聚类分析算法解析.doc
聚类分析_精品文档

1聚类分析内涵1.1聚类分析定义聚类分析(Cluste.Analysis)是一组将研究对象分为相对同质的群组(clusters)的统计分析技术.也叫分类分析(classificatio.analysis)或数值分类(numerica.taxonomy), 它是研究(样品或指标)分类问题的一种多元统计方法, 所谓类, 通俗地说, 就是指相似元素的集合。
聚类分析有关变量类型:定类变量,定量(离散和连续)变量聚类分析的原则是同一类中的个体有较大的相似性, 不同类中的个体差异很大。
1.2聚类分析分类聚类分析的功能是建立一种分类方法, 它将一批样品或变量, 按照它们在性质上的亲疏、相似程度进行分类.聚类分析的内容十分丰富, 按其聚类的方法可分为以下几种:(1)系统聚类法: 开始每个对象自成一类, 然后每次将最相似的两类合并, 合并后重新计算新类与其他类的距离或相近性测度. 这一过程一直继续直到所有对象归为一类为止. 并类的过程可用一张谱系聚类图描述.(2)调优法(动态聚类法): 首先对n个对象初步分类, 然后根据分类的损失函数尽可能小的原则对其进行调整, 直到分类合理为止.(3)最优分割法(有序样品聚类法): 开始将所有样品看成一类, 然后根据某种最优准则将它们分割为二类、三类, 一直分割到所需的K类为止. 这种方法适用于有序样品的分类问题, 也称为有序样品的聚类法.(4)模糊聚类法: 利用模糊集理论来处理分类问题, 它对经济领域中具有模糊特征的两态数据或多态数据具有明显的分类效果.(5)图论聚类法: 利用图论中最小支撑树的概念来处理分类问题, 创造了独具风格的方法.(6)聚类预报法:利用聚类方法处理预报问题, 在多元统计分析中, 可用来作预报的方法很多, 如回归分析和判别分析. 但对一些异常数据, 如气象中的灾害性天气的预报, 使用回归分析或判别分析处理的效果都不好, 而聚类预报弥补了这一不足, 这是一个值得重视的方法。
聚类分析法解读

②根据原始数据的性质,选择适当变换方法,再根 据不同的变换方法选择不同的距离或相似系数。如标准化变 换处理下,相关相似系数和夹角余弦一支;又如原始数据在 进行聚类分析之前已经对变量的相关性做了处理,则通常可 采用欧式距离而不必选用斜交空间距离。再如选择距离时,
2018/12/4 12
9.2 聚类分析的种类
1.2 聚类分析的种类
1.2.1 系统聚类分析法 1.2.2 动态聚类分析法
1.2.3 模糊聚类分析法
1.2.4 图论聚类分析法
2018/12/4 13
1.2.1 系统聚类分析法
1.基本思想和分析步骤 (1)基本思想 系统聚类分析的基本思想是,把n个样品看成p维(p个
指标)空间的点,而把每个变量看成p维空间的坐标轴,根据
2018/12/4 6
xij
1.1 聚类与聚类分析
1.1.2聚类分析的原理 ▪中心化变换 对于一个样本数据,观测p各指标,n个样品的数据资料 x12 x1 p x11 阵为 x x x 21 22 2p X x x x n2 np n1
2018/12/4
2018/12/4 3
1.1 聚类与聚类分析
1.1.2聚类分析的原理 (1)聚类分析的定义 聚类分析是将样品或变量按照它们性质上的亲疏程度 进行分类的多元统计分析方法。进行聚类分析时,用来描述 物品或变量的亲疏程度通常有两个途径,一个是把每个样品 或变量看成是多维空间上的一个点,在多维坐标中,定义点 与点,类与类之间的距离,用点与点间距离来描述作品或变 量之间的亲疏程度;二是计算样品或变量的相似系数,用相 似系数来描述样品或变量之间的亲疏程度。
聚类分析算法实验报告(3篇)

第1篇一、实验背景聚类分析是数据挖掘中的一种重要技术,它将数据集划分成若干个类或簇,使得同一簇内的数据点具有较高的相似度,而不同簇之间的数据点则具有较低相似度。
本实验旨在通过实际操作,了解并掌握聚类分析的基本原理,并对比分析不同聚类算法的性能。
二、实验环境1. 操作系统:Windows 102. 软件环境:Python3.8、NumPy 1.19、Matplotlib 3.3.4、Scikit-learn0.24.03. 数据集:Iris数据集三、实验内容本实验主要对比分析以下聚类算法:1. K-means算法2. 聚类层次算法(Agglomerative Clustering)3. DBSCAN算法四、实验步骤1. K-means算法(1)导入Iris数据集,提取特征数据。
(2)使用Scikit-learn库中的KMeans类进行聚类,设置聚类数为3。
(3)计算聚类中心,并计算每个样本到聚类中心的距离。
(4)绘制聚类结果图。
2. 聚类层次算法(1)导入Iris数据集,提取特征数据。
(2)使用Scikit-learn库中的AgglomerativeClustering类进行聚类,设置链接方法为'ward'。
(3)计算聚类结果,并绘制树状图。
3. DBSCAN算法(1)导入Iris数据集,提取特征数据。
(2)使用Scikit-learn库中的DBSCAN类进行聚类,设置邻域半径为0.5,最小样本数为5。
(3)计算聚类结果,并绘制聚类结果图。
五、实验结果与分析1. K-means算法实验结果显示,K-means算法将Iris数据集划分为3个簇,每个簇包含3个样本。
从聚类结果图可以看出,K-means算法能够较好地将Iris数据集划分为3个簇,但存在一些噪声点。
2. 聚类层次算法聚类层次算法将Iris数据集划分为3个簇,与K-means算法的结果相同。
从树状图可以看出,聚类层次算法在聚类过程中形成了多个分支,说明该算法能够较好地处理不同簇之间的相似度。
聚类分析的结果分析和编程

聚类分析方法一:最短距离法程序:proc cluster data=work.zidan outtree=tree method=sin notie;id g;proc tree horizontal;id g;run;结果:Eigenvalue Difference Proportion Cumulative1 29740435.8 25650007.9 0.8718 0.87182 4090427.9 3812173.1 0.1199 0.99173 278254.8 274011.4 0.0082 0.99994 4243.4 4242.5 0.0001 1.00005 0.9 0.9 0.0000 1.00006 0.0 0.0 0.0000 1.00007 0.0 0.0000 1.0000Cluster HistoryNormMinNCL ------Clusters Joined------- FREQ Dist5 26 2 0.28954 1 CL5 3 0.33333 CL4 4 4 0.50012 3 5 2 0.70171 CL3 CL2 6 1.0154方法二:最长距离法程序:proc cluster data=work.zidan outtree=tree method=complete;id g;proc tree horizontal;id g;run;结果:Eigenvalues of the Covariance MatrixEigenvalue Difference Proportion Cumulative1 29740435.8 25650007.9 0.8718 0.87182 4090427.9 3812173.1 0.1199 0.99173 278254.8 274011.4 0.0082 0.99994 4243.4 4242.5 0.0001 1.00005 0.9 0.9 0.0000 1.00006 0.0 0.0 0.0000 1.00007 0.0 0.0000 1.0000Root-Mean-Square Total-Sample Standard Deviation = 2207.564Mean Distance Between Observations = 7428.48(变量之间的平均距离)Cluster HistoryNorm TMax iNCL ------Clusters Joined------- FREQ Dist e5 26 2 0.28954 1 4 2 0.50013 3 5 2 0.70172 CL4 CL5 4 0.72971 CL2 CL3 6 1.7694此结果表示的是:类的个数,合并类的名字,新类中的变量个数和最长正规化距离。
聚类分析算法

第二章聚类分析2·4 聚类的算法2.4.1 聚类的技术方案⑴简单聚类根据相似性阈值和最小距离原则聚类∀x i∈Ω={ x1,x2,…,x n} = ω1⋃ω2⋃…⋃ωc;if D(xi ,mj)≤T, mj=(1/nj)∑xi(j),xi(j)∈ωj,nj是ωj中的样本个数,T是给定的阀值。
Then xi ∈ωi类心一旦确定将不会改变。
⑵谱系或层次聚类按最小距离原则不断进行两类合并类心不断地修正,但模式类别一旦指定后就不再改变。
⑶依据准则函数动态聚类影响聚类结果的主要因数:类心、类别个数、模式输入顺序。
所谓动态聚类,是指上述因数在聚类过程中是可变的。
规定一些分类的目标参数,定义一个能刻划聚类过程或结果优劣的准则函数,聚类过程就是使准则函数取极值的优化过程。
这类方法有—均值法、ISODATA法、近邻函数法以及运用图论理论的最小张树法。
2.4.2 简单聚类方法㈠根据相似性阈值和最小距离原则的简单聚类方法⒈条件及约定设待分类的模式为,选定类内距离门限。
⒉算法思想计算模式特征矢量到聚类中心的距离并和门限比较而决定归属该类或作为新的一类中心。
通常选择欧氏距离。
⒊算法原理步骤⑴取任意的一个模式特征矢量作为第一个聚类中心。
例如,令第一类的中心。
⑵计算下一个模式特征矢量到的距离。
若,则建立新的一类,其中心;若,则。
⑶假设已有聚类中心,计算尚未确定类别的模式特征矢量到各聚类中心的距离,如果,则作为新的一类的中心,;否则,如果( 2-4-1)则指判。
检查是否所有的模式都分划完类别,如都分划完了则结束;否则返到⑶。
⒋性能●计算简单。
●聚类结果很大程度上依赖于距离门限的选取、待分类特征矢量参与分类的次序和聚类中心的选取。
当有特征矢量分布的先验知识来指导门限及初始中心的选取时,可以获得较合理结果。
⒌改进通常采用试探法,选用不同的门限及模式输入次序来试分类,并对聚类结果。
例如,计算每一聚类中心与该类中最远样本点进行检验,即用聚类准则函数J1的距离,或计算类内及类间方差,用这些结果指导及的重选。
聚类分析详解

聚类分析详解sklearn—聚类分析详解(聚类分析的分类;常⽤算法;各种距离:欧⽒距离、马⽒距离、闵式距离、曼哈顿距离、卡⽅距离、⼆值变量距离、余弦相似度、⽪尔森相关系数、最远(近)距离、重⼼距离)这⼀章总结的很痛苦,打公式费时费⼒。
⽂章⽬录1.聚类分析1.1聚类⽅法1.2 常见聚类算法:1.3 cluster提供的聚类算法及其使⽤范围2. 各种距离2.1 连续性变量的距离2.1.1 欧⽒距离2.1.2 曼哈顿距离2.1.3 切⽐雪夫距离2.1.4 闵可夫斯基距离2.1.5 标准欧式距离2.1.6 马⽒距离2.1.7 补充:距离判别法,同样⽤到马⽒距离2.2 离散型变量距离2.2.1 卡⽅距离2.2.2 Phi距离2.2.3 ⼆值变量距离2.2.4 Jaccard系数2.3基于相似系数的相似性度量(⽤相似度表⽰距离)2.3.1 余弦相似度2.3.2 汉明距离2.3.3 Jaccard相似系数2.3.4 ⽪尔森相关系数2.4 个体与类以及类间的亲疏关系度量2.4.1 最远(近)距离2.4.2 组间平均链锁距离2.4.3 组内平均链锁距离2.4.4 重⼼距离2.4.5 离差平⽅和距离(Ward⽅法)3. 常⽤的聚类⽬标函数3.1 连续属性的SSE3.2 ⽂档数据的SSE计算公式:3.3 簇$E_i$的聚类中⼼$e_i$计算公式:1.聚类分析1.1聚类⽅法类别包括的主要算法划分(分裂)⽅法K-Means算法(均值)、K-medoids算法(中⼼点)、K-modes算法(众数)、k-prototypes算法、CLARANS(基于选择)层次分析BIRCH算法(平衡迭代规约)、CURE算法(点聚类)、CHAMELEON(动态模型)基于密度DBSCAN(基于⾼密度连接区域)、DENCLUE(密度分布函数)、OPTICS(对象排序识别)基于⽹格STING(统计信息⽹络)、CLIOUE(聚类⾼维空间)、WAVE-CLUSTER(⼩波变换)基于模型统计学⽅法、神经⽹络此外还有,最优分割法(有序样本聚类)、模糊聚类法(应⽤模糊集理论)、图论聚类…这个⽔太深了,看了半天是不是发现⾃⼰就只会k均值和birch系统聚类啊…真真真的学⽆⽌境1.2 常见聚类算法:算法名称描述K-Means K均值算法是⼀种快速聚类算法,在最⼩化误差函数的基础上将数据划分为预定的K簇。
聚类分析方法详细介绍和举例

聚类分析⽅法详细介绍和举例聚类分析例如:下表是1999年中国省、⾃治区的城市规模结构特征的⼀些数据,可通过聚类分析将这些省、⾃治区进⾏分类,具体过程如下:省、⾃治区⾸位城市规模(万⼈)城市⾸位度四城市指数基尼系数城市规模中位值(万⼈)京津冀699.70 1.4371 0.9364 0.7804 10.880 ⼭西179.46 1.8982 1.0006 0.5870 11.780 内蒙古111.13 1.4180 0.6772 0.5158 17.775 辽宁389.60 1.9182 0.8541 0.5762 26.320 吉林211.34 1.7880 1.0798 0.4569 19.705 ⿊龙江259.00 2.3059 0.3417 0.5076 23.480⼀、聚类分析的数据处理1、地理数据的对数变换:原始数据⾃然对数变换省、⾃治区⾸位城市规模(万⼈)城市⾸位度四城市指数基尼系数城市规模中位值(万⼈)⾸位城市规模(万⼈)城市⾸位度四城市指数基尼系数城市规模中位值(万⼈)京津冀699.7 1.4371 0.9364 0.7804 10.88 6.5507 0.3626 -0.0657 -0.2479 2.3869 ⼭西179.46 1.8982 1.0006 0.587 11.78 5.1900 0.6409 0.0006 -0.5327 2.4664 内蒙古111.13 1.418 0.6772 0.5158 17.775 4.7107 0.3492 -0.3898 -0.6620 2.8778 辽宁389.6 1.9182 0.8541 0.5762 26.32 5.9651 0.6514 -0.1577 -0.5513 3.2703 吉林211.34 1.788 1.0798 0.4569 19.705 5.3535 0.5811 0.0768 -0.7833 2.9809 ⿊龙江259 2.3059 0.3417 0.5076 23.48 5.5568 0.8355 -1.0738 -0.6781 3.1561 2、地理数据标准化:⾃然对数变换标准差标准化数据⾸位城市规模(万⼈)城市⾸位度四城市指数基尼系数城市规模中位值(万⼈)⾸位城市规模(万⼈)城市⾸位度四城市指数基尼系数城市规模中位值(万⼈)6.5507 0.3626 -0.0657 -0.2479 2.3869 1.5572 -1.1123 0.4753 1.7739 -1.30255.1900 0.6409 0.0006 -0.5327 2.4664 -0.5698 0.3795 0.6309 0.2335 -1.08204.7107 0.3492 -0.3898 -0.6620 2.8778 -1.3189 -1.1841 -0.2851 -0.4660 0.05935.9651 0.6514 -0.1577 -0.5513 3.2703 0.6419 0.4356 0.2594 0.1330 1.14835.3535 0.5811 0.0768 -0.7833 2.9809 -0.3142 0.0588 0.8096 -1.1218 0.34535.5568 0.8355 -1.0738 -0.6781 3.1561 0.0037 1.4225 -1.8900 -0.5526 0.8316⼆、采⽤欧⽒距离,求出欧式距离系数欧式距离系数表d1 d2 d3 d4 d5 d6 d1 0d2 1.3561 0d3 1.7735 1.0618 0d4 1.5479 1.1484 1.2891 0d5 1.7936 0.9027 0.9235 0.8460 0d6 2.2091 1.5525 1.5312 1.1464 1.4006 0三、最短距离法进⾏聚类分析如下:第⼀步:以欧式距离作为分类统计量,得出初始距离矩阵D(0)D(0)表G1 G2 G3 G4 G5 G2 1.3561G3 1.7735 1.0618G4 1.5479 1.1484 1.2891G5 1.7936 0.9027 0.9235 0.8460G6 2.2091 1.5525 1.5312 1.1464 1.4006第⼆步:在D(0)中,最⼩元素为D54=0.846,将G5与G4合并成⼀新类G7,G7={G5,G4},然后在计算新类G7与其它各类间的距离D7,1= min(d41,d51)=min(1.5479, 1.7936)= 1.5479D7,2= min(d42,d52) = min(1.1484,0.9027)= 0.9027D7,3= min(d43,d53) = min(1.2891, 0.9235)= 0.9235D7,6= min(d64,d65) = min(1.1464, 1.4006)= 1.1464第三步:作D (1)表,先从D(0)表中删除G4,G5类所在⾏列的所有元素,然后再把新计算出来的G7与其它类间的距离D71,D72,D73填到D (0)中,得D(I)表第四步:在D (1)中,最⼩元素为D72=0.9027,将G7与G2合并成⼀新类G8,G8={G2,G7}={G2,G4,G5},然后在计算新类G8与其它各类间的距离D8,1= min(d21,d71)= min(1.3561, 1.5479)= 1.3561 D8,3= min(d23,d73) = min(1.0618, 0.9235)= 0.9235 D8,6= min(d62,d76)= min(1.5525, 1.1464)= 1.1464第五步:作D (2)表,先从D(1)表中删除G2,G7类所在⾏列的所有元素,然后再把新计算出来的G8与其它类间的距离D81,D83,D86填到D (2)中,得D(2)表D (2)表G1 G3 G6 G3 1.7735 G6 2.2091 1.5312 G81.35610.92351.1464第六步:在D (2)中,最⼩元素为D38=0.9235,将G8与G3合并成⼀新类G9,G9={G3,G8},然后在计算新类G9与其它各类间的距离D9,1= min(d13,d18) = min(1.7735, 1.3561)= 1.3561 D9,6= min(d36,d86) = min(1.5312, 1.1464)= 1.1464第七步:作D (3)表,先从D(2)表中删除G3,G8类所在⾏列的所有元素,然后再把新计算出来的G9与其它类间的距离D91 ,D96填到D (3)中,得D(3)表第⼋步:在D (3)中,最⼩元素为D69= 1.1464,将G6与G9合并成⼀新类G10,G10={G6,G9},然后在计算新类G10与其它各类间的距离D10,1= min(d16,d69) = min(2.2091, 1.1464)= 1.1464第九步:作D (4)表,先从D(3)表中删除G6,G9类所在⾏列的所有元素,然后再把新计算出来的G10与其它类间的距离D10,1填到D (4)中,得D(4)表D (1)表G1 G2 G3G6G2 1.3561 G3 1.7735 1.0618 G6 2.2091 1.5525 1.5312 G71.54790.90270.9235 1.1464D (3)表G1 G6 G6 2.2091 G9 1.3561 1.1464D(4)表G1G10 1.1464G11={G10.G1}由此表可知,G10和G1类最后合成了⼀类,计算过程结束。
聚类分析的算法及应用共3篇

聚类分析的算法及应用共3篇聚类分析的算法及应用1聚类分析的算法及应用聚类分析(Cluster Analysis)是一种数据分析方法,它根据数据的相似度和差异性,将数据分为若干个组或簇。
聚类分析广泛应用于数据挖掘、文本挖掘、图像分析、生物学、社会科学等领域。
本文将介绍聚类分析的算法及应用。
聚类分析的算法1. 基于距离的聚类分析基于距离的聚类分析是一种将数据点归类到最近的中心点的方法。
该方法的具体实现有单链接聚类(Single-Linkage Clustering)、完全链接聚类(Complete-Linkage Clustering)、平均链接聚类(Average-Linkage Clustering)等。
其中,单链接聚类是将每个点最近的邻居作为一个簇,完全链接聚类是将所有点的最小距离作为簇间距离,平均链接聚类是将每个点和其他点的平均距离作为簇间距离。
2. 基于密度的聚类分析基于密度的聚类分析是一种将数据点聚集在高密度区域的方法。
该方法的主要算法有密度峰(Density Peak)、基于DBSCAN的算法(Density-Based Spatial Clustering of Applications with Noise)等。
其中,密度峰算法是通过计算每个点在距离空间中的密度,找出具有局部最大密度的点作为聚类中心,然后将其余点分配到聚类中心所在的簇中。
而基于DBSCAN的算法则是将高密度点作为聚类中心,低密度点作为噪声,并将边界点分配到不同的聚类簇中。
3. 基于层次的聚类分析基于层次的聚类分析是通过不断将相似的点合并为一个组或将簇一分为二的方法。
该方法的主要算法有自顶向下层次聚类(Top-Down Hierarchical Clustering)和自底向上层次聚类(Bottom-Up Hierarchical Clustering)。
其中,自顶向下层次聚类从所有数据点开始,将数据点分为几个组,并不断通过将组合并为更大的组的方式,直到所有的数据点都被合并。
聚类分析解析课件

类间距的度量
类:一个不严格的定义
定义9.1:距离小于给定阀值的点的集合 类的特征
◦ 重心:均值 ◦ 样本散布阵和协差阵 ◦ 直径
类间距的定义
最短距离法 最长距离法 重心法 类平均法 离差平方和法 等等
最小距离法(single linkage method)
极小异常值在实际中不多出现,避免极 大值的影响
类的重心之间的距离
对异常值不敏感,结果更稳定
离差平方和法(sum of squares
method或ward method)
W代表直径,D2=WM-WK-WL
即
DK2L
nL nk nM
XK XL XK XL
Cluster K
Cluster M
Cluster L
◦ 对异常值很敏感;对较大的类倾向产生较大的距 离,从而不易合并,较符合实际需要。
如表9.2所示,每个样品有p个指标,共 有n个样品
每个样品就构成p维空间中的一个点
:第i个样品的第k个指标对应的取值
◦ i=1……n; k=1……p
:第i个样品和第j个样品之间的距离
◦ i=1……n; j=1……n
点间距离测量问题
样品间距离与指标间距离 间隔尺度、有序尺度与名义尺度 数学距离与统计距离 相似性与距离:一个硬币的两面
类图上发现相同的类
饮料数据
16种饮料的热量、咖啡因、钠及价格四种变量
SPSS实现
选择Analyze-Classify-Hierarchical Cluster, 然 后 把 calorie ( 热 量 ) 、 caffeine ( 咖 啡
因)、sodium(钠)、price(价格)选入 Variables, 在Cluster选Cases(这是Q型聚类:对观测 值聚类),如果要对变量聚类(R型聚类) 则选Variables, 为 了 画 出 树 状 图 , 选 Plots , 再 点 Dendrogram等。 可以在Method中定义点间距离和类间距 离
机器学习之聚类分析

机器学习之聚类分析什么是聚类分析?聚类分析是机器学习领域中常用的无监督学习方法之一。
它的目标是将一组样本分成若干个不相交的子集,使得同一个子集中的样本彼此相似,不同子集中的样本差异较大。
聚类算法通过找到样本之间的相似性或距离来完成这一任务。
聚类分析的常见应用聚类分析在许多领域都有广泛的应用,下面介绍一些常见的应用场景:社交网络分析在社交网络分析中,聚类分析可以用于识别相关的用户群体。
通过对用户的社交行为进行聚类,可以发现潜在的社交圈子或兴趣群体。
这对于社交网络推荐系统和社交媒体营销策略非常有用。
市场细分在市场营销中,聚类分析可以用于将消费者划分为不同的市场细分。
通过对消费者的购买行为、喜好和偏好进行聚类分析,可以发现不同市场细分的消费者群体,从而制定针对性的营销策略。
图像分析在图像处理和计算机视觉领域,聚类分析可以用于图像分割和目标检测。
通过对图像像素进行聚类,可以将图像分成不同的区域,从而实现图像分割。
对于目标检测,聚类算法可以帮助识别图像中的不同目标,并将它们分成不同的类别。
聚类分析的常用算法在聚类分析中,有多种算法可以用于找到样本之间的相似性或距离。
下面介绍一些常用的聚类算法:K-均值聚类K-均值聚类是最常用的聚类算法之一。
它将样本划分为 K 个不相交的簇,每个簇的中心的是该簇中所有样本的均值。
该算法的主要思想是通过迭代的方式不断优化样本到簇中心的距离,直到达到收敛。
层次聚类层次聚类是一种将样本逐步合并或分裂的聚类算法。
它可以得到一个树状的聚类结构,可以根据需要选择不同的聚类数量。
层次聚类算法有两种常见的方法:自下而上的凝聚层次聚类和自上而下的分裂层次聚类。
密度聚类密度聚类是一种基于样本之间密度的聚类算法。
它通过找到样本密度较高的区域,并将其作为簇的中心。
密度聚类算法对于数据分布不规则或包含噪声的情况下表现较好。
使用Python进行聚类分析在 Python 中,有许多库可以用于聚类分析。
其中,scikit-learn 是一个常用的机器学习库,它提供了丰富的聚类算法实现。
第8章聚类分析基本概念和算法

C2
C5
...
p1
p2
p3
p4
p9
p10 p11 p12
中间步骤
我们希望合并两个最邻近的cluster (C2 and C5) 并更新临近度
矩阵
C1 C2 C3 C4 C5
C1
C3 C4
C1
C2 C3
C4 C5
Proximity Matrix
二元数据的相似性度量
两个仅包含二元属性的对象之间的相似性度量 也称相似系数
两个对象的比较导致四个量
f00 = x取0并且y取0的属性个数 f01 = x取0并且y取1的属性个数 f10 = x取1并且y取0的属性个数 f11 = x取1并且y取1的属性个数
简单匹配系数
SMC = 值匹配的属性个数 / 属性个数 = (f11 +f00) / (f01 + f10 + f11 + f00)
二分k均值算法
初始化簇表,使之包含由所有的点组成的簇。 Repeat
从簇表中取出一个簇。 for i=1 to 实验次数 do
使用基本k均值,二分选定的簇。 end for 从二分实验中选择具有最小总sse的两个簇。 将这两个簇添加到簇表中。 Until 簇表中包含k个簇。
K means 的优点与缺点
proximity matrix
p1 p2 p3 p4 p5 . . .
p1
p2 p3
p4
p5
.
.
. Proximity Matrix
...
p1
p2
p3
p4
p9
p10 p11 p12
中间步骤
生物大数据技术中的聚类分析方法

生物大数据技术中的聚类分析方法生物大数据技术的迅猛发展为科研人员提供了丰富的数据资源,然而如何从庞大的数据中提取有意义的信息,成为了生物信息学研究的重要课题之一。
在生物大数据分析中,聚类分析方法被广泛应用,它通过将相似的样本或数据点归为一类,从而揭示数据集中的隐藏模式和结构。
本文将介绍生物大数据技术中的聚类分析方法,并剖析其应用于生物学研究的意义。
聚类分析是一种无监督学习的方法,其目标是根据数据点的相似性,将它们划分为不同的群组或簇。
在生物领域,聚类分析方法适用于多种应用场景,如基因表达数据的分类、蛋白质序列的聚类和进化树的构建等。
下面将介绍几种常用的生物大数据技术中的聚类分析方法。
1. K-means聚类算法:K-means算法是一种常见的聚类分析方法,其基本思想是将数据集分成K个簇,使得簇内的数据点相互之间的距离最小,并使得簇与簇之间的距离最大化。
在生物学研究中,K-means算法可以用于基因表达数据的分类。
通过将基因表达矩阵中的每一行看作一个数据点,将基因按照表达水平划分为不同的簇,可以帮助研究人员发现基因表达的模式和规律。
2. 层次聚类分析:层次聚类分析是一种基于样本之间相似性的聚类方法,其通过计算数据点之间的距离或相异度,并逐步将相似的数据点合并为一个簇。
这种方法能够形成一棵聚类树,可以将样本按照不同的层次进行分类。
在生物学研究中,层次聚类分析可以应用于多个领域,如RNA测序数据的组织分类、蛋白质序列的聚类和系统发育关系的推断等。
3. 基于密度的聚类方法:基于密度的聚类方法将样本点集划分为不同的簇,其中每个簇代表着一个高密度的区域,被低密度或离群点所包围。
这种聚类方法适用于具有复杂形状和不规则分布的数据集。
在生物学研究中,基于密度的聚类方法可以用于DNA甲基化数据的挖掘、蛋白质相互作用网络的簇划分等。
4. 基于模型的聚类方法:基于模型的聚类方法是在给定概率模型的情况下,将数据点分配到不同的簇中。
聚类分析讲义范文

聚类分析讲义范文一、聚类分析的基本原理聚类分析的基本原理是通过计算数据点之间的相似性度量,将相似性高的数据点归为一类,从而形成簇(cluster)。
相似性度量方法主要包括欧几里得距离、曼哈顿距离、切比雪夫距离、闵可夫斯基距离等。
欧几里得距离是最常用的相似性度量方法,其计算公式为:d(x, y) = sqrt((x1-y1)^2 + (x2-y2)^2 + ... + (xn-yn)^2)聚类分析的基本步骤包括:1.选择合适的相似性度量方法和聚类算法。
2.初始化聚类中心,将数据点分配到最近的聚类中心。
3.更新聚类中心,重新计算每个聚类中心的位置。
4.重复第2步和第3步,直到聚类中心的位置不再变化为止。
5.输出聚类结果。
二、聚类分析的常用算法1. K-means算法:K-means算法是最常用的聚类算法之一、其核心思想是在每次迭代中,计算每个数据点到所有聚类中心的距离,并将每个数据点分配到距离最近的聚类中心。
然后,重新计算每个聚类中心的位置。
重复执行这两个步骤,直到聚类中心的位置不再变化。
K-means算法的优势是简单快速,但对初始聚类中心的选择较为敏感。
2.层次聚类算法:层次聚类算法通过计算所有数据点之间的相似性,构建一个层次性的聚类结果。
这个结果可以表示为一个树状结构,其中每个节点代表一个聚类。
层次聚类算法的优势是不需要预先指定聚类个数,但计算复杂度较高。
3.密度聚类算法:密度聚类算法将聚类看作是在数据空间中找到高密度区域的过程。
该算法通过计算每个数据点在其邻域内的密度,将高密度区域作为簇的候选。
然后,通过在高密度区域之间构建连接来将簇进行合并。
密度聚类算法的优势在于可以发现任意形状和大小的簇,对于噪声和异常点具有较好的鲁棒性。
三、聚类分析的评估方法四、聚类分析的应用聚类分析在市场调研中可以帮助企业识别相似的顾客群体,从而定制合适的产品和推广策略。
在社交网络分析中,聚类分析可以用来发现具有相似兴趣和行为的用户群体,从而进行精准广告投放。
聚类分析

聚类分析算法作为一种有效的数据分析方法被广泛应用于数据挖掘、机器学习、图像分割、语音识别、生物信息处理等,聚类算法还可以应用于商业分析,它可以帮助市场决策人员从消费者数据库中区分出不同的消费群体,并且概括出每一类消费者的消费模式或习惯。
从本质来说聚类算法是将总体中的个体分类以发现数据中的结构,希望一个类中的个体彼此接近或相似,而与其它类中的个体相异,这样就可以对划分出来的每一类进行深入的分析从而概括出每一类的特点。
1 . 聚类分析算法的基本定义给定一个对象集合X = {x 1 , x2 , , xn},假设每个对象 xi , i = 1...n 含有 m 个特征,在此用向量的方式来表示对象的特征,)....,(2,1m i l l l x =,聚类分析的过程就是根据对象的特征来分析对象之间的相似程度,并根据某种聚类决策准则来获得聚类结果。
通常聚类分析算法一般包含四个部分:(1)特征获取与选择;(2)计算相似度;(3)分组;(4)聚类结果展示。
特征获取与选择就是为了获得能够恰当表示对象属性的数据,并且减少数据的冗余度。
计算相似度就是根据对象的特征来计算对象之间的相似程度,在聚类过程中可能一次性的计算所有对象之间的相似度信息,也可能在聚类分析的过程中按需要来计算对象之间的相似度信息。
分组就是根据对象之间的相似程度来判断对象之间的类别信息,将类似的对象分组到同一个类中,不相似的对象分组到不同的类中。
聚类结果展示可以只是简单的输出对象分组信息,也可以用图形化的方式来展示聚类结果,但是目前的研究趋势要求算法能够自动的归纳出每个不同的分组所代表的类信息并抽象出类中对象的共同特性。
2. 聚类分析方法聚类算法主要分为层次化聚类方法,划分式聚类方法,基于密度的聚类方法,基于网格的聚类方法,基于核的聚类算法,基于谱的聚类方法,基于模型的聚类方法,基于遗传算法的聚类方法,基于 SVM 的聚类方法,基于神经网络的聚类方法等等.1)划分式聚类方法对于一个给定的 n 个数据对象的数据集,采用目标函数最小化的策略,初始时选择一定量的聚类中心或数据点,通过某种原则把数据划分到各个组中,每个组为一个簇。
聚类分析.
➢ 总体平方误差: E2= e12+ e22=19.36+8.12=27.48
➢ 依据距离重心M1和M2的最小距离,再分配所 有的样本时,类内样本重新分布将是:
d(M ,x )=((0-1.66) +(2-0.66) ) =2.14 x1=(0,2),x2=(0,0),x计算的好处是提高了分治方法的效率。
SMC(Y,X3)=2/6=0.
d(M1,x2)=1. 例如:仍然采用上例的数据集。
x4 → C2 ={x4} →M2={5,0} 第一样本x1为第一个类C1={x1}。 通常情况下,当样本从一个类被分配到另一个类时,如果不会出现总体误差减小的情况,便满足收敛准则。
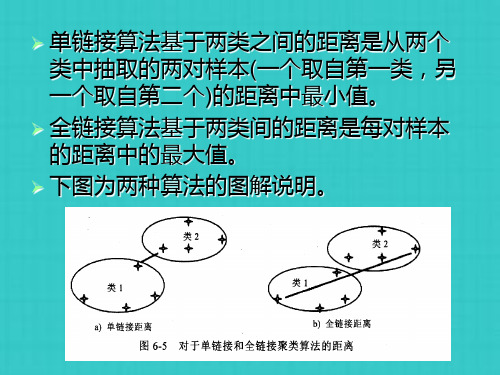
➢ 第二步:单链接算法。
按最小距离合并x2和x3,生成新类 {x2,x3},其距离为1.5。 x4和x5合并成 一个新类{x4,x5},其距离为2。同时, 类{x2,x3}和{x1}间的最小距离也是2.0, 将其合并成一个新类{x1,x2,x3} ,其距 离为2。最后,两个类{x1,x2,x3}和 {x4,x5}可以以更高的级别进行合并,其 最小单链接距离为3.5。树状图如下:
➢ 第一步:计算欧氏距离。
d(x1,x2)=2, d(x1,x3)=2.5 d(x1,x4)=5.4 d(x1,x5)=5 d(x2,x3)=1.5, d(x2,x4)=5, d(x2,x5)=5.29 d(x3,x4)=3.5, d(x3,x5)=4.03 d(x4,x5)=2
按升序排列:
d(x2,x3)=1.5,d(x1,x2)=2, d(x4,x5)=2, d(x1,x3)=2.5, d(x3,x4)=3.5,d(x3,x5)=4.03,d(x2,x4)=5,d(x1,x5)=5, d(x2,x5)=5.29, d(x1,x4)=5.39
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
聚类分析算法解析一、不相似矩阵计算1.加载数据data(iris)str(iris)分类分析是无指导的分类,所以删除数据中的原分类变量。
iris$Species<-NULL2. 不相似矩阵计算不相似矩阵计算,也就是距离矩阵计算,在R中采用dist()函数,或者cluster包中的daisy()函数。
dist()函数的基本形式是dist(x, method = "euclidean", diag = FALSE, upper = FALSE, p = 2)其中x是数据框(数据集),而方法可以指定为欧式距离"euclidean", 最大距离"maximum", 绝对值距离"manhattan", "canberra", 二进制距离非对称"binary" 和明氏距离"minkowski"。
默认是计算欧式距离,所有的属性必须是相同的类型。
比如都是连续类型,或者都是二值类型。
dd<-dist(iris)str(dd)距离矩阵可以使用as.matrix()函数转化了矩阵的形式,方便显示。
Iris数据共150例样本间距离矩阵为150行列的方阵。
下面显示了1~5号样本间的欧式距离。
dd<-as.matrix(dd)二、用hclust()进行谱系聚类法(层次聚类)1.聚类函数R中自带的聚类函数是hclust(),为谱系聚类法。
基本的函数指令是结果对象 <- hclust(距离对象, method=方法)hclust()可以使用的类间距离计算方法包含离差法"ward",最短距离法"single",最大距离法"complete",平均距离法"average","mcquitty",中位数法 "median" 和重心法"centroid"。
下面采用平均距离法聚类。
hc <- hclust(dist(iris), method="ave")2.聚类函数的结果聚类结果对象包含很多聚类分析的结果,可以使用数据分量的方法列出相应的计算结果。
str(hc)下面列出了聚类结果对象hc包含的merge和height结果值的前6个。
其行编号表示聚类过程的步骤,X1,X2表示在该步合并的两类,该编号为负代表原始的样本序号,编号为正代表新合成的类;变量height表示合并时两类类间距离。
比如第1步,合并的是样本102和143,其样本间距离是0.0,合并后的类则使用该步的步数编号代表,即样本-102和-143合并为1类。
再如第6行表示样本11和49合并,该两个样本的类间距离是0.1,合并后的类称为6类。
head (hc$merge,hc$height)下面显示的聚类结果对象hc包含的merge和height结果值的50~55步的结果。
第50步结果表明样本43与13类(即第13步的聚类合类结果)合并。
所有的类编号负数j表示原数据的样本编号,正数i表示聚类过程的第i步形成的新类。
再如54步,表示聚类过程第7步和第37步形成的类合并为新类,新类成为第54类,其类间距离是0.2641715。
data.frame(hc$merge,hc$height)[50:55]3.绘制聚类图聚类完成后可以使用plot()绘制出聚类的树图。
plot(hc, hang = -1, labels=iris$Species)4.指定分类和类中心聚类通过观察树形图,可见由于数据例很多,使图形很乱,不容易确定合理的分类,为简化图形,使用cutree()来确定最初的分类结果,先初步确定各个样本的最初分类的类数后,然后用hclust()再次聚类,重新聚类后,得出最后的分类结果。
memb <- cutree(hc, k = 10) #确定10个分类table(memb) #各类中的样本数为了进行重新聚类,需要计算各类的类中心的类中心,这里用各类的均向量来代表各类的类中心,计算程序如下。
cent <- NULLfor(k in 1:10){cent <- rbind(cent, colMeans(irisSample[memb == k, , drop = FALSE]))}有了各类的类中心后,再次使用hclust()函数,从10类起重新开始聚类。
hclust()函数这时需要指定各类的类中心,并采用中心法来聚类。
hc1 <- hclust(dist(cent), method = "centroid", members = table(memb))plot(hc1)hc1再次聚类的树形图就很精简了。
从树形图上看,确定为三类是比较合适的。
4.输出最终分类结果使用cutree()来确定输出各个样本的最后分类。
memb <- cutree(hc, k =3 #确定3个分类table(memb) #各类中的样本数下面将聚类结果和原始的分类比较可见聚类结果是不错的。
错误率约为14/150=9.33%。
5.直接数据距离阵聚类可以直接输入距离矩阵,然后用as.dist()函数转为hclust()可以使用的距离阵对象,然后再用hclust()再进行聚类分析。
x<-read.table(text="id BA FI MI VO RM TOBA 0 662 877 255 412 996FI 662 0 295 468 268 400MI 877 295 0 754 564 138VO 255 468 754 0 219 869RM 412 268 564 219 0 669TO 996 400 138 869 669 0 ", header=T) s(x)<-x$idx$id<-NULLx转换为距离阵。
x<-as.dist(x)hc <-hclust(x)显示聚类过程。
data.frame(hc$merge,hc$height)绘制聚类图。
plot(hc2)从聚类图可见聚为两类比较合适。
cutree(hc2,2)所以BA、VO、RM聚为1类而TO、FI、MI聚为另一类。
四、使用cluster包进行聚类分析1.计算距离矩阵除了可以使用dist()函数外,加载了cluster包后还可以使用daisy()函数,该函数可以使用的数据类型更多一些。
当原始数据表中包含的数据是混合型即属性变量既有有连续又有分类的时候,dist()计算就不够完美,而采用daisy()函数就可以毫无问题。
使用daisy()必须加载cluster包。
第一次使用还必须安装。
install.packages(‘cluster’,repos=’htt//192.168.3.225/rpkg)library(cluster)daisy()函数的形式和dist()相似,可以采用的距离包含欧式距离"euclidean", 绝对值距离和"manhattan", "gower"。
dd2<-daisy(iris)dd2<-as.matrix(dd1)2.使用agens()聚类cluster包提供了两种层次聚类的方法,一种是a bottom-up 自底而上,采用先将数据样本先各自看成一类,然后通过合并法的聚类过程,最终形成一个大类,包含全部样本,agnes()函数完成这个功能,这个称为合并层次聚类。
而diana()则采用相反的方法,即 a top-down 自顶而下,先将所有样本看成一类,然后通过分裂类的过程将样品最终分为各自样本类,这个称为分裂法。
agens()函数的形式是agnes(x, diss = TRUE|FALSE, metric = "euclidean", stand = FALSE|TRUE,method = "average", par.method, keep.diss = n < 100, keep.data = !diss) 其中的diss指定x对象是否是不相似矩阵对象,与hclust()函数不同,agnes()既可以使用原始数据也可以采用距离阵。
stand是要求对数据进行标准化,而可以采用的类间距离计算方法有“average”平均法,“UPGMA”,“single”最短距离法,“complete”最大距离法,“ward”离差法,“weighted”权重平均距离法和“flexible”弹性法。
keep.diss和keep.data指定在结果中是否保留不相似矩阵和数据,保留这些结果需要更多的内存。
ag <- agnes(iris, diss=F, metric="euclidean", stand=F, method ="single")data.frame(ag$merge,ag$height)[50:55,]plot(ag, ask = FALSE, which.plots = NULL)cutree(ag,k=3)装载数据load'adultuci.rdata')str(AdultUCI)删除原来的目标变量AdultUCI$income<-NULL删除原始据中数据缺失的数据例AdultUCI<-na.omit(AdultUCI)。