非无菌产品微生物限度标准修订
对非无菌剂型的注册药剂制品微生物质量的要求背景

对非无菌剂型的注册药剂制品微生物质量的要求背景根据《药剂业及毒药条例》(第138 章)及其附属规例(第138A 章),药剂制品应符合安全、效能及素质方面的标准,并获药剂业及毒药管理局注册,方可于本港销售。
2. 在非无菌制剂中,某些微生物的存在可使产品减低甚至失去治療作用,亦对病人的健康构成潜在风险。
因此,非无菌剂型的药剂制品的微生物限量应受监控。
3. 非无菌剂型的药剂制品的微生物检查已被编入不同的药典,如美国药典、英国药典、欧洲药典及日本药典等。
在2009年5月,上述药典中的微生物质量要求均已订立相同的标准。
注册要求4. 为确保注册药剂制品在微生物质量方面的安全及素质,药剂业及毒药(药剂制品及物质注册:临床试验及药物测试证明书)委员会(委员会)决定从2019年10月1日起,所有非无菌剂型的注册药剂制品(包括正在申请注册的新药和已注册药剂制品),在整个保质期内必须符合上述第3段的主要药典订明的微生物限度标准(节录于表一)。
表一: 非无菌剂型的微生物限度标准给药途径需氧菌总数(cfu/g 或cfu/ml)霉菌和酵母菌总数(cfu/g 或cfu/ml)控制菌口服给药固体制剂103102大肠埃希菌阴性(1 g or 1 mL) 口服给药液体制剂102101大肠埃希菌阴性(1 g or 1 mL) 直肠给药制剂103102-口腔黏膜给药制剂齿龈给药制剂皮肤给药制剂鼻用制剂耳用制剂102101金黄色葡萄球菌阴性(1 g or 1 mL)铜绿假单胞菌阴性(1 g or 1 mL)阴道给药制剂102101铜绿假单胞菌阴性(1 g or 1 mL)金黄色葡萄球菌阴性(1 g or 1 mL)白色念珠菌阴性(1 g or 1 mL)透皮贴剂(对一个含有黏着层与背衬的贴剂限量)102101金黄色葡萄球菌阴性(1 patch)铜绿假单胞菌阴性(1 patch)呼吸道吸入给药制剂(适用于雾化用液体制剂的特别要求)102101金黄色葡萄球菌阴性(1 g or 1 mL)铜绿假单胞菌阴性(1 g or 1 mL)耐胆盐革兰阴性菌阴性(1 g or 1 mL)备注:非无菌产品的微生物检验方法应参考美国药典、英国药典、欧洲药典或日本药典。
(整理)年药典通则1106非无菌产品微生物限度检查.

通则1106 非无菌产品微生物限度检查:控制菌检查法控制菌检查法系用于在规定的试验条件下,检查供试品中是否存在特定的微生物。
当本法用于检查非无菌制剂及其原、辅料等是否符合相应的微生物限度标准时,应按下列规定进行检验,包括样品取样量和结果判断等。
供试品检出控制菌或其他致病菌时,按一次检出结果为准,不再复试。
供试液制备及实验环境要求同“非无菌产品微生物限度检查:微生物计数法”。
如果供试品具有抗菌活性,应尽可能去除或中和。
供试品检查时, 若使用了中和剂或灭活剂,应确认有效性及对微生物无毒性。
供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。
培养基适用性检查和控制菌检查方法适用性试验供试品控制菌检查中所使用的培养基应进行适用性检查。
供试品的控制菌检查方法应进行方法适用性试验,以确认所采用的方法适合于该产品的控制菌检查。
若检验程序或产品发生变化可能影响检验结果时,控制菌检查方法应重新进行适用性试验。
菌种及菌液制备菌种试验用菌株的传代次数不得超过5 代(从菌种保藏中心获得的干燥菌种为第0 代),并采用适宜的菌种保藏技术进行保存,以保证试验菌株的生物学特性。
金黄色葡萄球菌(Staphylococcus aureus)〔CMCC(B)26 003〕铜绿假单胞菌(Pseudomonas aeruginosa)〔CMCC(B)10 104〕大肠埃希菌(Escherichia coli)〔CMCC(B)44 102〕乙型副伤寒沙门菌(Salmonella paratyphi B)〔CMCC(B)50 094〕白色念珠菌(Candida albicans)〔CMCC(F)98 001〕生孢梭菌(Clostridium sporogenes)〔CMCC(B)64 941〕菌液制备将金黄色葡萄球菌、铜绿假单胞菌、大肠埃希菌、沙门菌分别接种于胰酪大豆胨液体培养基中或在胰酪大豆胨琼脂培养基上,30~35℃培养18~24 小时;将白色念珠菌接种于沙氏葡萄糖琼脂培养基上或沙氏葡萄糖液体培养基中,20~25℃培养2~3 天;将生孢梭菌接种于梭菌增菌培养基中置厌氧条件下30~35℃培养24~48 小时或接种于硫乙醇酸盐流体培养基中30~35℃培养18~24 小时。
欧洲药典-2.6.12非无菌产品的微生物限度检查

2.6.12非无菌产品的微生物限度检查 (有活力的需氧菌总数的计数)本附录给出了两种测试方法第一种给出与专论相符合的相关测试方法除非说明采用第2种测试。
参照本章的内容采用第1种测试方法。
考虑到申请上市许可的需要。
第2种测试方法也是欧洲药典正式方法的组成部分。
一旦相关部分修改,用第2种测试方法代替第1种。
第二种方法为了与日本药局方和美国药典的要求一致A .欧洲药典的方法下述检验方法是对有氧条件下生长的嗜温细菌和真菌数的计数检查。
这些试验设计主要用来检查样品是否符合药典对微生物限度的要求,用于这一目的时要遵守以下指导原则,包括样品的抽样数选取及结果的解释。
这些试验也可以用于药典中抗菌防腐剂效力的检验(5.1.3)。
它们可进一步用来监测原料质量,药物制剂的微生物质量监测(5.1.4)。
当用于这些目的时,例如生产者用于原料和/或成品的检测或者用于认证过程评价时,进行测试的方法包括抽取样品数及结果的解释是生产者和主管当局间协议中的事项。
检定应该在样品不会被污染的条件下进行。
用来防止污染的预防措施必须不影响在试验中微生物的检出。
如果被检测的产品含有抗菌活性,它必须被充分中和,并证明所用的灭活剂是有效的和对微生物是无毒性的本章描述了薄膜滤过法,培养板记数法测定有活力的需氧菌总数当没有其它方法进行细菌计数时可用最大可能数法。
方法的选择基于产品的性质及预期的微生物数量这些因素。
任何被选用的方法必须被适当地验证。
当5.1.3章或5.1.4章联合应用时,可以使用平皿法(pour—plate),表面涂布平皿计数法(surface—spread)及薄膜滤过法。
供试品的准备抽样计划:抽样必须遵循明确的抽样计划。
抽样计划由一批样品的数量,与不被接受的高污染产品相关的健康危险,产品的特性,预期污染水平等因素而定。
除另有规定外,取10g或10ml样品或制剂,按上述方法预处理。
从原料中或从制剂包装中随机地选取样品。
根据样品(原料或制剂)的特性,必要时可从多个包装中取样,混合后,得到所需检品量。
USP2022年版〈61〉非无菌产品微生物限度检查:微生物计数法-中文版

USP43 <61>非无菌产品微生物限度检查:微生物计数法介绍下文所述的试验将允许在有氧条件下生长的嗜温细菌和真菌的定量计数。
这些测试主要用于确定一种物质或制剂是否符合既定的微生物质量标准。
当用于此类目的时,应按以下的规定进行,包括要样品取样量,和解释判断如下所述结果。
本方法不适用于含有活微生物作为活性成分的产品。
可采用包括自动化方法在内的替代微生物学方法,前提是已经证明与药典方法等效。
一般程序在避免待检产品的受到外来微生物污染的条件下进行测定。
防止污染的措施必须确保不会影响待检产品中微生物的检出。
如果待检产品具有抗菌活性,则应尽可能将其去除或中和。
如果灭活剂用于此目的,必须证明其微生物有效性和无毒性。
如果表面活性物质用于样品制备,应确认其对微生物无毒性,并且与所用的任何灭活剂相容性。
计数方法按照说明,使用薄膜过滤法或平板计数法之一。
最可能数(MPN)法通常用于微生物计数准确度较差;但是,对于某些生物负荷非常低的产品,这可能是最合适的方法。
方法的选择是基于产品的性质和微生物限度标准等因素。
所选方法必须允许测试足够的样本量,以判断是否符合质量标准。
所选方法的适用性必须经确认。
生长促进试验、计数方法和阴性对照的适用性一般要求必须建立检测待测产品中微生物的能力。
如果引入了可能影响测试结果的测试性能或产品变化,则必须确认其适用性。
试验菌株的制备使用试验菌株的标准化稳定悬浮液或按如下所述进行制备。
采用适宜的菌种保藏技术,以确保用于接种的微生物的传代次数不超过5代。
如表1所述,分别培养每种细菌和真菌测试菌株。
霉孢子悬浮,可将0.05%的聚山梨醇酯80加入该缓冲液中。
悬浮液在2h之内使用,或存放在2~8℃条件下24h内使用。
作为制备并稀释黑曲霉或枯草杆菌营养细胞的新鲜悬浮液的替代方法,可制备稳定的孢子悬浮液,然后将适量的孢子悬浮液用于试验接种。
稳定的孢子悬浮液可在2~8℃下保存,保存期需经过时间验证。
非无菌产品微生物限度标准修订

路漫漫其修远兮, 吾将上下而求索
2020年4月9日星期四
2015年版药典药品微生物度标准修订
我国药品微生物限度标准的历史沿革 2015版微生物限度标准增修订内容 非无菌药品微生物限度标准 微生物限度标准的表述 需氧菌总数、真菌数数值的表述 微生物限度标准的解释
路漫漫其修远兮, 吾将上下而求索
标准 2、品种正文项下:
【 无菌 】 受热不稳定、制剂不能进行最终灭菌的无菌工艺产品 用于手术、烧伤或严重创伤的局部给药制剂 应符合无菌检查法规定。 3、非无菌化学药品及生物制品制剂的微生物限度标准(见表1)
按10个给药途径,以固体、液体性状分别给出具体的限度标准要求。
路漫漫其修远兮, 吾将上下而求索
路漫漫其修远兮, 吾将上下而求索
《 2015版药典 》抑菌效力检查法修订内容
路漫漫其修远兮, 吾将上下而求索
抑菌剂 是指抑制微生物生长的化学物质,有时也称防腐剂。 ⒈抑菌效力检查法系用于测定无菌及非无菌制剂的抑菌活性,以评价最终产 品的抑菌效力, ⒉用于指导生产企业在研发阶段制剂中抑菌剂浓度的确定。 以防止制剂在正常贮藏或使用过程中可能发生的微生物污染和繁殖使药物变 质而对使用者造成危害,尤其是多剂量包装的制剂。
路漫漫其修远兮, 吾将上下而求索
微生物限度标准解释
⑷制剂通则项下微生物限度检查项目 必检项目 制剂通则项下有微生物限度要求的制剂为必检项目 原则性要求 化学药品丸剂、口服 片剂、胶囊剂、颗粒剂;应对其被微生 物污染的风险进行评估,在保证患者用药安全安全的前提下,通过历史回顾 分析可不进行批批检验。 ⑸如果上述制剂的性质及工艺的原因导致产品易受微生物污染,应在品 种项下列出微生物限度检查项及微生物限度标准,如 生化类制剂。
1105非无菌产品微生物限

1105非无菌产品微生物限度检查:微生物计数法中菌药典2015年版表3供试品的最少检验量供试品供试品装量每支供试品接人每种培养基的最少量液体制剂^I m llm l<C V^40m l40m K V< 100m lV>100m l全量半量,但不得少于l m l20m l10%但不少于20m l固体制剂M〈50m g50m g^M<C300m g300m g^M<C5g全量半量150m g500m g半量(生物制品)生物制品的原液及半成品半量医疗器具外科用敷料棉花及纱布缝合线、一次性医用材料带导管的一次性医疗器具(如输液袋)其他医疗器具取lOOmg 或lc m X 3cm整个材料①二分之一内表面积整个器具®(切碎或拆散开)①如果医用器械体积过大,培养基用量可在2000m l以上,将其完全浸没。
1105非无菌产品微生物限度检查:微生物计数法微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
当本法用于检查非无菌制剂及其原、辅料等是否符合相应的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。
除另有规定外,本法不适用于活菌制剂的检査。
微生物计数试验环境应符合微生物限度检查的要求。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
单向流空气区域、工作台面及环境应定期进行监测。
如供试品有抗菌活性,应尽可能去除或中和。
供试品检査时,若使用了中和剂或灭活剂,应确认其有效性及对微生物无毒性。
供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。
计数方法计数方法包括平皿法、薄膜过滤法和最可能数法(M ost-P rob a b l e-Numb e r M e tho d,简称MP N法)。
MP N法用于微生物计数时精确度较差,但对于某些微生物污染量很小的供试品,M P N法可能是更适合的方法。
1106 非无菌产品微生物限度检查:控制菌检查法

2、大肠埃希菌 (Escherichia coli)
大肠埃希菌是两端钝圆的短小直杆菌 革兰阴性 无芽孢,多有鞭毛 培养最适温度为37℃ 可在15-46℃生长 兼性厌氧
大肠埃希菌检查流程
结果判断 若麦康凯琼脂平板上有菌落生长,应鉴定 若麦康凯琼脂平板上没有菌落生长,或虽有菌落生长 但鉴定结果为阴性,判供试品未检出大肠埃希菌
供试液制备及实验环境要求同微生物计数法
若使用了中和剂或灭活剂,应确认有效性及无毒性
如果使用了表面活性剂,应确认其无毒性以及相容性
可采用替代的微生物检查法,包括自动检测方法,但 必须证明替代方法等效于药典规定的检查方法。 ——新增内容
3、结果判断方式
在各控制菌项下:较少的生化实验或“进一步进行 适宜的鉴定试验” 没长:未检出
5、金黄色葡萄球菌 (Staphylococcus aureus)
属于葡萄球菌属 菌体呈球形或略呈椭圆 形,菌体较小 需氧或兼性厌氧 最适生长温度为37℃, 最适生长pH为7.4
金黄色葡萄球菌检查流程
结果判断 若甘露醇氯化钠琼脂平板上有黄色菌落或外周有黄色 环的白色菌落生长,应鉴定 若没有疑似的菌落生长,或虽有相符或疑似的菌落生 长但鉴定结果为阴性,判供试品未检出金黄色葡萄球 菌
琼脂 方法:涂布 至少2皿 ①是否生长 ②比较菌落特征
液体 方法:直接接种 培养基是否浑浊、 比较浑浊程度
③要求: 菌株:根据表格要求接种 接种量:根据测试指标而定 ①+&指示:不大于100cfu ②—:不少于100cfu,也不能过多 ③涂布:不少于0.1mL接种,也不能过多 培养时间 ① +&指示:不长于规定的最短培养时间 ②—:不短于规定的最长培养时间
*铜绿假单胞菌(Ps.aeruginosa) 铜绿假单胞菌(Ps.aeruginosa) *金黄色葡萄球菌(S.aureus) 梭菌(Clostridia) 白色念珠菌(C.albicans) 金黄色葡萄球菌(S.aureus) 梭菌(Clostridia) 白色念珠菌(C.albicans)
【2020版中国药典】通则-非无菌微生物限度检查

【2020版中国药典】通则-非无菌微生物限度检查1105非无菌产品微生物限度检查:微生物计数法
微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。
除另有规定外,本法不适用于活菌制剂的检查。
学习:将旧版的“相应”更换为“规定”,更便于按照1107进行判定执行。
微生物计数试验环境应符合微生物限度检查的要求。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
洁净空气区域、工作台面及环境应定期进行监测。
学习:将旧版的“单向流空气区域”更换为“洁净空气区域”。
计数方法
……供试品检查时,应根据供试品理化特性和微生物限度标准等因素选择计数方法,检测的样品量应能保证所获得的试验结果能够判断供试品是否符合规定。
所选方法的适用性须经确认。
……
提示:后文增加了对于“贵重药品、微量包装药品”的检验量的更全面的表述,因注意结合此处的要求。
计数培养基适用性检查和供试品计数方法适用性试验
……菌液制备……取黑曲霉的新鲜培养物加人适量含0.05%(ml/ml)聚山梨酯80的pH7.0无菌氯化钠-蛋白胨缓冲液或……
学习:此处改动同无菌检查法,将“3~5ml”的具体量调整为“适量”,便于根据孢子的量灵活掌握菌液制备方法。
培养基适用性检查
微生物计数用的商品化的预制培养基、由脱水培养基或按处方配制的培养基均应进行培养基适用性检查。
学习:类似于无菌检查法,此处将“成品培养基”修改为“商品。
非无菌药品微生物限度检查指导原则

非无菌药品微生物限度检查指导原则微生物限度检查是药品质量控制中的重要环节,尤其对于非无菌药品而言更为重要。
本文将对非无菌药品微生物限度检查的指导原则进行详细介绍,旨在提高药品的质量安全性。
一、检验项目及标准非无菌药品微生物限度检查的主要检验项目包括总生菌数、大肠菌群、霉菌和酵母菌等。
每一项检验项目都有相应的标准来衡量合格与否。
以下是常用的标准:1. 总生菌数:根据药典要求,大部分非无菌药品每克不得超过1000 CFU(菌落形成单位)。
2. 大肠菌群:大肠菌群是肠道中的常见菌种,其存在可能暗示有肠源性污染。
检验结果一般要求不得检出大肠菌群。
3. 霉菌和酵母菌:霉菌和酵母菌是环境中广泛存在的微生物,在非无菌药品中的存在可能引发变质,甚至导致严重的药品质量问题。
一般情况下,每克药品中不得检出霉菌和酵母菌。
二、样品的选择和采集在进行微生物限度检查前,需选择合适的样品,并采取正确的样品采集方式。
以下是一些常用的样品选择和采集方法:1. 样品选择:根据药品的特性,选择代表性的样品进行检测。
选取多个批次的不同规格的样品进行检验更有利于全面评估该药品的微生物污染水平。
2. 样品采集:在采集样品前,先进行适当的表面消毒,以避免外源性污染。
采集时应遵循严格的无菌操作,确保样品的真实性和可靠性。
常用的样品采集方法包括划线法、切割法、稀释法等。
三、检验方法和操作流程微生物限度检查需要使用一系列严格的操作流程和检验方法,以保证结果的准确性和可比性。
以下是一般的操作流程:1. 样品预处理:根据药品的特性,选择适当的预处理方法,如溶解、稀释、震荡等,以提高微生物的检出率。
2. 培养基选择:根据不同的菌种需求,选择适宜的培养基进行菌落的培养。
常用的培养基有营养琼脂平板、大肠埃希菌选择平板、马铃薯葡萄糖琼脂平板等。
3. 培养条件:根据菌种的生长特性和检验项目的要求,设定适当的温度、时间和培养条件,以促进菌落的生长。
4. 菌落计数:通过目视或自动计数法,对培养基上的菌落进行计数。
非无菌药品微生物限度标准

或
cfu/10cm2)
霉菌和酵母菌总数 (cfu/g、cfu/ml 或 cfu/10cm2)
控制菌
口服给药★
固体制剂
103
液体制剂
102
不得检出大肠埃希菌(1g 或 1ml);含脏器提取物的制剂 102 还不得检出沙门菌(10g 或 101 10ml)
口腔黏膜给药制剂
齿龈给药制剂
102
鼻用制剂
不得检出大肠埃希菌、金黄
或 cfu/10cm2)
或 cfu/10cm2)
固体口服给药制剂
不得检出大肠埃希菌(1g);
不含豆豉、神曲 104(丸剂 3×104)
102
不得检出沙门菌(10g);耐
等发酵原粉
胆盐革兰阴性菌应小于
含豆豉、神曲等
105
5×102不得检出大肠埃希菌(1ml);
102
铜 绿 假 单 胞 菌 ( 1g 或
10cm2);阴道、尿道给药制
102
剂还不得白色念珠菌、梭菌
(1g 或 10cm2)
不得检出金黄色葡萄球菌、
102
铜绿假单胞菌(1ml);阴道、
尿道给药制剂还不得白色
102
念珠菌、梭菌(1ml)
5. 非无菌的药用原料及辅料微生物限度标准(见表 3)
表 3 非无菌药用原料及辅料微生物限度标准
10cm2)
化学药品制剂和生物制品制剂若含有未经提取的动植物来源的成份及矿物质还
不得检出沙门菌(10g 或 10ml)。
4. 非无菌含药材原粉的中药制剂微生物限度标准(见表 2)
表 2 非无菌含药材原粉的中药制剂微生物限度标准
给药途径
需氧菌总数 霉菌和酵母菌总数
控制菌
2015版中国药典微生物限度

1.4.2供试品检查
• 供试液制备
– ⑵ 水不溶性非油脂类供试品
• 取供试品, 用 pH7.0 无菌氯化钠-蛋白胨 缓冲液,或 pH7.2 磷酸盐缓冲液,或胰酪 大豆胨液体培养基制备成 1:10 供试液。 分散力较差的供试品,可在稀释液中加入 表面活性剂如 0.1%的聚山梨酯 80,使供 试品分散均匀。若需要,调节供试液 pH 值至 6~8。必要时,用同一稀释液将供 试液进一步 10倍系列稀释。
1.3.3计数方法适用性试验
1. 供试液制备 2. 接种和稀释 3. 抗菌活性的去除与灭活 4. 供试品中微生物的回收
– 平皿法 – 薄膜过滤法 – 最可能数法(MPN 法)
5. 结果判断
1.4 供试品检查
• 1.4.1检验量
– 检验量即一次试验所用的供试品量(g、ml
或cm²)。
– 除另有规定外,一般供试品的检验量为10g 或
• 需氧菌总数是指胰酪大豆胨琼脂培养基上生长的 总菌落数(包括真菌菌落数);
• 霉菌和酵母菌总数是指沙氏葡萄糖琼脂培养基上 生长的总菌落数(包括细菌菌落数)。
• 若因沙氏葡萄糖琼脂培养基上生长的细菌使霉菌 和酵母菌的计数结果不符合微生物限度要求,可 使用含抗生素(如氯霉素、庆大霉素)的沙氏葡 萄糖琼脂培养基或其他选择性培养基(如玫瑰红 钠琼脂培养基)进行霉菌和酵母菌总数测定。
1.4.2供试品检查
• 供试液制备
– ⑷需用特殊方法制备供试液的供试品
• 膜剂供试品 • 肠溶及结肠溶制剂供试品 • 气雾剂、喷雾剂供试品 • 贴膏剂供试品
1.4.2供试品检查
1. 平皿法
– 平皿法包括倾注法和涂布法。 – 除另有规定外,取规定量供试品,按方法适用性
试验确认的方法进行供试液制备和菌数测定,每 稀释级每种培养基至少制备2个平皿。 – 培养和计数 除另有规定外,胰酪大豆胨琼脂培养 基平板在30~35℃培养3~5天,沙氏葡萄糖琼脂 培养基平板在20~25℃培养5 ~7天, 观察菌落 生长情况,点计平板上生长的所有菌落数,必要时 可适当延长培养时间至7 天进行菌落计数并报告 。菌落蔓延生长成片的平皿不宜计数。点计菌落 数后,计算各稀释级供试液的平均菌落数,按菌 数报告规则报告菌数。 – 若同稀释级两个平皿的菌落数平均值不小于15, 则两个平皿的菌落数不能相差1 倍或以上。
2020年版《中国药典》通则 —“非无菌药品微生物限度标准”

2020年版《中国药典》通则“非无菌药品微生物限度标准”(蓝色字体表示新增内容,红色字体表示删减内容)非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及、中药提取物及中药饮片的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、严重烧伤、严重创伤的局部给药制剂应符合无菌检查法规定。
3.非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表 1。
表 1 非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准给药途径需氧菌总数(cf u/g、cf u/m l或cf u/10c m2)霉菌和酵母菌总数(cf u/g、c fu/m l或cfu/10cm2)控制菌口服给药①固体制剂液体及半固体制剂103102102101不得检出大肠埃希菌(1g 或1ml);含脏器提取物的制剂还不得检出沙门菌(10g 或10ml)口腔黏膜给药制剂齿龈给药制剂鼻用制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或 10cm2)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或10cm2)呼吸道吸入给药制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、或 10ml ) 阴道、尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g 、1ml 或 10cm 2);中药制剂还不得检出梭菌(1g 、 1ml 或 10cm 2)直肠给药 固体制剂103102 不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 或 1ml )其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 、1ml 或 10cm 2)注 ①化学药品制剂和生物制品制剂若含有未经提取的动植物来源的成份及矿物质还不得检出沙门菌(10g 或 10ml )。
生物制品原液微生物限度标准

生物制品原液微生物限度标准
生物制品原液的微生物限度标准主要依据《中国药典》和《药品生产质量管理规范》进行制定。
以下是具体的标准:
1. 对于非无菌的生物制品,其微生物限度标准通常包括细菌、霉菌和酵母菌总数以及控制菌的检查。
2. 细菌、霉菌和酵母菌总数的标准:对于每1ml生物制品原液,细菌数不
得超过100cfu,霉菌和酵母菌数不得超过100cfu。
同时,对于每1g生物
制品原液,细菌数不得超过1000cfu,霉菌和酵母菌数不得超过1000cfu。
3. 控制菌检查的标准:对于生物制品原液,应按照《中国药典》和《药品生产质量管理规范》的规定进行控制菌检查,如大肠杆菌、金黄色葡萄球菌等。
控制菌检查应为阴性。
4. 对于有特殊用途或规定的生物制品,如用于免疫治疗的制品、基因治疗制品等,其微生物限度标准应根据相关规定进行制定。
此外,在生物制品的生产过程中,应遵循无菌操作规程,采用经过验证的无菌工艺和设备,并进行严格的质量控制,确保产品的安全性和有效性。
非无菌产品微生物限度标准
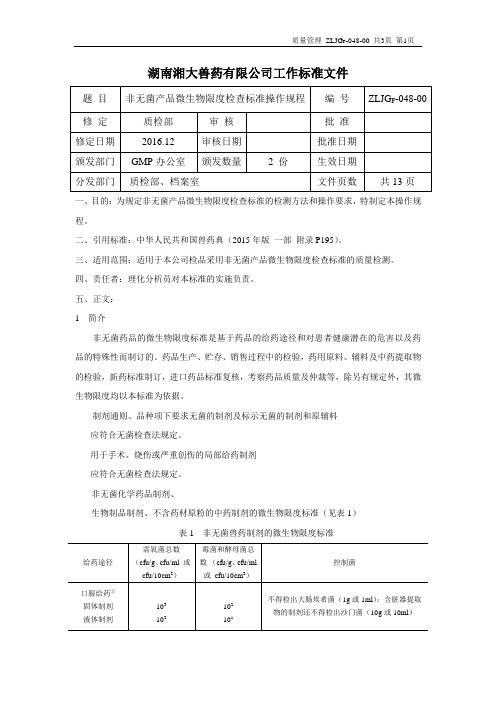
湖南湘大兽药有限公司工作标准文件题目非无菌产品微生物限度检查标准操作规程编号ZLJG F-048-00 修定质检部审核批准修定日期2016.12 审核日期批准日期颁发部门GMP办公室颁发数量 2 份生效日期分发部门质检部、档案室文件页数共13页一、目的:为规定非无菌产品微生物限度检查标准的检测方法和操作要求,特制定本操作规程。
二、引用标准:中华人民共和国兽药典(2015年版一部附录P195)。
三、适用范围:适用于本公司检品采用非无菌产品微生物限度检查标准的质量检测。
四、责任者:理化分析员对本标准的实施负责。
五、正文:1 简介非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及中药提取物的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
制剂通则、品种项下要求无菌的制剂及标示无菌的制剂和原辅料应符合无菌检查法规定。
用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。
非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准(见表1)表1 非无菌兽药制剂的微生物限度标准给药途径需氧菌总数(cfu/g、cfu/ml 或cfu/10cm2)霉菌和酵母菌总数(cfu/g、cfu/ml或cfu/10cm2)控制菌口服给药①固体制剂液体制剂103102102101不得检出大肠埃希菌(1g或1ml);含脏器提取物的制剂还不得检出沙门菌(10g或10ml)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g、1ml或10cm2)直肠给药固体制剂液体制剂103102102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g或1ml)其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)注:①兽药制剂若含有未经提取的动植物来源的成分及矿物质,还不得检出沙门菌(10g或10ml)。
2015版中国药典微生物限度

1.4.2供试品检查
• 供试液制备
– ⑶油脂类供试品
• 取供试品,加入无菌十四烷酸异丙酯使溶 解,或与最少量并能使供试品乳化的无菌 聚山梨酯 80或其他无抑菌性的无菌表面 活性剂充分混匀。表面活性剂的温度一般 不超过 40℃(特殊情况下,最多不超过 45℃),小心混合,若需要可在水浴中进 行,然后加入预热的稀释液使成 1∶10供 试液,保温,混合,并在最短时间内形成 乳状液。必要时,用稀释液或含上述表面 活性剂的稀释液进一步 10倍系列稀释。
– 培养和计数 培养条件和计数方法同平皿计数 法,每张滤膜上的菌落数应不超过100cfu。
• 菌数报告规则
– 以相当于 1g、1ml 或10cm2 供试品的菌落数 报告菌数;若滤膜上无菌落生长,以﹤1 报 告菌数(每张滤膜过滤1g、1ml 或10cm2 供 试品),或﹤1 乘以最低稀释倍数的值报告菌 数。
1.4.2供试品检查
• 供试液制备
– ⑷需用特殊方法制备供试液的供试品
• 膜剂供试品 • 肠溶及结肠溶制剂供试品 • 气雾剂、喷雾剂供试品 • 贴膏剂供试品
1.4.2供试品检查
1. 平皿法
– 平皿法包括倾注法和涂布法。 – 除另有规定外,取规定量供试品,按方法适用性
试验确认的方法进行供试液制备和菌数测定,每 稀释级每种培养基至少制备2个平皿。 – 培养和计数 除另有规定外,胰酪大豆胨琼脂培养 基平板在30~35℃培养3~5天,沙氏葡萄糖琼脂 培养基平板在20~25℃培养5 ~7天, 观察菌落 生长情况,点计平板上生长的所有菌落数,必要时 可适当延长培养时间至7 天进行菌落计数并报告 。菌落蔓延生长成片的平皿不宜计数。点计菌落 数后,计算各稀释级供试液的平均菌落数,按菌 数报告规则报告菌数。 – 若同稀释级两个平皿的菌落数平均值不小于15, 则两个平皿的菌落数不能相差1 倍或以上。
欧洲药典 7.0 _非无菌产品的微生物限度检查

2.6.12非无菌产品的微生物限度检查:微生物计数试验1.简介:该法所描述的是在需氧条件下生长的嗜温性细菌和真菌类微生物的定量检测。
设计该试验的最初目的是为了确定一种物质或制剂是否符合微生物质量的既定标准。
如果是这种目的,则需要遵从下面的说明,包括所需样品数,从而根据下面所提到的方法来进行结果解释。
该法不适用于包含微生物作为活性成分的产品。
如果与药典的等效性作了相关说明,可以使用自动收集法作为替代法。
2.一般步骤设计方法时所用条件要避免外在微生物对待测产品的污染。
必须釆取措施,使其不影响任何待测微生物。
如果待测产品有抗菌活性,必须将这种活性去除或中和掉。
如果因此使用了灭活剂,必须证明它们对微生物的高效性和无毒性。
如果在样品制备中加了表面活性剂,必须证明它们对微生物的无毒性和与灭活剂的兼容性。
3.计数方法按规定用微孔滤膜法或平板计数法。
MPN法对微生物测定来说是最不精确的方法,但如果某些产品所含微生物的量极少,则该法是最合适的。
方法选择基于下列因素:如产品的特性和所要求的微生物限量。
所选方法必须能够测定充足的样品从而来判断其是否符合标准。
所选方法的适用性必须建立。
4.促生长实验、计数法的适用性和阴性对照4-1 总则该试验检测产品中微生物的检测能力必须被验证。
如果测试条件改变或产品改变,必须证实方法的适适用性,因为这些改变可能影响试验的最终结果。
4-2 试验菌株的准备使用测试菌株标准稳定的悬浮液或按下面方法制备。
使用批菌种培养技术(seed-lot systems)使得用于接种的微生物从最初的主批种子传代不多于5次。
细菌和真菌试验株的生长分别见表2.6.12-2。
使用氯化钠蛋白胨(pH 7.0)缓冲液或pH 7.2的磷酸缓冲液制作试验用悬浮液。
对于黑曲霉菌需要加入0.05%聚山梨醇酯80到缓冲液中。
在两小时内使用该菌悬液或2-8℃下可保存24小时。
作为一种可替代的制备方法,稀释含有黑曲霉菌或枯草芽孢杆菌的新鲜悬浮液,该悬浮液中加有营养细胞,从而可以制得一种稳定的孢子悬浮液,对于接种试验来说需要将该孢子悬浮液调整到一个合适的体积下。
非无菌产品微生物限度检查标准

附录ⅪJ 非无菌药品微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。
3.非无菌化学药品及生物制品制剂的微生物限度标准(见表1)表1 非无菌化学药品及生物制品制剂的微生物限度标准给药途径需氧菌总数(CFU/g、CFU/ml或CFU/10cm2)霉菌及和酵母菌总数(CFU/g、 CFU/ml或CFU/10cm2)控制菌口服固体给药制剂103102不得检出大肠埃希菌(1g);含脏器提取物的制剂还不得检出沙门菌(10g)口服液体给药制剂102101不得检出大肠埃希菌(1ml);含脏器提取物的制剂还不得检出沙门菌(10ml)口腔黏膜给药制剂齿龈给药制剂鼻用制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)呼吸道吸入给药制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、耐胆盐革兰阴性菌(1g或1ml)阴道、尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g、1ml或10cm2)直肠给药固体制剂液体制剂103102102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g或1ml)其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)4. 非无菌的药用原料及辅料微生物限度标准(见表2)表2 非无菌药用原料及辅料微生物限度标准需氧菌总数(CFU/g 或 CFU/ml)霉菌和酵母菌总数(CFU/g或CFU/mL)控制菌药用原料及辅料103102* 注*:限度未做统一规定。
3、(孙老师)1107 非无菌药品微生物限度标准解析

2、标准执行的注意事项
哪些需要做微生物限度检查 –正文项下【微生物限度】有要求 –制剂通则项下该品种对【微生物限度】有要 求 –“微生物限度检查法”标准规定
新药的微生物限度标准 基于数据分析和风险评估 原辅料、给药途径及对患者的潜在危害综合考虑控制 菌标准 -产品特性:是否适合微生物生长?是否有抑菌力 -用药对象:婴儿、体弱者、病患
原辅料、中药提取物、中药饮片 -参考制剂要求、生产工艺特点设置控制菌要求
4、标准限度放宽了? –标准限度放宽,但是培养基的营养能力增加
1107 非无菌药品微生物限度标准 解析
四川省食品药品检验检测院 孙雪奇 2016年6月
内
容
• 一、标准概况 • 二、修订依据 • 三、微生物限度标准 • 四、国外药典标准收载情况
一、标准概况
方 法 标 准
1995年Chp附录收载 微生物限度检查法
1986版《药品卫生标准》及 1989版《药品卫生补充规定》 2000版Chp收载限度标准 (按剂型划分限度标准,按 给药途径及制剂特点制定控 制菌标准。)
TSA 营养 琼脂
11600
90
4500
110
<10
5600
<10
110
10
<10
例:金葡计数比较 以同一金黄色葡萄球菌菌悬液分别在营养琼脂和 TSA上计数。
培养基 样品(cfu/ml)
TSA 营养 琼脂
127
121
103
101
四、国外药典标准收载情况
2015年版中国药典微生物限度检查法

1.1 总则:
• 环境: – 微生物计数试验环境应符合微生物限度检查的要 求。(在不低于GMP 现行版要求的D 级洁净环境 、局部洁净度不低于B 级的单向流空气区域内进 行)【10版:在环境洁净度10000级下的局部洁净 度100级的单向流空气区域内】。 – 检验全过程必须严格遵守无菌操作,防止再污染 ,防止污染的措施不得影响供试品中微生物的检 出。 – 单向流空气区域、工作台面及环境应定期进行监 测。
菌数报告规则
– 需氧菌总数测定宜选取平均菌落数小于 300cfu 的稀释级、霉菌和酵母菌总数测定宜 选取平均菌落数小于100cfu 的稀释级,作为 菌数报告(取两位有效数字)的依据。取最 高的平均菌落数,计算1g、1ml 或10 cm² 供 试品中所含的微生物数。 – 如各稀释级的平皿均无菌落生长,或仅最低 稀释级的平板有菌落生长,但平均菌落数小 于1 时,以﹤1 乘以最低稀释倍数的值报告菌 数。
1.4.2供试品检查
• 供试液制备 – ⑴ 水溶性供试品
• 取供试品,用 pH7.0 无菌氯化钠-蛋白胨 缓冲液,或pH7.2 磷酸盐缓冲液,或胰酪 大豆胨液体培养基溶解或稀释制成 1:10 供试液。若需要,调节供试液 pH 值至 6 ~8。必要时,用同一稀释液将供试液进 一步 10倍系列稀释。水溶性液体制剂也 可用混合的供试品原液作为供试液。
1.3.1菌液制备及使用
试验菌株 试验菌液的制备
金黄色葡萄球菌 〔CMCC(B) 26 003)〕
铜绿假单胞菌 〔CMCC(B)10 104〕 枯草芽孢杆菌 〔CMCC(B) 63 501〕 白色念珠菌 〔CMCC(F) 98 001〕
胰酪大豆胨琼脂培养基或胰酪大豆胨液体培 养基 【10版:营养肉汤或营养琼脂培养基】 30~35℃,18~24小时
非无菌产品微生物限度标准修订课件

目录 CONTENTS
• 非无菌产品微生物限度标准概述 • 非无菌产品微生物限度标准修订背景 • 非无菌产品微生物限度标准修订内容 • 非无菌产品微生物限度标准修订的影响 • 非无菌产品微生物限度标准修订的实施与监
管 • 非无菌产品微生物限度标准修订的意义与展
望
01
修订微生物限度标准可以促使企业加强生产过程中的卫生 控制,提高产品质量和稳定性,减少产品投诉和退货的情 况,提升企业的市场竞争力。
促进产业升级和转型
修订非无菌产品微生物限度标准可以推动企业加强技术创新和研发,提高产品的 技术含量和附加值,促进产业的升级和转型。
微生物限度标准的提高可以促使企业加强生产设备的更新和改造,提高生产效率 和产品质量,同时也可以带动相关产业的发展,推动产业链的完善和升级。
• 对违反标准的行为进行处罚和纠正,维护标准的严肃性和 权威性。
监管措施和责任分工
01
责任分工
02
03
04
药品监管部门负责制定和发布 标准,并监督标准的实施和执
行。
企业应按照标准要求生产和质 量控制,确保产品符合标准要
求。
行业协会和社会组织应积极参 与标准的宣传和推广,促进标
准的广泛应用和实施。
后续评估和改进建议
微生物限度标准的制定依据
科学依据
根据科学研究和实验数据,评估 不同微生物对人体的危害程度和 传播途径,从而制定相应的限度
标准。
法规要求
各国政府和国际组织会制定相关法 规和标准,要求企业按照规定执行 ,以确保产品的卫生质量和安全性 。
实践经验
在生产实践中,企业会根据产品的 特性和生产环境等因素,制定符合 实际情况的微生物限度标准,并不 断优化和改进。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
102
不得检出金黄色葡萄球菌、铜
102
绿假单胞菌(1g或1ml)
102
金黄色葡萄球菌、铜绿假单胞 菌不得检出(1g、1ml)或102cm 2)
表2 非无菌含药材原粉的中药制剂的微生物限度标准
给药途径
不含豆豉、神曲等 发酵类原粉的口服 制剂 固体制剂 液体制剂 含豆豉、神曲等发 酵类原粉的口服制 剂 固体制剂 液体制剂 用于表皮或粘膜完 整的局部给药制剂 固体制剂 液体制剂
分类及其标准
1、制剂通则、品种项下要求无菌的制剂及标示无菌的制剂和原辅料应符合无菌检查
法规定 2、用于手术、烧伤或严重创伤的局部给药制剂 应符合无菌检查法规定 3、非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标
准(见表1) 4、非无菌含药材原粉的中药制剂微生物限度标准(表2) 5、非无菌的药用原料及辅料微生物限度标准(表3) 6、中药提取物及中药饮片的微生物限度标准(表4) 7、有兼用途径的制剂 应符合各给药途径的标准 8、霉变、长螨者 以不合格论
给药途径
阴道尿道给药制 剂
直肠 给药 制剂
固体 液体
其他局部给药制
剂
需氧菌总数 CFU/g、
CFU/ml) 或 CFU/102cm)
102
103 102
102
霉菌酵母菌总
数
(CFU/gCFU/m
控制菌
l) 或CFU/102cm)
不得检出金黄色葡萄球菌、铜
101
绿假单胞菌、白色念珠菌 (1g、
1ml或102cm)
不得检出金黄色葡萄球菌、铜 绿假单胞菌 (1g、1ml或102cm) 阴道、尿道给药制剂还不得检 出 白 色 念 珠 菌 、 梭 菌 (1g 、 1ml 或102cm) 不得检出金黄色葡萄球菌、铜 绿假单胞菌 (1g、1ml或102cm) 阴道、尿道给药制剂还不得检 出 白 色 念 珠 菌 、 梭 菌 (1g 、 1ml 或102cm)
2015版药典通则非无菌产品微生物限度标准 增修订内容
2015年版药典药品微生物度标准修订
我国药品微生物限度标准的历史沿革 2015版微生物限度标准增修订内容 非无菌药品微生物限度标准 微生物限度标准的表述 需氧菌总数、真菌数数值的表述 微生物限度标准的解释
我国药品微生物限度标准
1973年 我国开展药品微生物限度检查 1978年 卫生部颁发《药品卫生试行标准》 1986年 卫生部颁布《药品卫生标准》 1995年版药典 首次收载微生物限度检查法 2000年版药典 收载微生物限度标准按(细菌数、酵母菌数和霉菌数按剂型制订); 控制菌(致病菌)按给药途径制订。 2005年版药典 收载方法验证要求(按给药途径制订限度标准)收载方法验证要求 2010年版药典 收载培养基适用性检查要求,第二增补本基本与ICH接轨
增修订内容
修订概况 1、以一部为限度标准的基础,参照ICH协调案和USP、JP、 EP 标准 增修订了2015版药典标准。 说明 1、一、二、三部附录中内容不同。 2、微生物限度标准以表格的形式描述。
增订内容 1、除中药、化学药和生物制品的限度标准外,增订了原敷料、中药提取物及饮片 的微生物限度标准。
标准
1、制剂通则、品种项下要求无菌的制剂及标示无菌的制剂 应符合无菌检查法规 定。例如: 附录Ⅰ制剂通则 : 【无菌】:注射剂、眼用制剂 植入剂、冲洗剂。 【无菌】或【微生物限度】: 散剂、凝胶剂 、软膏剂、搽剂 、洗剂 、涂膜剂 、鼻用制剂、气雾剂、喷雾剂 。
标准
2、品种正文项下:
【 无菌 】 受热不稳定、制剂不能进行最终灭菌的无菌工艺产品 用于手术、烧伤或严重创伤的局部给药制剂 应符合无菌检查法规定。 3、非无菌化学药品及生物制品制剂的微生物限度标准(见表1)
按10个给药途径,以固体、液体性状分别给出具体的限度标准要求。
表1 非无菌化学药品及生物制品制剂的微生物限度标准
给药途径
口服 给药 制剂
固体 液体
口腔黏膜给药制剂 齿龈给药制剂 鼻用制剂 耳用制剂 皮肤给药制剂
呼吸道吸入给药制剂
阴道尿道给药制剂
需氧菌总数 CFU/g、 CFU/ml)
或CFU/102cm) 103
102
102 102 102
102
霉菌酵母菌总数 (CFU/gCFU/ml) 或CFU/102cm)
102
101
101 101 101
101
控制菌
不得检出大肠埃希氏菌 (1g) 含脏器提取物的制剂不得检出沙门 菌(10g) 不得检出大肠埃希氏菌 (1ml) 含脏器提取物的制剂不得检出沙门 菌(10ml) 不得检出大肠埃希氏菌、金黄色葡 萄球菌、铜绿假单胞菌 (1g、1ml或 102cm)
不得检出大肠埃 希氏菌 (1g或 1ml) 不 得 检 出 沙 门 菌 ( 10g 或 10ml ) 耐 胆 盐 革 兰 阴 性 菌 应 小 于102(1g)或101(1ml)
不得检出大肠埃 希氏菌 (1g或 1ml) 不 得 检 出 沙 门 菌 ( 10g 或 10ml ) 耐 胆 盐 革 兰 阴 性 菌 应 小 于102(1g)或101(1ml)
修订内容
1、菌数计数结果以10n表示; 2、 以需氧菌总数取代细菌数。以耐胆盐革 兰阴性菌取代大肠菌群。
2015年版药典药品微生物度标准修订
基于药品的给药途径对患者健康 潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,化学药品原料药、中药提取物及 辅料的检验,新药标准制订,进口药品标准复核,考察药品质及仲裁等, 除另有规定外,其微生物限度均以Байду номын сангаас标准为依据。
不得检出金黄色葡萄球菌、铜绿假 单胞菌 (1g、1ml或102cm ) 不得检出大肠埃希菌、 金黄色葡萄球菌、铜绿假单胞菌、 耐胆盐革兰阴性菌(1g或1ml) 不得检出金黄色葡萄球菌、铜绿假 单胞菌、白色念 珠菌 (1g 、1ml 或 102cm)
续 表1 非无菌化学药品及生物制品制剂的微生物限度标准
需氧菌总数 CFU/g、 CFU/ml)
或CFU/2)
104,丸剂3×104 5×102
105 103
104 102
用于表皮或粘膜不
完整的局部给药制
剂
固体制剂
103
液体制剂
102
霉菌酵母菌总数 (CFU/gCFU/ml)
或CFU/2)
102 102
5×102 102
102 102
102 102
控制菌