基因靶向技术(gene targeting)的国际研究
gene-targeting

Development and re finement of a high-ef ficiency gene-targeting system for Aspergillus flavusPerng-Kuang Chang ⁎,Leslie L.Scharfenstein,Qijian Wei,Deepak BhatnagarSouthern Regional Research Center,Agricultural Research Service,U.S.Department of Agriculture,1100Robert E.Lee Boulevard,New Orleans,LA 70124,United Statesa b s t r a c ta r t i c l e i n f o Article history:Received 7January 2010Received in revised form 19February 2010Accepted 4March 2010Available online 16March 2010Keywords:Aspergillus flavus A flatoxinFunctional genomics pyrGGene targeting Conidial pigmentAn ef ficient gene-targeting system based on impairment of the nonhomologous end-joining pathway and the orotidine monophosphate decarboxylase gene (pyrG )in Aspergillus flavus was established.It was achieved by replacing the ku70gene with the Aspergillus oryzae pyrithiamine resistance (ptr )gene and by inserting the Aspergillus parasiticus cypA gene into the pyrG locus.The utility of this system was demonstrated by disruption of nine candidate genes for conidial pigment biosynthesis.The gene-targeting frequencies ranged from 80to 100%.Two linked genes on chromosome 4,wA and olgA ,were con firmed to be involved in pigment formation.In contrast to the parental strain which produced yellowish-green conidia,the knockout mutants produced white and olive-green conidia,respectively.The system was further re fined by restoring the pyrithiamine sensitivity and uracil auxotrophy in the A.flavus transformation recipient with an engineered pyrG marker.The improvement allowed gene manipulation using the reusable pyrG marker as shown by the restoration of laeA -mediated a flatoxin production in an A.flavus laeA -deleted mutant.Published by Elsevier B.V.1.IntroductionAspergillus flavus is a main producer of carcinogenic a flatoxins and is a pathogen of many agricultural commodities.It is also the second leading causative agent of invasive and non-invasive aspergillosis (Hedayati et al.,2007).A flatoxins,if ingested,pose a great risk to human and animal health,hence,their levels are stringently regulated (Guzman-de-Pena and Pena-Cabriales,2005;Otsuki et al.,2001).Of 77countries having regulations limiting mycotoxins,48have speci fic regulatory levels for total a flatoxins in foodstuffs and 21have regulations for a flatoxins in feedstuffs (FAO,1997).Signi ficant economic losses thus can result from a flatoxin contamination of food and feed.The estimated A.flavus genome is 37Mb and contains about 12,000genes (Payne et al.,2006).Genomic resources such as whole genome sequence and EST of A.flavus play an increasingly important role in the understanding of a flatoxin biosynthesis and fungus –plant interactions (Payne et al.,2006;Yu et al.,2004).Equally important is their impact on the understanding of A.flavus pathogenicity in humans and animals.The functions of the majority of A.flavus genes,however,are unknown.Although DNA microarrays allow genome-wide gene expression and association to be studied (Cary et al.,2007;Wilkinson et al.,2007),gene targeting along with subsequent genetic complementation to regain lost traits is still the best approach for understanding gene function.Historically,only Neurospora crassa orotidine monophosphate decarboxylase gene,pyr-4(Woloshuk et al.,1989),is frequently used for A.flavus transformation.A few others including A.flavus β-tubulin gene (Seip et al.,1990)and A.parasiticus nitrate reductase gene (Duran et al.,2007)are occasionally used.The ble gene for resistance to the antibiotic phleomycin has been demonstrated as a positive selectable marker for A.flavus (He et al.,2007);its ef ficacy is still under evaluation.Resistance markers developed from closely related A.oryzae ,such as the ptr pyrithiamine resistance gene (Kubodera et al.,2000)and a mutated succinate dehydrogenase gene,sdhB (cxr),resistant to carboxin,a systemic fungicide,developed for A.oryzae and A.parasiticus are likely applicable to A.flavus (Shima et al.,2009).High-throughput gene functional analysis requires an ef ficient system to reduce time and labor involved in identifying gene knockouts.Signi ficant increases in gene-targeting frequencies have been reported by disabling components of the nonhomologous end-joining (NHEJ)pathway,such as DNA-dependent protein kinase catalytic subunits of Ku70and Ku80,and DNA ligase IV (da Silva Ferreira et al.,2006;Meyer et al.,2007;Mizutani et al.,2008;Ninomiya et al.,2004;Takahashi et al.,2006).Adopting available protocols,demonstrating their ef ficacy and establishing a useful system for A.flavus remains a daunting task.This is particularly true when a system,in terms of selectable marker and recipient strain,is required for multiple rounds of gene-targeting experiments or for reintroducing targeted genes into a knockout strain.The inadequacy of current systems in these aspects has,in part,hampered the progress of the functional genomics in A.flavus .In this study,we first established a gene-targeting system for A.flavus by replacing ku70with A.oryzae ptr and by creating a uracilJournal of Microbiological Methods 81(2010)240–246⁎Corresponding author.Tel.:+15042864208;fax:+15042864419.E-mail address:perngkuang.chang@ (P.-K.Chang).0167-7012/$–see front matter.Published by Elsevier B.V.doi:10.1016/j.mimet.2010.03.010Contents lists available at ScienceDirectJournal of Microbiological Methodsj o u r na l h o m e p a g e :w w w.e l se v i e r.c o m /l o c a t e /j m i c m e t hauxotroph using a knock-in technique(Bardiya and Shiu,2007;Skory et al.,1990).We demonstrated its utility by disrupting several candidate genes and identified two genes involved in conidial pig-ment biosynthesis.We further refined this system by restoring pyrithiamine sensitivity with an engineered pyrG marker for subse-quent re-creation of the uracil auxotrophy.The incorporation of the reusable pyrG marker to the resulting recipient strain makes this improved system facilitate functional genomics study.2.Materials and methods2.1.Fungal strainsThe A.flavus CA14which produces aflatoxins and large sclerotia (Hua et al.,2007)was isolated from a pistachio bud in the Wolfskill Grant Experimental Farm(University of Davis,Winters,California, USA).CA14N1is a nitrate-nonutilizing strain derived from CA14.On a Difco™Czapek Solution Agar(CZ,Becton and Dickinson Company, Sparks,Maryland,USA)plate it exhibited expansive mycelial growth but barely produced conidia due to its inability to use nitrate,which is the sole nitrogen source of CZ.2.2.Construction of A.flavus ku70disruption vectorIn silico identification of the A.flavus ku70gene was performed initially on the5X draft database of the A.flavus genome sequence (http://www.aspergillusfl/genomics/).The ku70disruption vector was constructed in three steps.First,a0.5kb ku70coding region at the5′end(Fig.1A)was amplified by PCR using primers ku94H and ku600P(see Table1for all primers with designations and sequences).The PCR fragment after digestion with HindIII and PstI was cloned into the corresponding sites in pUC19.Second,a0.5kb coding region at the3′end was generated by PCR with ku1650P and ku2150K.The fragment was cloned into the vector obtained from the first step.Third,the2.0kb A.oryzae ptr marker amplified from pPTR1 (TaKarRa,Japan)with ptrU730P and ptr1230P was digested with PstI and cloned into the PstI site of the above construct.The disruption vector,pAfKuDV,was linearized by FastDigest®HindIII and KpnI in a universal buffer(Fermentas,Glen Burnie,Maryland,USA)prior to fungal transformation.Approximately5μg DNA was used in each of the two transformation experiments.2.3.Preparation of protoplasts and transformationIn the initial transformation for the disruption of the ku70gene, approximately108conidia harvested from V8agar plates(Chang and Hua,2007)were inoculated into100ml Czepak-Dox Broth(Becton and Dickinson)supplemented with0.5%casamino acids.The culture was shaken at150rpm for11–12h at30°C.Mycelia were collected on a 100μm nylon cell strainer,transferred to a50ml tube,and resuspended in 20ml offilter-sterilized enzyme solution that contained200mg lysing enzymes from Trichoderma harzianum(L1412,Sigma,St.Louis,Missouri, USA),50mg driselase,(Sigma),and40mg cell-wall digesting enzyme (Applied Plant Research,The Netherlands)in0.55M KC1,0.05M citric acid,pH5.8.The digestion was allowed to progress for2–3h at30°C with shaking(60rpm).Protoplasts were harvested byfiltering through a 40-μm nylon cell strainer and pelleted using a microcentrifuge.The protoplasts were washed twice with a solution of0.6M KC1,50mM CaC12and10mM Tris–HCl,pH8.0.Fungal transformation was performed as previously described with minor modifications(Horng et al.,1990). Polyethylene glycol(PEG)solution consisting of40%(w/v)PEG4000 (Fluka,Germany),0.6M KCl,50mM CaC12and10mM Tris–HC1,pH8.0 was used instead.The transformation mixture was added to CZ regeneration medium containing0.6M KCl,5mM ammonium sulfate, and0.1μg pyrithiamine(PT)/ml.Plates were incubated at30°C for up to 10days.2.4.Confirmation of gene disruption by PCRConidia of PT-resistant colonies were inoculated into1ml Potato Dextrose Broth(PDB;EMD Chemicals Inc.,Damstadt,Germany)in a 2-ml microfuge tube.The tube was incubated horizontally at30°C for 18to24h.Harvested mycelia were processed using a Scientific Industries'Disruptor Genie™(ZYMO RESEARCH,Orange County, California,USA).Genomic DNA was prepared using the ZR Fungal/ Bacterial DNA Kit™(ZYMO RESEARCH).Paired primers,based on specific locations in the expected genomic pattern generated after disruption of ku70by homologous recombination,were used in PCR (see Fig.1).They were set1,ku900and ku1500,set2,ku2210and ptr1,and set3,ku60and ptr800.PCR was carried out under the following conditions in a PERKIN ELMER GeneAmp PCR System2400. Fifty pmol of each primer and about10ng genomic DNA were added to50μl Platinum Blue PCR Supermix(Invitrogen,Carlsbad,California, USA)and subject to30cycles consisting of denaturation at94°C for 30s,annealing at55°C for30s and extension at72°C for2.0min. Similar PCR approaches were used to confirm other gene disruption events(seebelow).Fig.1.Replacement of A.flavus ku70by the ptr selectable marker.(A)Diagram depicting the replacement via double-crossover recombination.(B)PCR analyses of genomic DNA patterns of the recipient,R,and the ku70disruptant,D.The primers used were1:ku900 and ku1500,2:ptr1and ku2210,and3:ptr800and ku60.The DNA size markers(in kb) are lambda DNA/Hind III andØX174RF DNA/HaeIII fragments.241P.-K.Chang et al./Journal of Microbiological Methods81(2010)240–2462.5.Generation of a ku70-and pyrG-deleted strainA gene knock-in strategy(Bardiya and Shiu,2007)was adopted to generate a pyrG-deleted recipient rather than resorting to mutagen or UV treatment.Construction of the pyrG knock-in vector included cloning pyrG-associated fragments to theflanking regions of A. parasiticus cypA,a major part of which is missing in A.flavus(Ehrlich et al.,2004).A1.0kb5′-untranslated region(UTR)plus coding region and a1.0kb3′UTR plus downstream region of pyrG were generated by PCR using primers pg5HK and pg3Sp,and pg5Sp and pg3Sc,respec-tively.The primers cy280and cy550were used to amplify a2.7kb A. parasiticus cypA-containing fragment.These fragments containing tagged or native restriction sites were cloned sequentially into pUC19. The resulting vector was linearized by KpnI and SacI digestion prior to transformation.Preparation of protoplasts and fungal transformation were performed as described above except that,after transformation, PEG was removed by centrifugation.The protoplasts,after regeneration overnight at30°C in PDB containing0.6M KCl and uracil(2mg/ml), were spread onto PDA plates which contained0.6M KCl,uracil(2mg/ ml),and5-fluoroorotic acid(FOA,2mg/ml).Disruption of pyrG in a transformant was confirmed by PCR with the primers pg5HK and pg3Sc. The generatedΔpyrG strain lacked230nt in the pyrG3′coding region plus60nt in3′UTR.e of pyrG-based disruption vectors for identifying genes involved in conidial pigment biosynthesisOnly a few Aspergillus genes have been reported to be involved in conidial pigment biosynthesis,i.e.A.fumigatus alb1,abr1,abr2(a laccase gene=A.nidulans yA)and a yg1(Tsai et al.,1999)and A.nidulans wA (=A.fumigatus alb1)(Mayorga and Timberlake,1992).Homologues of the above genes and several genes encoding laccases that may be involved in pigment formation were identified by BLAST(Schaffer et al., 2001)search of the Aspergillus Comparative Database at Broad Institute (/annotation/genome/aspergil-lus_group/MultiHome.html).Restriction analyses on the identified genes and theirflanking regions were carried out using the DNAMAN software(Lynnon Soft,Vandreuil,Quebec,Canada).DNA fragments specific to theflanking regions of a gene to be targeted were amplified by PCR,digested with appropriate restriction enzymes(Fementas),and inserted into unique sites of pPG28,which contains the A.parasiticus pyrG gene in a 2.7-kb BamHI–SalI fragment(GenBank accession number:EU879956,Fig.2A).The PCR primers used are listed in Table2.Mycelia for the preparation of protoplasts were obtained from conidia of theΔpyrG mutant grown in PDB containing0.5mg uracil/ml shaken at150rpm for20h at30°C.The disruption vectors were linearized prior to transformation to yield DNA ends that are identical to the targeting sites.2.7.Replacement of the inserted ptr marker by a reusable pyrG selectable markerA gene replacement protocol was used to restore the pyrithiamine sensitivity of thefirst createdΔpyrG transformation recipient strain.A 1.4kb5′-UTR of ku70was amplified by PCR using primers ku5Sm and ku5X.A1.7kb3′-UTR of ku70was amplified using primers ku3X and ku3S.The two fragments were cloned sequentially into the corresponding sites in pUC19to give pKu5+3.A direct repeat-recombination strategy was the basis for generating a reusable pyrG marker used to replace the ptr dominant selectable marker.To this end,the R region in Fig.2A was amplified with primers pgXho and pgSal.The PCR fragment after digested by XhoI and SalI was used to replace the1.0kb XhoI–SalI region(right side of Fig.2A),which resulted in a pyrG markerflanked by two0.5kb direct repeats(R-pyr-R);a putative polyadenylation signal,AATAA,is located about10nt before the XhoI site.The R-pyr-R fragment generated after BamHI and SalI digestion was cloned into the XbaI site of pKu5+3by blunt-end ligation.The resulting vector was digested with HindIII and SmaI(Fementas)prior to transformation.Table1Oligoprimer designations and their sequences.Primer Sequenceku94H CTGAAAGCTTCGGAGGCTAACku600P ATACTGCAGGGTCATCATTATCGGTGACTku1650P CTTCCTGCAGAGAAACTCTGGku2150K TGAGGAACAGGTACCGTAAGC.ku60CGACGAGAGTGTACACACTTku270CGGATGAGTTGGAGCTGAAGku900TCAAGATCTGTCCCACGGku1500TGACGGACGTCATCAGCGku2210TCCACACGCTCAACGAGATCku2540GACTGCAACGTTGCTGGTACku5Sm CTTCGCCCGGGTACGGGTCACCTAATCku5X TATCTAGAGTCGTAAGTCATGAATTGCGTku3X ATTCTAGACAACGCTAGTATTGGTTACGAGku3S CTAGAACGAATTCGTGTCGACACTGAptrU730P ATACTGCAGACGGGCAATTGATTACGptr1230P TTACTGCAGCCGCTCTTGCATCTTTGptr1TGGCAGCTGGAGGAGACATGptr800CCTTCTGTGCGAAGCGCTTGpg5HK ATTAAGCTTATTGCTATGTCCCTGAAAGpg3Sp ATTGCATGCTAACTTCAGACTGAACCTCpg5Sp TATGCATGCACTCGAAATGACTACTACTATpg3Sc TGAGTCTAGCTGAGCTCGGCTCpgXho ATACTCGAGATCTCAGAACAATATACCAGpgSal ATAGTCGACCGGCTTATTCAGTAGATTcy280AATGCTAGCTTGTGTGGATTCGTGAGTGTCcy550ATAGCTAGCATTGCTCTGCATACTCGGAClaeA5Sp CGATTAGTTCGTTGAACTGTCAlaeA5S AATGTCGACTGTGAGTAGTACGAGTCGlaeA3B ATAGGATCCACA AATTATTCACGGTGlaeA3Sc TGGCACCACACAAGCTCATATClaeA243ATAGTCGACTTACCGGACAGTGCAAGGlaeA2897TAAGTCGACAAGAGCTGCATCGCGATGTAtfR5Sp CCGGCATGCCTAGACAGACAATCACtfR5S TATGTCGACCTCACGTCTGTGCAGGCCtfR3B GTCGGATCCACATCAAAGAGGGATACTtfR3SmATAGCCCGGGTAATGTCGTTGGTCFig.2.The original and the reusable pyrG markers for fungal transformation.(A)The2.7kb BamHI–SalI fragment that contains the A.parasiticus pyrG gene inserted into themultiple-cloning-site region of pUC19(pPG28).E,EcoRI*;Sc,SacI*;K,KpnI;Sm,SmaI*;B,BamHI*;P,PstI;Xh,XhoI*;S,SalI*;Sp,SphI*;H,HindIII*.The symbol*indicatesunique restriction sites.(B)Schematic representation of the forced recombinationbetween the R repeat regions under FOA positive selection to regain uracil auxotrophy. 242P.-K.Chang et al./Journal of Microbiological Methods81(2010)240–2462.8.Determination of frequencies of self-resolution of the reusable pyrG marker on different mediaConidia were harvested from a PT-sensitive strain,whose previously inserted ptr marker had been replaced by the reusable pyrG (R-pyr -R)marker.Different culture media were used to examine the selection ef ficiency for the resolved auxotrophic mutants.Approximately 105,106,and 107conidia were spread onto PDA plates which contained 2mg uracil/ml and 2mg FOA/ml,and CZ(NH 4+)plates which contained 2mg uracil/ml,2mg FOA/ml,and 5mM ammonium sulfate.The plates were incubated at 30°C for 7to 10days.The ku70-speci fic primers ku270and ku2540were used to con firm forced resolution (loop-out)of the pyrG marker leaving only one copy of the R region in the ku70locus (Fig.2B).2.9.Deletion of laeA by R-pyrG-R and re-creation of a uracil auxotrophic ΔlaeA mutantUsing similar steps described in Section 2.2,we constructed a laeA -disruption vector based on the R-pyr -R marker with the following primers:laeA5Sp,laeA5S,laeA3B,and laeA3Sc to amplify the two flanking regions used in targeting laeA via double-crossover recombi-nation.The resulting vector was linearized with SphI and SacI prior to transformation.Putative ΔlaeA mutants were selected based on changes in colony morphology and loss of a flatoxin production.Disruption of laeA was con firmed by PCR analyses of the genomic patterns of the transformants (data not shown).Approximately 2×106conidia of a con firmed ΔlaeA mutant were spread onto CZ(NH 4+)plates,which contained 2mg uracil/ml,2mg FOA/ml,and 5mM ammonium sulfate,for the generation of uracil auxotrophic ΔlaeA mutants.2.10.Reintroduction of full-length genomic laeA into an A.flavus ΔlaeA mutantThe laeA gene was reintroduced into the ctfR2gene locus (AFL2G_07245.2)located in a subtelomeric region of chromosome 3.Disruption of ctfR2had no effect on the production of a flatoxin,cyclopiazonic acid,nor on gross morphology (unpublished results).The primer pair,tfR5Sp and tfR5S,and the primer pair,tfR3B and tfR3Sm were used to amply a 5′and a 3′regions of ctfR2,respectively.The two PCR fragments were cloned into corresponding sites in pUC19followed by insertion of the A.parasiticus pyrG gene.The full-length laeA gene was ampli fied from A.flavus CA14genomic DNA template using Accu-Prime ™Pfx PCR Supermix (Invitrogen)with primers laeA243and laeA2897each tagged with a SalI site.The 2.7kb laeA -containing SalI –SalI fragment was cloning into the SalI –XhoI sites after the 1.2kb SalI –XhoI fragment downstream of the pyrG gene (see Fig.2A)was removed.The resulting targeting vector was digested with HindIII and SphI prior to transformation.The primers laeA243and laeA2897were used in PCR to con firm the presence of the full-length laeA gene in the comple-mented transformants.2.11.Thin-layer chromatography (TLC)analysis of a flatoxin B 1The transformants on the regeneration plates (PDA supplemented with 0.6M KCl)putatively complemented by the full-length laeA were transferred onto PDA plates for TLC analysis.One PDA agar plug was cored from a 5day-old transformant culture plate grown at 30°C,placed into a microfuge tube and extracted with 0.2ml of acetone for 1h.Ten microliter of each extracts was spotted onto a Si250TLC silica gel plate (J.T.BAKER,Phillipsburg,New Jersey,USA).The metabolites were resolved with a solvent system of toluene:ethyl acetate:acetic acid (80:10:10,vol/vol/vol).3.Results3.1.Replacement of ku70by the dominant ptr selectable marker Growth of A.flavus CA14and CA14N1was inhibited at 0.01μg PT/ml.Therefore,0.1μg PT/ml was chosen as the selection concentration.Two independent transformation experiments yielded a total of 102PT-resistance colonies.PCR analyses of the genomic DNA from 80transformants showed that two transformants were ku70knockouts.Deletion of ku70was con firmed with primers speci fic to the ku70coding region,ptr downstream and upstream regions,and regions beyond the expected homologous recombination sites (Fig.1A).No PCR products were generated from the genomic DNA of the ku70knockout when primers ku900and ku1500were used.In contrast,these primers yielded a 0.6kb PCR fragment from the recipient's genomic DNA (Fig.1B).As expected,primers ptr1and ku2210,which amplify a small region beyond the expected integration site i.e.,ku2150and the upstream region of ptr (Fig.1A),generated a 1.4kb PCR fragment from the genomic DNA of the ku70knockout.Likewise,primers ptr800and ku60yielded a 1.0kb PCR fragment,expected from another flanking region after ku70was targeted by ptr through homologous integration.The two regions were not present in the recipient's genome.These results showed that the ptr marker had replaced the ku70gene in the A.flavus genome.e of the Δku70ΔpyrG strain to identify conidial pigment biosynthetic genesThe ku70-defective genetic background and the positive FOA selection facilitated the generation of ΔpyrG mutants that produced white-colored conidia on regeneration plates (data not shown).A Δku70ΔpyrG strain was used as the transformation recipient in the identi fication of conidial pigment genes.Of the nine genes disrupted two genes,AFL2G_09923.2and AFL2G_09924.2,were con firmed to be involved in conidial pigment biosynthesis (Table 2).The AFL2G_09923.2knockouts produced expected white conidia while knockouts of AFL2G_09924.2,a homolog of A.fumigatus abr2(br:brown)and A.nidulans yA (y:yellow),produced olive-green conidia not reported before in Aspergillus species (Fig.3A).They were named wA and olgA ,Table 2PCR primers used in the construction of disruption vectors for identifying conidial pigment genes.Gene locusa5′Flanking region3′Flanking regionGeneAFL2G_00330.2cacggcccgggatgtcacta gtatcgggatccaccttccctcctt cggtttgagcaagcttagaatc gctggaagatcgtcgaccctgtgaa ayg1AFL2G_09923.2gcagcttataggagaattcact gccgactggatccctctgtgtattgtcgacttccgatactatcta caatgcatgcaatacttaccgatg alb1(wA )AFL2G_09924.2acgtattggtagcatgctcacgc tgcgtcgacagctttaagccacgcggc ttcgaagagctccggtctttccgt catggatccacgagcacccgatgt abr2(yA )A.flavus olgA AFL2G_09962.2atggaacatcacccgggtattggcca tatggatccaatgatgagatgtcag tacgtcgacttcaaacttgtccctg tcacttgcgtcgcatgcctg abr1AFL2G_08008.2cctatctggtgcatgcggtgaacttg gttgcattgagtcgacgccaggc gatgaggaggatccctacattg gtcgagtacagaattcgaact lac1AFL2G_09420.2cgcgtttaattcggcatgca ggaagtcgacacgggcagtg ctagtggatccatcccacaga cgagattggaattccccggg lac2AFL2G_10583.2ttgctgggagcatgcctgtatcac gttcatggtcgacaaccatac cgaacagcggatccactgtattg catgcttggcttgtttgatcglac3AFL2G_11132.2ctacggcatgcccgggtgacacag atcgcgtcgacacggatagg tataggatccgcgctgccaac atgggaattcccgggataagacatgg lac4AFL2G_11750.2cgtgcgtcactatgcatgcagg catcatgggtctgtcgacgccatcggtgatggtgacacagctggtcaa cacaagaactagaattcaagacgglac5a/annotation/genome/aspergillus_group/MultiHome.html .243P.-K.Chang et al./Journal of Microbiological Methods 81(2010)240–246respectively.The con firmed knockouts of each of the rest seven genes including homologs of A.fumigatus abr1and ayg1produced yellowish-green conidia same as those of the A.flavus parental strain (data not shown).The overall gene-targeting frequencies estimated from all the experiments ranged from 80%to 100%.The locations of these genes were tentatively assigned to the A.flavus genome using the chromo-somal map of A.oryzae (http://www.bio.nite.go.jp/dogan/MicroTop?GENOME_ID=ao ).The wA and olgA genes are clustered on chromosome 4,and AFLG_09962.2,a homolog of A.fumigatus abr1(Tsai et al.,1999),is located about 100kb away.Fig.3B shows the genomic patterns of some of the knockouts compared to those of the recipient strain as con firmed by PCR with primers encompassing the targeting region.They were 4.3kb vs.1.7kb for AFL2G_09962.2,4.1kb vs.1.5kb for olgA ,4.7kb vs.3.5kb for wA ,and 5.0kb vs.2.3kb for AFL2G_00330.2.3.3.Restoration of PT sensitivity and uracil auxotrophy in the original recipientA drawback of many developed transformation systems based on single selectable markers is the loss of auxotrophy or drug sensitivity after transformation.In this study,the ptr marker was readily replaced by the reusable pyrG marker (R-pyr -R)which contains two direct repeats of a 5′UTR region (Fig.2B),and the uracil auxotrophy wasrecovered from FOA selection.The estimated frequencies of self-resolution of the R-pyr -R marker reached 100%on CZ(NH 4+)and PDA media.The PTs Δku70ΔpyrG derivatives containing a single copy of the 5′UTR (R)were easily generated on both media (Fig.4A and B.Although CZ(NH 4+)yielded smaller colonies than PDA,it gave more than 5-fold higher ΔpyrG derivatives than PDA (Table 3).3.4.Demonstration of the ef ficacy of the improved system by reintroducing laeATo test the utility of the developed system,we disrupted the laeA gene,a major regulatory gene of secondary metabolism (Bok and Keller,2004),in a PTs Δku70ΔpyrG strain (data not shown),regenerated the uracil auxotrophy by forcing out the reusable pyrG marker,and restored the lost a flatoxin-producing ability by reintro-ducing an intact copy of laeA via a new round of transformation based on the pyrG selectable marker.Five of the six transformants examined produced a flatoxin B 1(Fig.5A),and the result was consistent with the presence of the intact laeA gene in the a flatoxin-producing transfor-mants (Fig.5B).4.DiscussionWe combined available selectable markers for fungal transforma-tion and formulated an ef ficient gene-targeting protocol for A.flavus .The demonstrated ef ficacy of this system indicates that the heterol-ogous genes are functional in A.flavus .An important consideration for high-throughput functional genomics is to minimize the time in selecting correct gene knockouts,that is,only a very small number of transformants should be examined before one is identi fied.The ku70gene is critical for DNA repair via the nonhomologous end-joining (NHEJ)pathway,and its impairment greatly reduces the frequency of heterologous integration of transforming DNA.The gene-targeting frequencies of the NHEJ-de ficient A.flavus system are well withintheFig.3.Identi fication of conidial pigment genes.(A)Colony morphology of AFL2G_09962.2(top),AFL2G_09924.2(olgA ,bottom left),and AFL2G_09923.2(wA ,bottom right)disruptants after growth at 30°C for 5days on PDA.(B)Con firmation of disruption of AFL2G_09962.2(9962),oligA ,wA and AFL2G_00330.2(0330).M:1kb DNA ladder,A:recipient strain,B:disruptant.Fig. 4.Determination of self-resolution frequencies on different media.(A)Colony morphology of FOA-resistant mutants.(B)PCR con firmation of deletion of the pyrG marker.C:PTs Δku70;1–4:independent derivatives.The lack of products in C was due to the presence of the R repeats in the R-pyrG -R marker which interfered PCR ampli fication.244P.-K.Chang et al./Journal of Microbiological Methods 81(2010)240–246ranges of what have been reported for a single homologous insertion in other fungi(da Silva Ferreira et al.,2006;Ninomiya et al.,2004; Takahashi et al.,2006).The generation of the A.flavus uracil auxotrophic mutant by the knock-in strategy based on FOA resistance can be easily adopted for other fungi.This approach eliminates the common practice of mutagen treatments which often result in genetic changes.Although these changes are not always morphologically apparent,they may complicate functional studies.A drawback of past developed transformation systems for A.flavus and many other fungi is the lack of double selectable markers and double mutants suitable for gene manipulation,for example gene knockout experiments followed by genetic complementation.The creation of necessary mutants by conventional approaches is a formidable task.The experimental steps can be adopted and used to readily generate a highly efficient system that consists of a reusable selection and second selection as the described uracil auxotrophy and pyrithiamine sensitivity to meet the prerequisites.It can be achieved byfirst knocking out an NHEJ-associated gene with either an auxotrophic marker,such as the nitrate reductase gene(niaD)in a spontaneous niaD mutant(Malardier et al., 1989;Pereira et al.,2004;Whitehead et al.,1990)or with a dominant marker,such as the aforementioned ptr,ble and sdhB(cxr)genes(He et al.,2007;Shima et al.,2009)to facilitate subsequent rounds of gene-targeting.The refined system has been used successfully to disrupt and reintroduce a gene,such as laeA,a regulatory gene of secondary metabolism(Bok and Keller,2004)into a specific genome locus to restore the lost aflatoxin production(Fig.5).The NHEJ-deficient background of the recipient stain also allows integration of a circular vector containing sequence regions identical to a genomic portion via singe-crossover recombination.Based on pyrG we used this approach and genetically complemented an A.flavus knockout mutant of msnA (unpublished results),the orthologue of Saccharomyces cerevisiae MSN2necessary for cells to cope with a broad range of stresses(Ruis and Schuller,1995).The added benefit of this system includes the restored pyrithiamine sensitivity,upon which the dominant ptr marker can be used as an alternative in double knockout or genetic complementation experiments to provide greater versatility. AcknowledgmentWe thank Alice Yeh of University of Virginia for technical assistance.ReferencesBardiya,N.,Shiu,P.K.,2007.Cyclosporin A-resistance based gene placement system for Neurospora crassa.Fungal Genet.Biol.44,307–314.Bok,J.W.,Keller,N.P.,eA,a regulator of secondary metabolism in Aspergillus spp.Eukaryot.Cell3,527–535.Cary,J.W.,O'Brian,G.R.,Nielsen,D.M.,Nierman,W.,Harris-Coward,P.,Yu,J.,Bhatnagar,D.,Cleveland,T.E.,Payne,G.A.,Calvo,A.M.,2007.Elucidation of veA-dependentgenes associated with aflatoxin and sclerotial production in Aspergillusflavus by functional genomics.Appl.Microbiol.Biotechnol.76,1107–1118.Chang,P.-K.,Hua,S.S.,2007.Molasses supplementation promotes conidiation but suppresses aflatoxin production by small sclerotial Aspergillusflavus.Lett.Appl.Microbiol.44,131–137.da Silva Ferreira,M.E.,Kress,M.R.,Savoldi,M.,Goldman,M.H.,Hartl,A.,Heinekamp,T., Brakhage, A.A.,Goldman,G.H.,2006.The akuB(KU80)mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus.Eukaryot.Cell5,207–211.Duran,R.M.,Cary,J.W.,Calvo,A.M.,2007.Production of cyclopiazonic acid,aflatrem, and aflatoxin by Aspergillusflavus is regulated by veA,a gene necessary for sclerotial formation.Appl.Microbiol.Biotechnol.73,1158–1168.Ehrlich,K.C.,Chang,P.-K.,Yu,J.,Cotty,P.J.,2004.Aflatoxin biosynthesis cluster gene cypA is required for G aflatoxin formation.Appl.Environ.Microbiol.70,6518–6524. FAO,1997.Worldwide Regulations for Mycotoxins1995.A Compendium.Rome,Italy, FAO Food and Nutrition.Paper64.Guzman-de-Pena,D.,Pena-Cabriales,J.J.,2005.Regulatory considerations of aflatoxin contamination of food in tinoam.Microbiol.47,160–164.He,Z.M.,Price,M.S.,Obrian,G.R.,Georgianna,D.R.,Payne,G.A.,2007.Improved protocols for functional analysis in the pathogenic fungus Aspergillusflavus.BMC Microbiol.7,104. Hedayati,M.T.,Pasqualotto,A.C.,Warn,P.A.,Bowyer,P.,Denning,D.W.,2007.Aspergillus flavus:human pathogen,allergen and mycotoxin producer.Microbiology153, 1677–1692.Horng,J.S.,Chang,P.-K.,Pestka,J.J.,Linz,J.E.,1990.Development of a homologous transformation system for Aspergillus parasiticus with the gene encoding nitrate reductase.Mol.Gen.Genet.224,294–296.Hua,S.S.,Tarun,A.S.,Pandey,S.N.,Chang,L.,Chang,P.-K.,2007.Characterization of AFLAV,a Tf1/Sushi retrotransposon from Aspergillusflavus.Mycopathologia163,97–104. Kubodera,T.,Yamashita,N.,Nishimura,A.,2000.Pyrithiamine resistance gene(ptrA)of Aspergillus oryzae:cloning,characterization and application as a dominant selectable marker for transformation.Biosci.Biotechnol.Biochem.64,1416–1421. Malardier,L.,Daboussi,M.J.,Julien,J.,Roussel,F.,Scazzocchio,C.,Brygoo,Y.,1989.Cloning of the nitrate reductase gene(niaD)of Aspergillus nidulans and its use for transformation of Fusarium oxysporum.Gene78,147–156.Mayorga,M.E.,Timberlake,W.E.,1992.The developmentally regulated Aspergillus nidulans wA gene encodes a polypeptide homologous to polyketide and fatty acid synthases.Mol.Gen.Genet.235,205–212.Meyer,V.,Arentshorst,M.,El-Ghezal,A.,Drews,A.C.,Kooistra,R.,van den Hondel,C.A., Ram,A.F.,2007.Highly efficient gene targeting in the Aspergillus niger kusA mutant.J.Biotechnol.128,770–775.Mizutani,O.,Kudo,Y.,Saito,A.,Matsuura,T.,Inoue,H.,Abe,K.,Gomi,K.,2008.A defect of LigD(human Lig4homolog)for nonhomologous end joining significantly improves efficiency of gene-targeting in Aspergillus oryzae.Fungal Genet.Biol.45,878–889. Ninomiya,Y.,Suzuki,K.,Ishii,C.,Inoue,H.,2004.Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining.Proc.Natl.Acad.Sci.USA101,12248–12253.Otsuki,T.,Wilson,J.S.,Sewadeh,M.,2001.What price precaution?European harmoniza-tion of aflatoxin regulations and African groundnuts exports.Eur.Rev.Agric.Econ.28, 263–284.Payne,G.A.,Nierman,W.C.,Wortman,J.R.,Pritchard, B.L.,Brown, D.,Dean,R.A., Bhatnagar,D.,Cleveland,T.E.,Machida,M.,Yu,J.,2006.Whole genome comparison of Aspergillusflavus and A.oryzae.Med.Mycol.44(Suppl),9–11.Table3Self-resolution frequencies of the R-pyrG-R selectable marker on media supplemented with FOA.Medium Number of spores FOA-resistant colony a Frequency(%)bCZ(NH4+)1051±1–c10612±3100107113±11100PDA1050±0–1061±1–10718±6100a Average±SD.b Frequency was estimated based on four independent colonies examined.c Notdetermined.Fig. plementation of aΔlaeA strain with an amplified genomic laeA fragment.(A)Restoration of aflatoxin in the transformants(B)Confirmation of the presence of the introduced laeA in the aflatoxin-producing transformants.M:DNA1-kb ladder.Transfor-mants1to6are the same in A and B.245P.-K.Chang et al./Journal of Microbiological Methods81(2010)240–246。
基因治疗的原理与研究概况PPT课件

*
*
(五)基因免疫治疗
通过将抗癌免疫增强的细胞因子或MHC基因导入肿瘤组织,以增强肿瘤微环境中的抗癌免疫反应。
*
*
二、基因转移技术
*
*
基因治疗的两种途径
载体
目的基因
in vivo
ex vivo
靶细胞
*
*
基因转移(gene transfer)技术:
1.病毒介导的基因转移 2.非病毒介导的基因转移
4.靶向性差。
1.不能整合到靶细胞的基因组DNA中。分裂增殖快的细胞,导入的重组病毒载体,随分裂而丢失的机会增多,表达时间相对较短。
(1)腺病毒产生过程中与293辅助细胞内E1区序列发生同源重组; (2)腺病毒载体与被治疗的患者体内已感染的野生型腺病毒,甚至乳头瘤病毒、巨细胞病毒发生重组。
*
*
(3)腺病毒相关病毒载体 腺病毒相关病毒(adenovirus associated virus,AAV)是一类单链线状DNA缺陷型病毒。其基因组DNA小于5 kb,无包膜,外形为裸露的20面体颗粒。AAV不能独立复制,只有在辅助病毒(如腺病毒、单纯疱疹病毒、痘苗病毒)存在时,才能进行复制和溶细胞性感染,否则只能建立溶源性潜伏感染。
③前病毒通过LTR高效整合至靶细胞基因组中,有利于外源基因在靶细胞中的永久表达。
④包装好的假病毒颗粒(携带目的基因的重组逆转录病毒载体)以出芽的方式分泌至辅助细胞培养的上清液中,易于分离制备。
*
*
逆转录病毒载体的主要缺点
随机整合,有插入突变、激活癌基因的潜在危险; 逆转录病毒载体的容量较小,只能容纳7 kb以下的外源基因。
*
*
1.病毒介导的基因转移系统
13分子习题答案

复习题:1、名词解释1、miRNA:----微小RNA,就是一种大小约为21-23个碱基单链小分子RNA,就是由具有发夹结构的约为70-90个碱基大小的单链RNA前体经过Dicer酶加工生成。
2、siRNA:----小干扰RNA(small interfering RNA),RNAi的关键效应分子,21-23个nt大小的双链RNA。
3、RNAi:----RNA干扰就是正常生物体内抑制特定基因表达的一种现象,它就是指当细胞中导入与内源性mRNA编码区同源的双链RNA(double stranded RNA,dsRNA)时,该mRNA发生降解而导致基因表达沉默的现象,这种现象发生在转录后水平,又称为转录后基因沉默(PTGS)4、PTGS:----转录后基因沉默,就是正常生物体内抑制特定基因表达的一种现象,它就是指当细胞中导入与内源性mRNA编码区同源的双链RNA(double stranded RNA,dsRNA)时,该mRNA发生降解而导致基因表达沉默的现象,这种现象发生在转录后水平,又称为转录后基因沉默(PTGS)5、Nucleosome:----核小体:染色质的基本结构亚基,由约200 bp的DNA与组蛋白八聚体所组成6、Informasome----信息体,真核细胞mRNA通常与一些蛋白质结合成核蛋白颗粒(RNP),构成信息体7、Chaperon: ----分子伴侣就是一类能介导蛋白质进行正确折叠、组装的蛋白质,能协助蛋白质获得正确的构型。
8、Ubiquitin: ----泛素,就是生物体内广泛存在的一类酸性蛋白质;含76个氨基酸,序列在进化过程中高度保守,C-端为Gly,且分子内部有多个Lys。
9、Capsase: ----(Cysteine-containing aspartate-specific proteases天冬氨酸特异性半胱氨酸蛋白酶/切冬酶/胱冬肽酶)富含半胱氨酸,以非活性状态的酶原存在,其肽链比有活性时长一些,将多出的部分切除,就转变成有活性的caspase。
crisper-cas 基因靶向技术
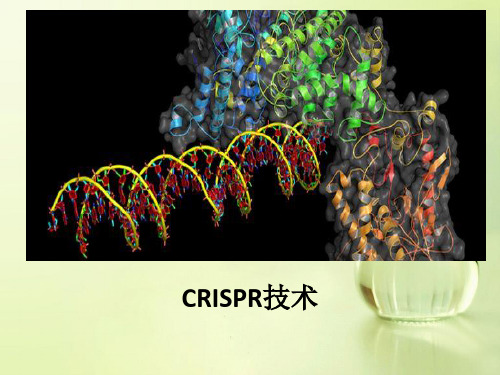
CRISPR担当细菌的防护罩
• CRISPR簇是一个广泛存在于细菌和古生菌基因组 中的特殊DNA重复序列家族,其序列由一个前导区 (Leader)、多个短而高度保守的重复序列区 (Repeat)和多个间隔区(Spacer)组成。 • 前导区一般位于CRISPR簇上游,是富含AT长度为 300~500bp的区域,被认为可能是CRISPR簇的启 动子序列。重复序列区长度为21~48bp,含有回 文序列,可形成发卡结构。重复序列之间被长度 为26~72bp的间隔区隔开。Spacer区域由俘获的 外源DNA组成,类似免疫记忆,当含有同样序列的 外源DNA入侵时,可被细菌机体识别,并进行剪切 使之表达沉默,达到保护自身安全的目的。
• 在II型系统中pre-crRNA的加工由Cas家族中的Cas9单独参 与。 • Cas9含有RuvC和HNH2个独特的活性位点,在crRNA成熟和 双链DNA剪切中发挥作用。此外,pre-crRNA转录的同时, 与其重复序列互补的反式激活crRNA(Trans-activating crRNA,tracrRNA)也转录出来,并且激发Cas9和双链RNA 特异性RNaseIII核酸酶对pre-crRNA进行加工。加工成熟 后,crRNA、tracrRNA和Cas9组成复合体,识别并结合于 crRNA互补的序列,然后解开DNA双链,形成R-loop,使 crRNA与互补链杂交,另一条链保持游离的单链状态,然 后由Cas9中的HNH活性位点剪切crRNA的互补DNA链,RuvC 活性位点剪切非互补链,最终引入DNA双链断裂(DSB)。
CRISPR的工作原理
• 当细菌抵御噬菌体等外源DNA入侵时,在前 导区的调控下,CRISPR被转录为长的RNA前 体(Pre RISPR RNA,pre-crRNA),然后加 工成一系列短的含有保守重复序列和间隔 区的成熟crRNA,最终识别并结合到与其互 补的外源DNA序列上发挥剪切作用。
关于靶基因说明

关于靶基因的说明"靶基因" :target gene"靶基因" 在学术文献中的解释1、在维甲酸诱导APL细胞分化的研究中,一般将受维甲酸及受体直接作用的基因称为靶基因,而将受靶基因蛋白质产物调控的基因称为受维甲酸调控基因 .2、靶基因是指编码特定蛋白质的结构基因,它能使转基因生物体产生新的表型.转基因鱼研究中所使用的靶基因大多是生长激素基因、抗冻蛋白基因、珠蛋白基因、金属硫蛋白基因和生长激素释放因子基因等.3、而欲进行杂交的实验样本(DNA或是RNA)称为靶基因,这指的是从试验样品中提取的丰度一定的未知序列的核苷酸链.基因靶技术(gene targeting)一、基因靶技术的原理基因靶技术的原理是设计一个靶载体,其内含有与靶位点相同的核苷酸序列,用基因转移的方法将载体导入靶细胞,通过载体与靶位点相同的核苷酸顺序间的同源重组,使外源DNA序列定点整合入靶位点,从而达到对靶位点进行定点修复或定点突变的目的。
载体分两种:一是序列插入型载体,通过与靶点序列的单交换导入外源序列;二是序列取代型载体,通过与靶点序列间的双交换,用外源序列取代靶序列。
二、基因靶技术的作用(一)确定基因表达水平目前应用最多的基因转移途径—逆转录病毒载体介导的基因转移存在二个问题,一是导入的外源基因难以生理方式进行表达;二是即使表达也不够完善。
采用基因同源重组的基因靶技术,可使病变的基因在原位得到改正,与正常基因的结构与调控环境一致,即以生理方式来进行表达。
1989年Thomphon采用小鼠胚干细胞HPRT基因的同源重组置换,使HPRT缺陷功能得到恢复。
(二)增强基因表达的稳定性与安全性通过逆转录病毒载体,将目的基因导入靶细胞后,随时间的推移,目的基因表达的稳定性会有所下降,表达的活性会慢慢的降低,有的甚至完全停止表达。
同样,外源基因与靶细胞染色体基因组的整合位点是随机的,常可导致基因的丢失和重排而失去表达活性。
2007年诺贝尔生理学或医学奖简介

・诺贝尔奖工作回顾・小鼠基因修饰基本原理及其在医学研究中的应用———2007年诺贝尔生理学或医学奖及其相关工作介绍汤富磊1 冯 娟2(1北京大学精神卫生研究所,北京100083;2北京大学医学部生理学与病理生理学系,北京100083) 2007年10月8日,瑞典皇家卡罗琳医学研究院诺贝尔生理学或医学奖评审委员会宣布,美国科学家Mari o R .Capecchi 、O liver S m ithies 和英国科学家Martin J.Evans 在“涉及使用胚胎干细胞进行小鼠特定基因修饰方面的一系列突破性发现”[1]而获得2007年度诺贝尔生理学或医学奖(图1)。
图1 2007年度诺贝尔生理学或医学奖获得者Mari o R.Capecchi 1937年出生于意大利。
1967年获哈佛大学生物物理学博士学位,长期担任美国犹他大学人类遗传学和生物学教授,同时在霍华德2休斯医学研究所(Howard 2Hughes Medical I nstitute )工作。
O liver S m ithies 1925年出生于英国。
1951年获牛津大学生物化学博士学位,现在美国北卡罗来纳大学教会山分校工作。
Mari o R.Capecchi 和O li 2ver S m ithies 分别独立地发现了利用两段DNA 片段的同源重组可以对哺乳动物基因组进行可控的基因修饰。
Martin J.Evans1941年出生于英国,1963年从剑桥大学毕业后进入伦敦学院解剖与胚胎系攻读博士学位。
现在英国加的夫大学担任哺乳动物遗传学教授。
1981年,Evans 从小鼠胚胎中成功地分离出未分化的胚胎干细胞,这些细胞是生物成体所有细胞的来源。
他还建立了一系列基本技术,包括对胚胎干细胞进行细胞培养、遗传操作,以及将遗传改造过的胚胎干细胞转入代孕母鼠体内以产生经遗传操作的后代。
上述三位科学家的工作,使人们可以在哺乳动物的生殖细胞中进行特定的基因改造,并繁殖出成功表达这种新基因的后代。
基因敲除小鼠在疾病研究中的应用_桑景荭

芳香物、卤化物和杀虫剂等,因此在污水的三级处理过程中也具有潜在应用价值。
研究显示,特氏杜氏藻中的金属结合肽和植物螯合肽可将无机砷代谢成砷聚合物,从而达到去除环境中重金属的目的。
6环境指示相对其他测试生物(如动物)而言,藻类对生活污水或工业废水有更高的敏感性。
利用藻类来指示环境毒性的研究,最近有很多报道。
Santin-Montanya等报道了7种微藻对海洋环境杀虫剂的测试方法,指出普氏杜氏藻是禾草灭、稀禾定、苯嗪草酮、二氯吡啶酸等杀虫剂的最佳指示剂。
黄小娟等[4]利用杜氏藻生长阻碍实验,测定排放到环境中特别是饮用水中的有机毒物,从而得知被测化合物的生物毒性。
这种行之有效的测试饮用水污染的方法,在水质污染监测和控制方面有着广阔的应用前景。
综上所述,杜氏藻是一种优越的经济藻类,具有独特的生理特性。
随着科技的发展以及消费者对天然产品的需求日益增加,杜氏藻的效能和应用价值将越来越广泛地被开发出来,进而在医药、食品、养殖业、化妆品等领域发挥越来越重要的作用。
主要参考文献1Leon R.,Vila M.,Hernanz D.et al.Production of phytoene by herbicide-treated microalgae Dunaliella bardawil in two-phase systems.Biotechnol Bioeng,2005,92:695—701.2尹鸿萍,盛玉青.盐藻多糖体内抑菌及抗炎作用的研究.中国生化药物杂志,2006,27(6):361—363.3朱红红,尹鸿萍.盐藻多糖抗病毒实验研究.南京中医药大学学报,2007,23(5):310—312.4黄小娟,沈洛夫,姜建国等.卤代物对盐藻生长抑制实验的联合效应的观察.癌变·畸变·突变,2005,17(1):27—28.(E-mail:chendefu@)基因敲除(gene knock-out)是指借助分子生物学、细胞生物学和动物胚胎学的方法,通过胚胎干细胞这一特殊的中间环节将模式生物正常的功能基因的编码区破坏,使特定基因失活,以研究该基因的功能,或者通过外源基因来替换宿主基因组中的相应部分,以便测定它们是否具有相同的功能,或者将正常基因引入宿主基因组中置换突变基因以达到靶向基因治疗的目的。
基因打靶技术.

四、外源DNA导人的方式
•
外源DNA导人的方式主要有显微注射法、电穿孔 法、精子载体法和逆转录病毒法等。目前应用最 广的是显微注射法。Capecchi报道,用显微注射 法导入可得到很高的转染效率,占接受外源DNA 细胞的10%~20%,但显微注射每次只能注射一个 细胞,而电穿孔法可同时使许多细胞得到转染。 Mansour等研究发现电穿孔可使1%的ES细胞稳定 转染。逆转录病毒载体法利用某些病毒与组织细 胞有特异的亲合力,可用于时空特异性基因打靶, 在人类疾病的基因治疗方面具有较大的发展潜力。
常用的选择标记基因
•
•
正选择标记基因有新霉素磷酸转移酶 (neo)、潮霉素B磷酸转移酶(hph)、黄 嘌呤/鸟嘌呤磷酸转移酶(gpt)、次黄嘌呤 磷酸转移酶(Hprt)、胸腺嘧啶激酶(tk) 及嘌呤霉素乙酰转移酶(puro)。 负选择标记基因有单纯疱疹病毒胸腺嘧啶 激酶(HSV-tk)、SacB、 rpsl(strA)、 tetAR、pheS、thyA、CacY、gata-I、 ccdB 等。
•
胚胎干细胞注入体内与完整胚胎形成嵌合 体后,可以发育形成包括生殖细胞在内的 一系列成体组织;正是由于胚胎干细胞的 特殊功能,ES细胞基因打靶技术已被广泛 地应用于建立转基因动物之中。从80年代 到90年代初,小鼠ES细胞基因打靶技术已 发展到成熟阶段。
•
1984年, Bradly等成功地用显微注射法将 ES细胞移入囊胚腔,并移植回假孕母鼠, 获得生殖系嵌合体,经过适当的交配,获 得了源于ES细胞系的纯系小鼠。此实验首 次证实体外培养的ES细胞能在体内分化发 育成生殖系嵌合体并可获得小鼠纯合体子 代。1987年,人们利用ES细胞技术建立了 次黄嘌呤磷酸核糖转移酶(hprt)基因敲除 (Gene knockout)的动物模型。此后,这项技 术得到了普遍应用和长足发展。
CRISPR技术的进展与未来展望

CRISPR技术的进展与未来展望近年来,CRISPR技术成为了生命科学领域的热点话题。
CRISPR是一种基因编辑技术,能够精确地改变DNA序列。
它的诞生标志着生命科学领域的一个巨大飞跃,对疾病治疗、新药开发等有着深远的影响。
本文将探讨CRISPR技术的进展与未来展望。
一、CRISPR技术的背景CRISPR技术源于自然界中细菌免疫系统的一种特殊机制,可将外来病毒基因特异性地剪切并摧毁。
如何将这一机制应用到人类基因领域,则是CRISPR首要面临的问题。
许多科学家在CRISPR上投入了大量时间和精力,致力于寻找新的方法和实践,以便能更好地应用CRISPR技术。
人们解锁了生命科学的新奥秘,启动了新的基因编辑革命的源头。
二、CRISPR技术的进展CRISPR技术的进展无处不在。
截至目前,全球已经有超过50个国家和地区的400多家学术机构和企业,开展了与CRISPR相关的科研工作。
下面,将从四个方面进行具体阐述。
1、基因诊断CRISPR技术可用于诊断基因突变等人类基因缺陷。
科研人员可以通过不到10美元的成本检测千兆碱基,并对其进行编辑。
CRISPR技术在基因诊断领域可谓是一个巨大的进展,有望缩短疾病的初期诊断时间。
2、基因治疗基因治疗是指通过基因编辑技术,对特定基因进行修正或加工,从而治疗一系列人类疾病。
CRISPR技术的出现使得基因治疗成为可能。
一些重大的疾病,比如癌症,可以被CRISPR技术治愈。
3、农业科技CRISPR技术在农业科技领域也有着有捷径的应用。
通过改变植物的基因,可以使其生长更快更健康,从而提高农业产量。
这对解决全球日趋严重的食品短缺问题是非常重要的。
4、新药开发新药的开发需要花费巨额的时间和成本。
CRISPR技术的出现为新药的开发带来了新的思考方式。
CRISPR技术可以帮助科学家更好地理解疾病发病机理,从而研发出更安全有效的药物。
三、CRISPR技术的未来展望CRISPR技术创造了一种全新的生命科学生态系统。
CRISPR-Cas9基因靶向修饰

有一种细菌编码的酶能够利用向导RNA(guide RNA)分子对特定的DNA片段进行定向切割,科学家们利用这种特点开发出了一种能够对基因组进行特异性定点改造的工具,对细胞乃至整个生物体进行基因组改造。
目前已经利用这种技术对细菌、人体细胞以及斑马鱼进行过成功的遗传学改造工作。
日本大阪大学(Osaka University)的科研人员们曾经在1987年发表过一篇论文,不过这篇论文在当时并没有引起太多人的注意,只不过是一篇普普通通的小文章。
这帮大阪大学的科学家当时正在对一种细菌编码的碱性磷酸酶(alkaline phosphatase)基因进行研究,不过他们在工作中发现,在这个基因编码区域的附近存在一小段不同寻常的DNA片段,这些片段是由简单的重复序列组成的,而且在片段的两端还存在一段不太长的特有的序列。
关于这样一个重复序列他们当时在论文中是这样评价的——―我们目前也不太清楚这些序列的生物学意义。
‖不过这个在差不多三十年之前取得的不起眼的―小发现‖在今天却绽放出了耀眼的光芒,因为如今科学家们正是利用这个小片段找到了一种可以对多种生物的基因组进行遗传改造的工具,而且这种方法操作起来还非常地简单。
就在短短的一个月之内,在包括《科学》(Science)和《自然生物技术》(Nature Biotechnology)等杂志上就已经发表了5篇相关的论文介绍这个现在被称作CRISPR–Cas系统(CRISPR–Cas systems)、简便而又实用的基因组改造新技术。
近几十年来,随着全基因组测序技术的不断成熟,我们在各种细菌和古细菌(archaea)中也陆续发现了很多成簇的、规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeat sequences,即CRISPR序列,这就是二十多年前日本科学家发现的那个序列)和CRISPR相关基因(CRISPR-associated genes, Cas gene)。
基因功能研究方法

3.2 反义RNA技术 反义RNA 技术是利用基因重组技术,构建人工表达载 体,使其离体或体内表达反义RNA ,反义RNA 能与靶mRN A形成较稳定的二聚体,从而抑制靶基因的表达。其作 用机理可能在DNA 复制、转录及翻译多水平上抑制靶 基因的表达。
23
3.3 核酶技术
核酶(Ribozyme) 技术是一类具催化活性的特殊RNA 分 子,通过碱基配对原则特异性灭活靶RNA 分子。可裂解 与其互补的mRNA及在DNA内插入DNA片段构成三链结构, 单个核酶分子可以结合多个mRNA 分子并使之在特定部 位断裂,而其本身具有较稳定的空间结构,不易受RNase 攻击,因而催化效率比反义RNA 高。常见的核酶有锤头 状、发夹状和斧头状三种,应用最多的是锤头状核酶。
5
芯片的制作
• 目前常用的基因芯片制作方法:
•
接触点样法、喷黑法、原位合成法。
• 接触点样法:是将样品直接点在基体上,其优点是仪器结 构简单、容易研制,是一种快速、经济、多功能的仪器, 可以在3.6cm2面积内点上10000个cDNA。不足之处是每个 样品都必须合成好、经过纯化、事先保存的。
6
• 喷黑法:是以定量供给的方式,通过压电晶体或其他推进 形式从很小的喷嘴内把生物样品喷射到玻璃载体上。同样 需要合成好的纯样品,包括cDNA、染色体DNA片段和抗体。 在1cm2面积上可喷射10000个点。
3
原理: 将成千上万条DNA片段(cDNA、表达序列标 签(expressed sequence tag ,EST) 或特异的寡核苷 酸片段) 按横行纵列方式有序点样在固相支持物上。 固相支持物为硝基纤维膜或尼龙膜时称为微阵列。固 相支持物改为指甲盖大小的玻片或硅片时所形成的微 阵列就称为DNA芯片。
基因靶向药物研究及临床应用的研究进展

基因靶向药物研究及临床应用的研究进展近年来,基因靶向治疗成为医学领域的热门话题,尤其是基于个体化医疗的需求,使得基因药物在治疗癌症、罕见病等疾病方面受到广泛关注。
本文将从研究现状、应用前景等方面探讨基因靶向药物的进展以及临床应用。
一、研究现状基因靶向药物是针对肿瘤等疾病中具有特殊表达的基因或基因产物的药物,与传统药物不同的是基因靶向药物是具有更精准的治疗效果。
在研究领域,基因靶向药物主要从以下几个方面进行探索:1、基因检测技术在基因靶向治疗过程中,基因检测技术的发展对治疗效果具有重要影响。
当前主要的基因检测技术包括NEXT-GENERATION SEQUENCING(NGS,下一代测序技术)、单基因测序 technology(Sanger测序)和荧光原位杂交等。
这些技术的出现,为精准医学提供了重要基础。
2、基因治疗研究基因治疗是基于一个人独特的基因组数据,去开发治疗方法和药物,是精准医疗的重要手段之一。
基因治疗的进展主要包括基因表达控制技术、RNA干扰技术、CRISPR/Cas9基因编辑技术等。
3、基于抗体的药物基于抗体的药物是指采用抗体技术制备的、具有高度特异性和亲和力的药物。
基于抗体的药物不仅在预防感染、治疗疼痛等方面发挥作用,更被广泛地应用于癌症免疫治疗领域。
该类药物目前仍处于研究阶段,但已经有一些药物进入了临床试验阶段。
二、应用前景基因靶向药物的研究取得了不小的进步,预示着其有着广阔的应用前景。
目前,基因靶向药物主要应用于癌症、罕见病、自身免疫等疾病的治疗中,具体如下:1、癌症治疗基因靶向药物在癌症治疗中,主要采用特异性小分子制剂和单克隆抗体制剂两种类型的药物。
小分子制剂因分子重量小,口服方便,不容易产生免疫反应而广泛应用于肺癌、乳腺癌、结直肠癌、胃癌等多种恶性肿瘤的治疗中。
单克隆抗体制剂因具有高度特异性、良好的稳定性和安全性等特点,也被广泛用于癌症治疗中。
2、罕见病治疗研究表明,基因靶向药物对于罕见病的治疗具有良好的效果。
基因敲除

二、基因敲除的方法
利用随机插入突变进行基因敲除。 特点:依赖于细胞内自然发生的同源染色体 的随机交换,但在体细胞内,基因同源重组 利用同源 大规模的随机插入突变理论上可实现在基因 的效率特别低( 低于 10- 6) 。增加了实际操 重组 作的工作量,限制了该项技术的应用 组范围内敲除任一基因目。 传统基因 敲除方法 该基因敲除( gene knock -out) 技术,是在转染 细胞中发生外源打靶基因与核基因组目标基因 之间的 DNA 同源重组,能够使外源基因定点 地整合到核基因组的特定位置上,从而达到改 应用随机 特点:可以分为 T-DNA 插入突变和转座子插 变细胞遗传特性的目的。 插入突变 入突变,两者是在植物中使用广泛的基因敲 除手段
二、基因敲除的方法
CRISPER/Cas系统的由来:
CRISPR(clustered regularly interspaced shortpalindromic repeats) 广泛存在于细菌和古细菌的基因组中,是细菌和古细菌的一种适应性免疫系统, 该系统可以介导外源 DNA 的降解,从而抵御病毒等外来入侵者。 1987年日本学者首次在大肠杆菌中发现该间隔重复序列。 2002年,Jansen 等将其正式命名为 CRISPR,基因编码的蛋白质统称为 CRISPR 附属蛋白(CRISPR-association proteins,Cas)。
CRISPR/Cas系统的分类:
TypeⅠ TypeⅡ TypeⅢ三种不同类型 TypeⅡ系统的主要特征是包含一个标志性的Cas9蛋白(分子质 量很大的多功能蛋白)参与crRNA的成熟以及降解入侵的噬菌 体DNA或是外源质粒。
CRISPR/Cas的基因座结构
5'端为tracrRNA基因
新型抗癌药物靶标的发现和研究

新型抗癌药物靶标的发现和研究近年来,随着人们生活方式的改变和环境污染程度的加剧,癌症已经成为了一种全球性的健康难题。
传统的癌症治疗方法往往存在着很多不足之处,如难以杀灭癌细胞和易导致严重的副作用等问题。
因此,研究新型抗癌药物的靶标成为了当下医学界的热门话题。
下面,本文将从四个方面来探讨新型抗癌药物靶标的发现和研究。
一、基因突变与癌症治疗癌症是由于某些基因突变引起的细胞恶性增殖所导致的一种严重疾病。
很多靶向治疗方案都是通过寻找癌细胞中的基因突变来治疗癌症。
此外,现在的单细胞分析技术也可以帮助科学家们更好地理解癌症发展的机制。
研究人员通过分析基因的表达谱和突变情况,可以发现很多与癌症发生和发展有关的蛋白质。
这些蛋白质可能会导致癌症的发生和发展,并且可以成为靶向治疗的目标。
一些患有特定基因突变的患者可以通过针对这些变异基因进行个性化治疗来控制疾病。
二、靶向治疗的进展和挑战靶向药物治疗已经成为了现代癌症疗法中的重要组成部分。
这些药物通过干扰癌细胞中的分子靶标,杀死癌细胞或减缓癌症的发展进程。
然而,靶向治疗面临着一些挑战。
其中一个挑战是癌细胞能够产生靶向药物的抗性。
这意味着药物可能会失去其功效,导致治疗失败。
为了克服这一问题,研究人员需要寻找新的靶向治疗方案,或者通过联合靶向治疗来提高药物的疗效。
另一个挑战是靶向药物可能会影响健康细胞,导致严重的副作用。
为了解决这个问题,科学家们需要更完善的分子靶标筛选方法,以便更好地选择特定的癌症细胞,以减少对健康细胞的影响。
三、新的治疗策略和开发新药在世界范围内,医学研究人员正在开发出新的癌症治疗策略和药物。
其中一些药物是基于针对特定细胞类型的基因编辑技术开发的,这些药物只能作用于癌细胞,并且具有更少的副作用。
这些新的治疗策略将会给癌症患者带来更好的治疗方式。
当然,新型药物开发也面临着严格的法规和法律制约,为了确保患者的安全和药物的有效性,开发药物的过程是十分复杂和耗时的。
靶向的名词解释

靶向的名词解释引言:靶向是一个在生物医学领域中经常出现的术语,它指的是通过特定的机制或方法,将治疗或诊断的重点集中在特定的靶点上。
在药物研发和临床实践中,靶向策略已经成为了一种重要的技术手段,能够提高治疗效果,减少毒副作用。
本文将对靶向的概念、分类及在不同领域中的应用进行探讨。
概念解释:靶向(Targeting)是一种针对生物体内的特定分子、细胞结构或生理过程的治疗或诊断方法,其目的是通过选择性地作用于特定靶点,实现对疾病或异常生理过程的干预。
靶向策略通常基于对病理生理过程的深入理解,通过针对特定的分子标志物或病理靶点,设计和开发具有高选择性和有效性的药物、治疗手段或诊断工具。
靶向分类:根据靶向策略所针对的对象不同,可以将其分为三大类:基因靶向、细胞靶向和分子靶向。
一、基因靶向:基因靶向是利用基因工程技术在DNA或RNA水平上作用于特定基因以调控其表达或功能的方法。
这一策略在基因治疗和基因编辑领域中得到广泛应用。
例如,通过针对癌症相关基因的抑制剂或激动剂,抑制或增加特定基因的表达,以达到治疗癌症的目的。
二、细胞靶向:细胞靶向是通过作用于特定的细胞或细胞亚群,达到调控其功能或干预病理过程的目的。
这一策略常用于免疫治疗、细胞治疗和组织工程等领域。
例如,采用经过修饰的免疫细胞,通过结合细胞表面的特异性抗原,实现对肿瘤细胞的杀伤作用。
三、分子靶向:分子靶向是基于细胞内或细胞外特定分子的存在或活性,通过设计和合成特定的抑制剂或激动剂来干预其功能。
这一策略广泛应用于药物研发和药理学研究领域。
例如,设计和合成特定的酶抑制剂,通过干扰酶的活性,达到调控疾病进程的目的。
靶向应用:靶向策略在各个领域中都有重要的应用,下面将以肿瘤治疗和药物传递为例来介绍其中的应用。
一、肿瘤治疗:靶向治疗在肿瘤治疗中具有重要的地位。
通过识别和利用肿瘤细胞表面特异性的标志物,可以设计和合成靶向抗癌药物。
这些药物能够选择性地在肿瘤细胞内发挥作用,减少对正常细胞的毒副作用。
分子生物学吕社民转基因生物与基因打靶

01
02
03
同源重组的分子过程
二、基因敲除的基本程序
1 、构建打靶载体 1)同源序列 2) 打靶载体常含两种筛选标志 neor :新霉素抗性基因,阳性筛选标志, neor基因插入用于打靶的外源DNA中,当重组后,细胞能在含新霉素的培养基中生长。
显微注射法 :
01
用显微注射仪将外源基因直接注射入实验动物受精卵中,利用外源基因整合到DNA中,从而得到转基因动物。
02
特点:导入速度快,操作简单,对DNA大小无限制,不需要载体,整合效率较高,它可以直接获得纯系。
03
缺点:需要贵重精密仪器,技术操作较难,并且外源基因的整合位点和整合的拷贝数都无法控制,易造成宿主动物基因组的插入突变,引起相应的性状改变,重则致死
法把外源基因导入精子,令其与卵子结合,受精。
接受外源基因的个体的产生
受精卵:受精卵→基因操作→注射假孕动物子宫→胚胎→个体。 胚胎干细胞:从着床前的动物胚胎中分离多功能胚胎干细胞 →基因操作→注射入动物囊胚→形成嵌合体胚胎→嵌合个体
转基因动物模型的建立
建立鼠系
染色体和基因水平:分离基因组DNA,进行斑点杂交,Southern杂交和PCR分析,原位杂交等以评估外源基因的整合情况。
基因打靶与肿瘤的研究 研究基因改变与肿瘤发生及治疗的关系
基因打靶可用于构建高效的动物反应器或植物反应器,用于药物生产。
02
03
三 、基因打靶在医学中的应用
转基因技术是在基因重组基础上建立新的多细胞真核生物的方法。这是一个具有重大意义的革命性的突破。它为我们培育新的物种提供了新的思路,为我们研究基因的功能,研究疾病发生的机理提供了有力的手段。
用于疾病相关的研究及建立疾病的动物模型
基因编辑CRISPR技术在海洋生物遗传育种的应用进展

第36卷第1期2021年2月Vol.36No.1Feb.2021大连海洋大学学报JOURNAL OF DALIAN OCEAN UNIVERSITYD0I:10.16535/ki.dlhyxb.2020-049文章编号:2095-1388(2021)01-0169-08基因编辑CRISPR技术在海洋生物遗传育种的应用进展于笛1,迟小妹,滕炜鸣打谢玺1,赵冲2,王庆志1*(1.辽宁省海洋水产科学研究院大连市海产贝类种质资源创新利用重点实验室,辽宁大连116023; 2.大连海洋大学水产与生命学院,辽宁大连116023)摘要:基因编辑(gene editing)是生命科学领域目前应用最广泛的技术之一,以其对生物内源基因改变的精确性极大地推动着生命科学的研究进程,而CRISPR技术则是目前适应范围最广、可靠性最高的一类基因编辑技术,与其他技术相比,该技术具有高效、简单等优点。
CRISPR等基因编辑技术已在动植物遗传育种、生物医疗等领域广泛应用,其中在海洋生物中的应用也日渐增多。
本文以基因编辑技术为切入点,综述了基因编辑技术的发展史、原理、应用过程,以及CRISPR技术在海洋生物遗传育种中的应用现状及发展前景,旨在为推动基因编辑技术在海洋生物资源保护与开发、遗传育种等领域的应用提供科学参考。
关键词:基因编辑;CRISPR;海洋生物;遗传育种中图分类号:Q789文献标志码:A基因编辑(gene editing)技术是指在基因组水平进行基因的定点插入/缺失突变、敲除、多位点同时突变和小片段删除等精确操作技术。
通过对基因编辑技术的研究,可以帮助人类探索生命本质,揭开疾病发生之谜,寻求疾病预防与治疗的有效途径[1]。
基因编辑的最初技术手段为同源重组介导的基因打靶技术(gene targeting),该技术虽可以准确对特定基因进行修饰,但在实际操作中存在效率低、耗时长且可能导致基因突变等问题,影响了该技术的实际应用。
基因靶向名词解释

基因靶向名词解释基因靶向是一种利用特定基因靶点进行疾病治疗的方法。
它通过识别特定的基因靶点,利用基因编辑工具对目标基因进行精确的编辑和调控,以达到治疗疾病的目的。
以下是关于基因靶向的主要名词解释:1. 靶点识别:靶点识别是基因靶向治疗的关键步骤,它涉及到确定与特定疾病相关的基因靶点。
这些靶点通常是与疾病发生、发展密切相关的基因位点。
2. 基因编辑工具:基因编辑工具是实现基因靶向的关键技术,包括CRISPR-Cas9系统、ZFNs和TALENs等。
这些工具能够精确地定位和编辑目标基因,实现对遗传信息的调控。
3. 遗传信息传递:遗传信息传递是指将治疗基因或编辑后的基因传递到患者体内,使其在体内发挥作用。
常用的传递方式包括病毒载体和非病毒载体。
4. 细胞筛选:细胞筛选是指在实验室内对细胞进行筛选,选择出具有特定特征或功能的细胞。
这些细胞可以用于后续的基因靶向治疗。
5. 疗效评估:疗效评估是指对基因靶向治疗效果的评价。
通常通过临床指标、影像学检查和实验室检查等方法进行评估。
6. 安全性评估:安全性评估是指对基因靶向治疗可能产生的副作用和不良反应进行评价。
这涉及到对患者进行长期的监测和评估。
7. 临床试验:临床试验是验证基因靶向治疗安全性和有效性的重要步骤。
通常分为Ⅰ、Ⅱ、Ⅲ期临床试验,以逐步评估治疗的安全性和有效性。
8. 基因靶向治疗:基因靶向治疗是指利用特定的基因编辑工具和传递方法,将治疗基因或编辑后的基因传递到患者体内,实现对特定疾病的精准治疗。
总之,基因靶向是一种具有巨大潜力的治疗方法,它通过对特定基因的精确调控和编辑,为许多难治性疾病提供了新的治疗手段。
关于靶基因说明
关于靶基因的说明"靶基因" :target gene"靶基因" 在学术文献中的解释1、在维甲酸诱导APL细胞分化的研究中,一般将受维甲酸及受体直接作用的基因称为靶基因,而将受靶基因蛋白质产物调控的基因称为受维甲酸调控基因 .2、靶基因是指编码特定蛋白质的结构基因,它能使转基因生物体产生新的表型.转基因鱼研究中所使用的靶基因大多是生长激素基因、抗冻蛋白基因、珠蛋白基因、金属硫蛋白基因和生长激素释放因子基因等.3、而欲进行杂交的实验样本(DNA或是RNA)称为靶基因,这指的是从试验样品中提取的丰度一定的未知序列的核苷酸链.基因靶技术(gene targeting)一、基因靶技术的原理基因靶技术的原理是设计一个靶载体,其内含有与靶位点相同的核苷酸序列,用基因转移的方法将载体导入靶细胞,通过载体与靶位点相同的核苷酸顺序间的同源重组,使外源DNA序列定点整合入靶位点,从而达到对靶位点进行定点修复或定点突变的目的。
载体分两种:一是序列插入型载体,通过与靶点序列的单交换导入外源序列;二是序列取代型载体,通过与靶点序列间的双交换,用外源序列取代靶序列。
二、基因靶技术的作用(一)确定基因表达水平目前应用最多的基因转移途径—逆转录病毒载体介导的基因转移存在二个问题,一是导入的外源基因难以生理方式进行表达;二是即使表达也不够完善。
采用基因同源重组的基因靶技术,可使病变的基因在原位得到改正,与正常基因的结构与调控环境一致,即以生理方式来进行表达。
1989年Thomphon采用小鼠胚干细胞HPRT基因的同源重组置换,使HPRT缺陷功能得到恢复。
(二)增强基因表达的稳定性与安全性通过逆转录病毒载体,将目的基因导入靶细胞后,随时间的推移,目的基因表达的稳定性会有所下降,表达的活性会慢慢的降低,有的甚至完全停止表达。
同样,外源基因与靶细胞染色体基因组的整合位点是随机的,常可导致基因的丢失和重排而失去表达活性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
?2000 667
?1999 602
?1998 542
?2009 526
?1997 502
?1996 404
?1995 331
?1994 181
?2010 128
?1993 94
?1992 49
E05.393.335
历史注释:
95
主题词详解:
The integration of exogenous DNA into the genome of an organism at sites where its expression can be suitably controlled. This integration occurs as a result of homologous recombination.
1 2 3 ...70
Top Journals Publications
?P Natl Acad Sci Usa 435
?Mol Cell Biol 425
?Science 247
?Gene Ther 222
?Nature 220
?J Biol Chem 201
?Tokyo 244
?Bethesda 216
?London 188
?Philadelphia 153
?Toronto 151
?San Francisco 131
?Baltimore 131
?Cambridge, USA 127
?Paris 118
?Orkin S 30
?Wakeham A 29
?Kmiec E 28
?Coffman T 26
?Akira S 26
?Yoshida N 26
?Alt F 25
?Deng C 25
?Penninger J 22
?Takeda S 22
?China 283
?Australia 212
?Netherlands 204
?Switzerland 167
?Italy 132
?Sweden 110
?South Korea 76
?Spain 75
?Belgium 70
?Austria 64
?1991 42
1 2
1 2 3
Top Countries Publications
?USA 4,736
?Japan 1,109
?Germany 790
?United Kingdom 652
?Canada 380
?France 353
树形结构1
研究技术
遗传学技术
基因打靶
/web/gopubmed/4?WEB01nvefjsp87l0mI4I1I00f01000j10040001&rl
Gene Targeting "Gene Targeting"[mesh]
?DNA 2,096
?Phenotype 1,922
?Recombination, Genetic 1,664
?Gene Expression 1,633
?gene expression 1,582
?Cell Line 1,547
?Tissues 1,488
在公报中,诺贝尔奖评审委员会指出,“基因靶向”的应用为人类的胚胎发育以及人类对抗衰老和疾病带来了希望。“在老鼠体内进行的‘基因靶向’已经渗透到生物医学的各个领域。在未来许多年内,它将继续帮助人类理解基因功能,继续造福人类。”公报中这样写道。
2007年度诺贝尔奖评选活动于8日在瑞典首都斯德哥尔摩拉开帷幕。瑞典卡罗林斯卡医学院当日宣布,将2007年度诺贝尔奖首个奖项——诺贝尔生理学/医学奖授予美国人马里奥·卡佩基、奥利弗·史密斯和英国人马丁·埃文斯 ,以表彰他们在干细胞研究方面尤其是“基因靶向”技术的发明方面所作的贡献。
?Nucleic Acids Res 179
?Development 163
?J Immunol 158
?J Neurosci 146
?Methods Mol Biol 141
?Genesis 135
?Embo J 129
?Gene Dev 123
?Smithies O 48
?Curiel D 47
?Bradley A 47
?Rajewsky K 46
?Mak T 42
?Capecchi M 34
?Schütz G 33
?Chambon P 32
?Noda T 32
?Maeda N 32
?Israel 47
?Taiwan 41
?Denmark 40
?Finland 38
1 2 3
1 2 3 ...35
Top Cities Publications
?Boston 332
?New York 309
?Houston 252
?St. Louis 114
?Seattle 112
?Chapel Hill 108
?San Diego 107
?Cincinnati 105
?Cambridge 102
?Kyoto 98
?Los Angeles 98
1 2 3 ...35
基因靶向技术(gene targeting)的国际研究 1985 - 2010年
已有 673 次阅读 2010-4-8 13:59 |个人分类:热点前沿|系统分类:论文交流|关键词:基因靶向技术
主题词:
基因打靶
英文名称:
Gene Targeting
树状结构号:
?Dev Biol 108
?J Clin Invest 108
?Cell 104
?Biochem Bioph Res Co 104
?Blood 93
?J Exp Med 93
1 2 3 ...70
1 2 3 ...1294
Top Terms Publications
Synonyms: Gene Targetings
1 2
Top Years Publications
?2004 805
?2001 777
?2002 773
?2006 771
?2003 747
?2005 718
?2007 711
?Genome 1,444
?Genomics 1,432
?Mice, Inbred C57BL 1,409
?Transgenes 1,397
?Base Sequence 1,395
1 2 3 ...1294
1 2 3 ...2092
Top Authors Publications
?Gene Targeting 10,088
?Genes 8,218
?Animals 7,999
?Mice 6,285
?Humans 3,933
?Proteins 3,648
?Mutation 2,441
?Mice, Knockout 2,371
1 2 3 ...2092
“基因靶向”技术,也通常被称作基因敲除。“基因靶向”技术利用胚胎干细胞改造老鼠体内的特定基因。在“基因靶向”技术的帮助下,科学家可以使实验鼠体内的一些“不活跃”基因失去作用,从而发现这些基因的实际功能。科学家希望借此发现人类一些疑难杂症在分子水平上的发病原因,并最终找到治疗途径。目前,“基因靶向”已被应用于对囊肿性纤维化、心脏病、糖尿病、阿尔茨海默症和癌症的研究。
10,088 of 33,384 documents semantically analyzed
Term: Gene Targeting
Description: The integration of exogenous DNA into the genome of an organism at sites where its n can be suitably controlled. This integration occurs as a result of homologous recombination.