负责药品、医疗器械、化妆品和消费环节食品安全的监督管理。
CFDA于2013年3月22日正式挂牌成立,食品药品监督管理局的官网也同步进行了更名,一律改成国家食品药品监督管理总局,英文简称由“SFDA”变成“CFDA”,就连原先的官方微博“中国药监”也改成了“中国食药监”美国食品和药物管理局(Foodand Drug Administration)简称FDA,FDA 是美国政府在健康与人类服务部(DHHS) 和公共卫生部 (PHS) 中设立的执行机构之一。
作为一家科学管理机构,FDA 的职责是确保美国本国生产或进口的食品、化妆品、药物、生物制剂、医疗设备和放射产品的安全。
它是最早以保护消费者为主要职能的联邦机构之一。
TGA[1] 是TherapeuticGoods Administration的简写,全称是治疗商品管理局,它是澳大利亚的治疗商品(包括药物、医疗器械、基因科技和血液制品)的监督机构。
依据1989年的治疗商品法案,TGA是递属于澳大利亚政府健康和老龄部下的一个部门。
TGA开展一系列的评审和监督管理工作,以确保在澳大利亚提供的治疗商品符合适用的标准,并保证澳大利亚社会的治疗水平在一个较短的时间内达到较高的水平。
欧洲药品质量理事会(European Directorate for Quality Medicines)简称EDQM,是欧洲理事会下属的药品管理系统的核心,旨在保证在欧洲生产和销售的药品具有同等优良的品质。
EDQM的职能是建立药品的质量标准以供欧洲药典委员会使用,制备标准品CRS,执行COS程序最终颁发COS证书等等。
欧洲药物管理局(European Medicines Agency)简称EMA,是欧洲官方药管机构之一,EMA主要负责欧盟市场药品的审查、批准上市,评估药品科学研究,监督药品在欧盟的安全性、有效性。
加拿大卫生部允许Plan B作为非处方药物出售

加拿大卫生部允许Plan B作为非处方药物出售
黄敏燕(摘)
【期刊名称】《国外药讯》
【年(卷),期】2005(000)009
【摘要】加拿大卫生部已经修改了该国的食品与药物管理规定,允许Barr制药公司的紧急避孕药Plan B(Levonorgestrel,左旋甲炔诺酮)(Ⅰ)作为非处方药物出售。
【总页数】1页(P12)
【作者】黄敏燕(摘)
【作者单位】无
【正文语种】中文
【中图分类】R95
【相关文献】
1.科学性是加拿大食品药物法的核心--访加拿大卫生部长Ujjal Dosanjh先生 [J], 刘怡
2.药物安全性监察-加拿大卫生部警告:mycophenolate mofetil导致纯红细胞再障 [J], 无
3.加拿大卫生部批准了20年来首个新的房颇治疗药物Multaq [J], 无
4.首个干细胞治疗药物Prochymal获加拿大卫生部批准上市 [J],
5.加拿大卫生部批准Raltegravir为成人多药耐药HIV治疗药物 [J], 姚瑜
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
May 2002COPYRIGHT 1999, 2002 BY EASTMAN CHEMICAL COMPANYRegulatory Information SheetEASTMAN TXIB PlasticizerEASTMAN TXIB Plasticizer (2,2,4-trimethyl-1,3-pentanediol diisobutyrate, CAS Reg. No. 6846-50-0) is lawful for use in certain food-contact applications which are described in the U.S. Food and Drug Administration regulations listed in the following table. The reader should refer to each appropriate regulation before using this product. The use of our product is subject to the limitations stated in the respective regulations.FCN No. 224 For use as a plasticizer in vinyl chloride polymers for use in repeated-use food-contact applications and in vinyl chloride polymer-based gloves used to process food.Limitations: For use at a level not to exceed 10 percent by weight of the finishedplasticized vinyl chloride polymer formulation and for use at temperatures not toexceed 40 degrees C. Effective Date of FCN: June 7, 2002. Refer to FCN inventorylist at FDA website: /~dms/opa-fcn.html.21 CFR 175.105 Adhesives: This regulation specifies that the adhesive shall be separated from the food by a functional barrier, or used subject to the following additional limitations: (a) Theamount of adhesive that contacts packaged dry foods shall not exceed the limits of goodmanufacturing practice; (b) The quantity of adhesive that contacts packaged fatty andaqueous foods shall not exceed the trace amount at seams and at edge exposurebetween packaging laminates that may occur within the limits of good manufacturingpractice, and under normal conditions of use the packaging seams or laminates willremain firmly bonded without visible separation.21 CFR 177.1200Cellophane 21 CFR 178.3740 Plasticizers in Polymeric Substances: TXIB may be used only in cellulosic packagingfor aqueous and dry foods and in amounts not to exceed 15 wt % of the food contactarticle.It is the responsibility of our customers to determine that their use of our product is safe, lawful, and technically suitable in the intended applications. We suggest that you be guided by the advice of your own regulatory or legal counsel in determining whether your specific application complies with applicable FDA regulations. Because of possible changes in the law and in regulations, as well as possible changes in our products, we cannot guarantee that the status of this product will remain unchanged. We recommend that customers continuing to use this product verify its status periodically.For additional information about this product, please contact your Eastman sales representative or visit our website at .Neither Eastman Chemical Company nor its marketing affiliates shall be responsible for the use of this information, or of any product, method or apparatus mentioned, and you must make your own determination of its suitability and completeness of your own use, for the protection of the environment, and for the health and safety of your employees and purchasers of your products. No warranty is made of the merchantability or fitness of any product; and nothing herein waives any of the Seller's conditions of sale.Neither Eastman Chemical Company nor its marketing affiliates shall be responsible for the use of this information, or of any product, method or apparatus mentioned, and you must make your own determination of its suitability and completeness of your own use, for the protection of the environment, and for the health and safety of your employees and purchasers of your products. No warranty ismade of the merchantability or fitness of any product; and nothing herein waives any of the Seller's conditions of sale.• NORTHAMERICA Eastman Chemical CompanyCorporate HeadquartersP.O. Box 431 Kingsport, TN 37662-5280 U.S.A.Telephone: U.S.A. and Canada, 1-800-EASTMAN (1-800-327-8626)Telephone: Other Locations, (1) 423-229-2000Fax: 423-229-1673• LATIN AMERICA• EUROPE/MIDDLE EAST/AFRICA Eastman Chemical Latin America, Inc.Eastman Chemical, Europe, Middle East, 2333 Ponce de Leon Blvd.and Africa, Ltd. Suite R-20Tobias Asserlaan 5 Coral Gables, FL 33134 U.S.A2517 KC The Hague Telephone: (1) 305-461-8240NETHERLANDS Fax: (1) 305-461-8254Telephone: (31) 70 370 1711Fax: (31) 70 370 1704• ASIAPACIFIC Eastman Chemical Japan Ltd. Eastman Chemical Asia Pacific Pte. Ltd. AIG Aoyama Building 5F #05-04 Winsland House2-11-16 Minami Aoyama 3 Killiney RoadMinato-ku, Tokyo 107-0062 SINGAPORE 239519JAPAN Telephone: (65) 738-4877Telephone: (81) 3-3475 9510 Fax: (65) 732-4930Fax: (81) 3-3475 9515Eastman Chemical Ltd. Shanghai Rep Office1206, CITIC Square,No. 1168 Nanjing Road West,Shanghai, 200041,P.R. CHINATelephone : (86) 21-6120 8700Fax : (86) 21-5292 9366© Eastman Chemical Company, 2002.。