QTL IciMapping3.0 简单教程
Qtxml文件常用的操作(读写,增删改查)

Qtxml⽂件常⽤的操作(读写,增删改查)项⽬配置pro⽂件⾥⾯添加QT+=xmlinclude <QtXml>,也可以include <QDomDocument>项⽬⽂件:.pro ⽂件1 QT += core xml23 QT -= gui45 TARGET = xmltest6 CONFIG += console7 CONFIG -= app_bundle89 TEMPLATE = app101112 SOURCES += main.cpp主程序:main.cpp1 #include <QCoreApplication>2 #include <QtXml> //也可以include <QDomDocument>34//写xml5void WriteXml()6 {7//打开或创建⽂件8 QFile file("test.xml"); //相对路径、绝对路径、资源路径都可以9if(!file.open(QFile::WriteOnly|QFile::Truncate)) //可以⽤QIODevice,Truncate表⽰清空原来的内容10return;1112 QDomDocument doc;13//写⼊xml头部14 QDomProcessingInstruction instruction; //添加处理命令15 instruction=doc.createProcessingInstruction("xml","version=\"1.0\" encoding=\"UTF-8\"");16 doc.appendChild(instruction);17//添加根节点18 QDomElement root=doc.createElement("library");19 doc.appendChild(root);20//添加第⼀个⼦节点及其⼦元素21 QDomElement book=doc.createElement("book");22 book.setAttribute("id",1); //⽅式⼀:创建属性其中键值对的值可以是各种类型23 QDomAttr time=doc.createAttribute("time"); //⽅式⼆:创建属性值必须是字符串24 time.setValue("2013/6/13");25 book.setAttributeNode(time);26 QDomElement title=doc.createElement("title"); //创建⼦元素27 QDomText text; //设置括号标签中间的值28 text=doc.createTextNode("C++ primer");29 book.appendChild(title);30 title.appendChild(text);31 QDomElement author=doc.createElement("author"); //创建⼦元素32 text=doc.createTextNode("Stanley Lippman");33 author.appendChild(text);34 book.appendChild(author);35 root.appendChild(book);3637//添加第⼆个⼦节点及其⼦元素,部分变量只需重新赋值40 time=doc.createAttribute("time");41 time.setValue("2007/5/25");42 book.setAttributeNode(time);43 title=doc.createElement("title");44 text=doc.createTextNode("Thinking in Java");45 book.appendChild(title);46 title.appendChild(text);47 author=doc.createElement("author");48 text=doc.createTextNode("Bruce Eckel");49 author.appendChild(text);50 book.appendChild(author);51 root.appendChild(book);5253//输出到⽂件54 QTextStream out_stream(&file);55 doc.save(out_stream,4); //缩进4格56 file.close();5758 }5960//读xml61void ReadXml()62 {63//打开或创建⽂件64 QFile file("test.xml"); //相对路径、绝对路径、资源路径都⾏65if(!file.open(QFile::ReadOnly))66return;6768 QDomDocument doc;69if(!doc.setContent(&file))70 {71 file.close();72return;73 }74 file.close();7576 QDomElement root=doc.documentElement(); //返回根节点77 qDebug()<<root.nodeName();78 QDomNode node=root.firstChild(); //获得第⼀个⼦节点79while(!node.isNull()) //如果节点不空80 {81if(node.isElement()) //如果节点是元素82 {83 QDomElement e=node.toElement(); //转换为元素,注意元素和节点是两个数据结构,其实差不多84 qDebug()<<e.tagName()<<""<<e.attribute("id")<<""<<e.attribute("time"); //打印键值对,tagName和nodeName是⼀个东西85 QDomNodeList list=e.childNodes();86for(int i=0;i<list.count();i++) //遍历⼦元素,count和size都可以⽤,可⽤于标签数计数87 {88 QDomNode n=list.at(i);89if(node.isElement())90 qDebug()<<n.nodeName()<<":"<<n.toElement().text();91 }92 }93 node=node.nextSibling(); //下⼀个兄弟节点,nextSiblingElement()是下⼀个兄弟元素,都差不多94 }9596 }9798//增加xml内容99void AddXml()100 {101//打开⽂件102 QFile file("test.xml"); //相对路径、绝对路径、资源路径都可以103if(!file.open(QFile::ReadOnly))106//增加⼀个⼀级⼦节点以及元素107 QDomDocument doc;108if(!doc.setContent(&file))109 {110 file.close();111return;112 }113 file.close();114115 QDomElement root=doc.documentElement();116 QDomElement book=doc.createElement("book");117 book.setAttribute("id",3);118 book.setAttribute("time","1813/1/27");119 QDomElement title=doc.createElement("title");120 QDomText text;121 text=doc.createTextNode("Pride and Prejudice");122 title.appendChild(text);123 book.appendChild(title);124 QDomElement author=doc.createElement("author");125 text=doc.createTextNode("Jane Austen");126 author.appendChild(text);127 book.appendChild(author);128 root.appendChild(book);129130if(!file.open(QFile::WriteOnly|QFile::Truncate)) //先读进来,再重写,如果不⽤truncate就是在后⾯追加内容,就⽆效了131return;132//输出到⽂件133 QTextStream out_stream(&file);134 doc.save(out_stream,4); //缩进4格135 file.close();136 }137138//删减xml内容139void RemoveXml()140 {141//打开⽂件142 QFile file("test.xml"); //相对路径、绝对路径、资源路径都可以143if(!file.open(QFile::ReadOnly))144return;145146//删除⼀个⼀级⼦节点及其元素,外层节点删除内层节点于此相同147 QDomDocument doc;148if(!doc.setContent(&file))149 {150 file.close();151return;152 }153 file.close(); //⼀定要记得关掉啊,不然⽆法完成操作154155 QDomElement root=doc.documentElement();156 QDomNodeList list=doc.elementsByTagName("book"); //由标签名定位157for(int i=0;i<list.count();i++)158 {159 QDomElement e=list.at(i).toElement();160if(e.attribute("time")=="2007/5/25") //以属性名定位,类似于hash的⽅式,warning:这⾥仅仅删除⼀个节点,其实可以加个break 161 root.removeChild(list.at(i));162 }163164if(!file.open(QFile::WriteOnly|QFile::Truncate))165return;166//输出到⽂件167 QTextStream out_stream(&file);168 doc.save(out_stream,4); //缩进4格169 file.close();172//更新xml内容173void UpdateXml()174 {175//打开⽂件176 QFile file("test.xml"); //相对路径、绝对路径、资源路径都可以177if(!file.open(QFile::ReadOnly))178return;179180//更新⼀个标签项,如果知道xml的结构,直接定位到那个标签上定点更新181//或者⽤遍历的⽅法去匹配tagname或者attribut,value来更新182 QDomDocument doc;183if(!doc.setContent(&file))184 {185 file.close();186return;187 }188 file.close();189190 QDomElement root=doc.documentElement();191 QDomNodeList list=root.elementsByTagName("book");192 QDomNode node=list.at(list.size()-1).firstChild(); //定位到第三个⼀级⼦节点的⼦元素193 QDomNode oldnode=node.firstChild(); //标签之间的内容作为节点的⼦节点出现,当前是Pride and Projudice 194 node.firstChild().setNodeValue("Emma");195 QDomNode newnode=node.firstChild();196 node.replaceChild(newnode,oldnode);197198if(!file.open(QFile::WriteOnly|QFile::Truncate))199return;200//输出到⽂件201 QTextStream out_stream(&file);202 doc.save(out_stream,4); //缩进4格203 file.close();204 }205206int main(int argc, char *argv[])207 {208209 qDebug()<<"write xml to file...";210 WriteXml();211 qDebug()<<"read xml to display...";212 ReadXml();213 qDebug()<<"add contents to xml...";214 AddXml();215 qDebug()<<"remove contents from xml...";216 RemoveXml();217 qDebug()<<"update contents to xml...";218 UpdateXml();219return0;220221 }写xml1 <?xml version="1.0" encoding="UTF-8"?>2 <library>3 <book id="1" time="2013/6/13">4 <title>C++ primer</title>5 <author>Stanley Lippman</author>6 </book>7 <book id="2" time="2007/5/25">8 <title>Thinking in Java</title>9 <author>Bruce Eckel</author>10 </book>11 </library>1 <?xml version='1.0' encoding='UTF-8'?>2 <library>3 <book time="2013/6/13" id="1">4 <title>C++ primer</title>5 <author>Stanley Lippman</author>6 </book>7 <book time="2007/5/25" id="2">8 <title>Thinking in Java</title>9 <author>Bruce Eckel</author>10 </book>11 <book time="1813/1/27" id="3">12 <title>Pride and Prejudice</title>13 <author>Jane Austen</author>14 </book>15 </library>删除xml1 <?xml version='1.0' encoding='UTF-8'?>2 <library>3 <book time="2013/6/13" id="1">4 <title>C++ primer</title>5 <author>Stanley Lippman</author>6 </book>7 <book time="2007/5/25" id="2">8 <title>Thinking in Java</title>9 <author>Bruce Eckel</author>10 </book>11 <book time="1813/1/27" id="3">12 <title>Pride and Prejudice</title>13 <author>Jane Austen</author>14 </book>15 </library>更新xml1 <?xml version='1.0' encoding='UTF-8'?>2 <library>3 <book id="1" time="2013/6/13">4 <title>C++ primer</title>5 <author>Stanley Lippman</author>6 </book>7 <book id="3" time="1813/1/27">8 <title>Emma</title>9 <author>Jane Austen</author>10 </book>11 </library>。
QTLIciMapping3.0简单教学教程

QTL IciMapping3.0 定位简单应用教程张茜中国农科院2012.6.14主要步骤•数据准备•新建project•导入数据•构建图谱•QTL定位准备数据•.map格式将txt格式后缀名改成.map即可(表头信息不能动),一个map文件中包括General Information、Marker Types 、Information for Chromosomes and Markers三部分信息主要更改数据:7为F2群体;1一般不动;Marker space type 选1或2均可,只要保持数据对应Maker Types带型统计方法这些数据是标记在第几条染色体(group)上,未构建图谱侧全为0点File 选New Project新建一个工作项命名保存路径点File 选*map导入构建准备好的map格式图谱的数据打开,完成数据导入点击分组,在此处出现group群点可以看到一个group下所含标记,右键点击一个标记可以对其位置调动或者删除完成分组后,点击ordering,转换成染色体组再点此按钮完成沟通准备工作,工具栏上的map图标变蓝可以点击构图了点击map 按钮出现图谱(右)点击即可出现下一个染色体图谱点击出现整体图谱Save 可以保存各种格式的图QTL定位数据准备将构图所得结果F2bip(在project-map-result文件下)先复制一份,再用txt打开方式打开所复制文件。
Bip文件中包含5部分General Information、Information for Chromosomes andMarkers、Linkage map (Marker namefollowed by position or the interval length)、 Marker Type 、Phenotypic Data更改数据:0改成1选File-open file-*bip打开更改保存好的bip格式文件选ICIM-ADD添加的下框(一般都默认),此时start按钮从灰色变黑色,单击即可进行定位Start 完了点ADD即出现下图加性效应图显性效应图总染色体添加lod值线下一个染色体在Graph 下可以选择连锁图和lod (上)或者连锁图和QTL(下)图谱结果信息在map目录下QTL结果信息在BIP目录下信息栏补充•QTL ICIMapping是在*map(oppen file子菜单下)下完成构件图谱,在*bip(oppen file子菜单下)下完成QTL定位。
GlobalMapper软件操作教程

Global Mapper软件操作教程一软件简介Global Mapper软件可支持多类型,多数据读取,且支持不同格式的数据输出,对矢量数据的编辑和管理简便的特点,其作为Lidar数据的质检环节,所带来的和易操作性,更适应于高效生产中。
二软件工具介绍1 . 软件界面Global Mapper软件界面简洁、友好,提供多途径常用的选项,且软件的大部分菜单和工具项配置了快捷键,所以操作很方便。
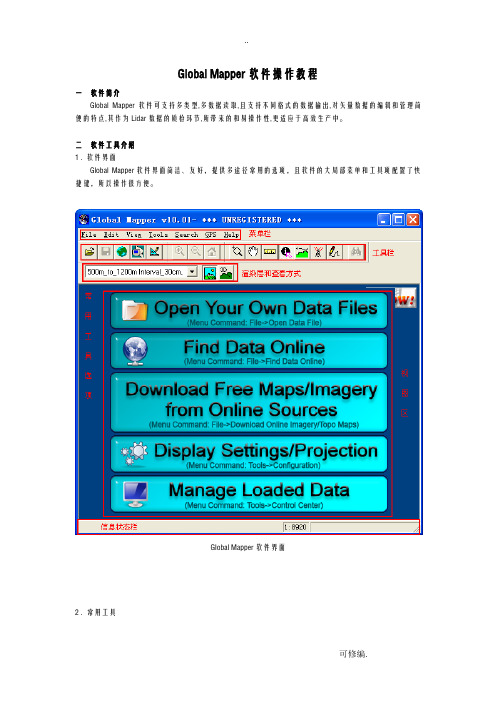
Global Mapper软件界面2 . 常用工具开启\关闭山丘阴影Show 3D ViewCtrl+3 数据三维视图查看,可于数据同步缩放,移动和旋转操作显示3D 视图3 . 菜单介绍Files(文件)菜单Eait(编辑)菜单View (查看)菜单Tools(工具)菜单Search(查询)菜单Digitizer Tool (矢量化工具)右键菜单三QC操作流程1.拷贝(颜色表文件)和(质检类型层文件)文件至Global Mapper软件安装目录C:\ProgramFiles\GlobalMapper10\下。
2.启动Global Mapper软件,首先查看渲染层处是否已经成功加载,如果未加载成功,重新启动软件。
已成功加载的颜色表项已成功记载的质检类型层(Configuration)3.打开数据文件。
可通过的点击视图区的Open Your Own data Files选项打开读取,或通过菜单Files-Open Data file(s) / Ctor+O,或通过工具栏打开读取数据。
可支持多数据添加4.通过菜单File-Save Workspace / Ctrl+S,或通过工具栏保存个人工作空间。
下次退出软件后,直接打开工作空间(*.gmw)即可。
5.在工具栏选择对读取数据进行渲染。
数据渲染后的效果注意:(1).可通过的其他颜色表分别检查数据。
(2).可以通过再在点击关闭对数据高程夸张。
6.通过菜单Tools-Digitizer /Alt+D或通过点击工具栏,使此工具处于激活状态,在视图区直接点击鼠标右键选择Greate New Area Feature,即可对问题区域进行区域标示操作。
GlobalMapper软件操作教程
Global Mapper软件操作教程一软件简介Global Mapper软件可支持多类型,多数据读取,且支持不同格式的数据输出,对矢量数据的编辑和管理简便的特点,其作为Lidar数据的质检环节,所带来的和易操作性,更适应于高效生产中。
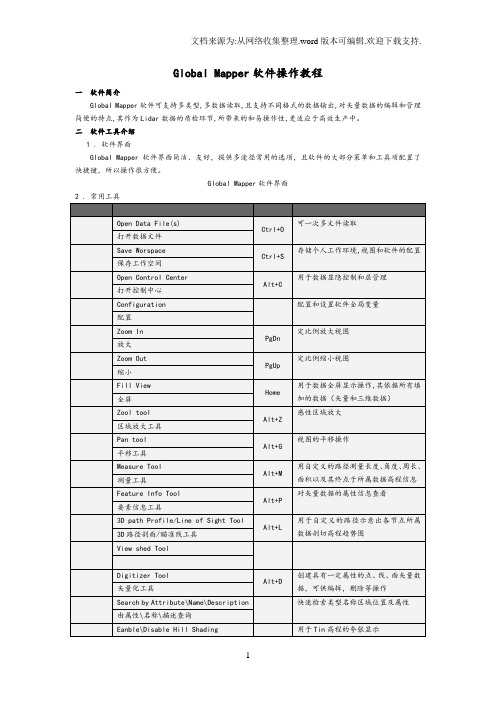
二软件工具介绍1 . 软件界面Global Mapper软件界面简洁、友好,提供多途径常用的选项,且软件的大局部菜单和工具项配置了快捷键,所以操作很方便。
Global Mapper软件界面2 . 常用工具3 . 菜单介绍Files〔文件〕菜单Eait 〔编辑〕菜单View 〔查看〕菜单Tools〔工具〕菜单Search 〔查询〕菜单Digitizer Tool 〔矢量化工具〕右键菜单三QC操作流程1.拷贝custom_shaders.txt 〔颜色表文件〕和custom_area_types.txt 〔质检类型层文件〕文件至GlobalMapper软件安装目录C:\Program Files\GlobalMapper10\下。
2.启动Global Mapper软件,首先查看渲染层处是否已经成功加载,如果未加载成功,重新启动软件。
已成功加载的颜色表项已成功记载的质检类型层〔Configuration〕3.翻开数据文件。
可通过的点击视图区的Open Your Own data Files选项翻开读取,或通过菜单Files-Open Data file(s) / Ctor+O,或通过工具栏翻开读取数据。
可支持多数据添加4.通过菜单File-Save Workspace / Ctrl+S,或通过工具栏保存个人工作空间。
下次退出软件后,直接翻开工作空间〔*.gmw〕即可。
5.在工具栏选择对读取数据进展渲染。
数据渲染后的效果注意:〔1〕.可通过的其他颜色表分别检查数据。
〔2〕.可以通过再在点击关闭对数据高程夸。
6.通过菜单Tools-Digitizer /Alt+D 或通过点击工具栏,使此工具处于激活状态,在视图区直接点击鼠标右键选择Greate New Area Feature,即可对问题区域进展区域标示操作。
GlobalMapper软件操作教程
Global Mapper软件操作教程一软件简介Global Mapper软件可支持多类型,多数据读取,且支持不同格式的数据输出,对矢量数据的编辑和管理简便的特点,其作为Lidar数据的质检环节,所带来的和易操作性,更适应于高效生产中。
二软件工具介绍1 . 软件界面Global Mapper软件界面简洁、友好,提供多途径常用的选项,且软件的大部分菜单和工具项配置了快捷键,所以操作很方便。
Global Mapper软件界面2 . 常用工具开启\关闭山丘阴影Show 3D ViewCtrl+3 数据三维视图查看,可于数据同步缩放,移动和旋转操作显示3D 视图3 . 菜单介绍Files(文件)菜单Eait (编辑)菜单View (查看)菜单Tools(工具)菜单Search (查询)菜单Digitizer Tool (矢量化工具)右键菜单三 QC操作流程1.拷贝custom_shaders.txt (颜色表文件)和custom_area_types.txt (质检类型层文件)文件至 Global Mapper软件安装目录C:\Program Files\GlobalMapper10\ 下。
2.启动Global Mapper软件,首先查看渲染层处是否已经成功加载,如果未加载成功,重新启动软件。
已成功加载的颜色表项已成功记载的质检类型层(Configuration)3.打开数据文件。
可通过的点击视图区的Open Your Own data Files选项打开读取,或通过菜单Files-Open Data file(s) / Ctor+O,或通过工具栏打开读取数据。
可支持多数据添加4.通过菜单File-Save Workspace / Ctrl+S,或通过工具栏保存个人工作空间。
下次退出软件后,直接打开工作空间(*.gmw)即可。
5.在工具栏选择对读取数据进行渲染。
数据渲染后的效果注意:(1).可通过的其他颜色表分别检查数据。
QTL_IciMapping3.0_简单教程

QTL IciMapping3.0 定位简单应用教程张茜中国农科院2012.6.14主要步骤•数据准备•新建project•导入数据•构建图谱•QTL定位准备数据•.map格式将txt格式后缀名改成.map即可(表头信息不能动),一个map文件中包括General Information、Marker Types 、Information for Chromosomes and Markers三部分信息主要更改数据:7为F2群体;1一般不动;Marker space type 选1或2均可,只要保持数据对应Maker Types带型统计方法这些数据是标记在第几条染色体(group)上,未构建图谱侧全为0点File 选New Project新建一个工作项命名保存路径点File 选*map导入构建准备好的map格式图谱的数据打开,完成数据导入点击分组,在此处出现group群点可以看到一个group下所含标记,右键点击一个标记可以对其位置调动或者删除完成分组后,点击ordering,转换成染色体组再点此按钮完成沟通准备工作,工具栏上的map图标变蓝可以点击构图了点击map 按钮出现图谱(右)点击即可出现下一个染色体图谱点击出现整体图谱Save 可以保存各种格式的图QTL定位数据准备将构图所得结果F2bip(在project-map-result文件下)先复制一份,再用txt打开方式打开所复制文件。
Bip文件中包含5部分General Information、Information for Chromosomes andMarkers、Linkage map (Marker namefollowed by position or the interval length)、 Marker Type 、Phenotypic Data更改数据:0改成1选File-open file-*bip打开更改保存好的bip格式文件选ICIM-ADD添加的下框(一般都默认),此时start按钮从灰色变黑色,单击即可进行定位Start 完了点ADD即出现下图加性效应图显性效应图总染色体添加lod值线下一个染色体在Graph 下可以选择连锁图和lod (上)或者连锁图和QTL(下)图谱结果信息在map目录下QTL结果信息在BIP目录下信息栏补充•QTL ICIMapping是在*map(oppen file子菜单下)下完成构件图谱,在*bip(oppen file子菜单下)下完成QTL定位。
(仅供参考)Icimapping 连锁图中文操作说明
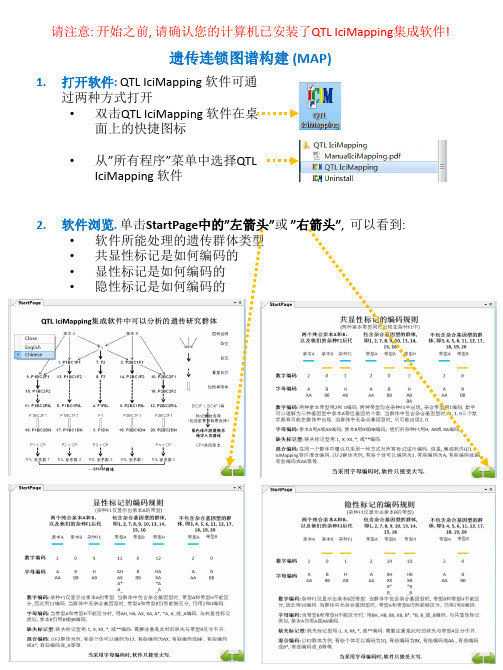
• 对连锁群”Chromsome4”再排序: 鼠标指向连锁群”Chromsome4”, 然后右击, 从弹出的快捷菜单中选择”Ordering”实现对”Chromosome4”的重排序
参数选择/设置窗口
4
6. 标记分群
遗传连锁图谱构建
• 单击Grouping下的 箭头, 软件将利用默 认/设定参数对标记 进行分群, 例如图例 中, 按LOD临界值 3.00 的标准分群
7. 标记排序
• 单击Ordering下的箭 头, 软件将利用默认 /设定参数对标记进 行排序, 例如图例中, 利用nnTwoOpt算法 排序
• 对连锁群重命名: 鼠标指向连锁群”Chromsome4”, 然后右击, 从弹出的快捷菜 单中选择”Rename”实现对”Chromosome4”的重命名, 或者…
• 将连锁群上移或下移
• 删除连锁群内的所有标记
• 改变连锁群首尾标记的循序
12
遗传连锁图谱构建
15. 高级用户 – 在EXCEL中管理遗传群体的信息 • 工作表”GeneralInfo”定义遗传群体的一些基本信息, 每项信息占1行.
5
遗传连锁图谱构建
8. 标记排序调整 (此选 项可跳过, 调整的目 的是获得更短的图 谱)
• 单击Rippling下的箭 头, 软件将利用默认 /设定参数对标记顺 序进行调整, 例如图 例中, 利用SARF做标 准, 窗口大小为5
第13章数量性状基因定位

自然群体与关联分析
• 对动物和人类的遗传研究来说,可供利用的只是自然条件 下的随机交配群体。利用自然群体中的剩余连锁不平衡开 展QTL作图研究,又称关联分析(association mapping)。
一个标记座位上2种标记型MM和 mm的性状分布
A
标记型MM
B 标记型mm
标记型MM
标记型mm
标记与性状基因存在连锁 标记与性状基因不存在连锁
标记和QTL两个座位上的基因型和频率
• 假定两个纯合亲本P1和P2的基因型分别为 MMQQ和mmqq。DH群体中,标记基因型 有MM和mm两种,QTL的基因型有QQ和 qq两种。
10个DH家系在1H染色体的14个标记型和平均粒重
亲本‘Harrington’用2编码,亲本‘TR306’用0编码,-1表 示缺失基因型。表型无缺失
分子标记 位置/cM
Act8A
0
OP06
10.9
aHor2
18.5
MWG943 78.2
ABG464 91.2
Dor3
111.2
iPgd2
114.7
cMWG733A 121.7
• 标记与QTL结合起来共有4种基因型,即 MMQQ、MMqq、mmQQ和mmqq。假定标 记与QTL间的重组率为r,4种基因型的频 率分别为 (1-r)/2、 r/2 、 r/2和(1-r)/2 。
QTL的基因型值
• 标记一般是中性DNA水平的多态性,标记本身并 不会产生任何表型效应。基因型MMQQ和mmQQ 的遗传效应是由QQ决定的;基因型MMqq和mmqq 的遗传效应是由qq决定的。因此,在加显性模型 下,4种基因型具有两种不同的均值:
QTL.gCIMapping V3.4 定位生物学质谱线性映射软件说明书

Package‘QTL.gCIMapping’October12,2022Type PackageTitle QTL Genome-Wide Composite Interval MappingVersion3.4Date2022-02-24Author Zhou Ya-Hui,Zhang Ya-Wen,Wen Yang-Jun,Wang Shi-Bo,and Zhang Yuan-MingMaintainer Yuanming Zhang<******************>Description Conduct multiple quantitative trait loci(QTL)and QTL-by-environment interac-tion(QEI)mapping via ordinary or compressed variance component mixed models with random-orfixed QTL/QEI effects.First,each position on the genome is detected in order to obtain a neg-ative logarithm P-value curve against genome position.Then,all the peaks on each effect(addi-tive or dominant)curve or on each locus curve are viewed as potential main-effect QTLs and QEIs,all their effects are included in a multi-locus model,their effects are esti-mated by both least angle regression and empirical Bayes(or adaptive lasso)in back-cross and F2populations,and true QTLs and QEIs are identified by likelihood ra-dio test.See Zhou et al.(2022)<doi:10.1093/bib/bbab596>and Wen et al.(2018)<doi:10.1093/bib/bby058>. Encoding UTF-8Depends R(>=3.5.0)License GPL(>=2)Imports Rcpp(>=0.12.17),methods,openxlsx,readxl,lars,stringr,data.table,glmnet,doParallel,foreach,MASS,qtlLinkingTo RcppNeedsCompilation yesRoxygenNote7.1.2Repository CRANDate/Publication2022-02-2418:30:02UTCR topics documented:DHdata (2)Dodata (2)F2data (3)12Dodata markerinsert (4)QTL.gCIMapping (5)Readdata (6)WangF (7)WangS (8)WenF (10)WenS (11)ZhouF (12)ZhouMethod (13)ZhouMethod_single_env (15)Index18 DHdata DH example dataDescriptionGCIM format of DH dataset.Usagedata(DHdata)DetailsInputfile for WangF function.Author(s)Maintainer:Yuanming Zhang<******************>Dodata Process raw dataDescriptionProcess raw dataUsageDodata(fileFormat=NULL,Population=NULL,method=NULL,Model=NULL,readraw=NULL,MultiEnv=FALSE)F2data3 ArgumentsfileFormat Format of dataset.Population Population type.method Method"GCIM"or method"GCIM-QEI"Model Random orfixed model.readraw Raw data.MultiEnv Whether to perform multi-environment analysisValuea listExamplesdata(F2data)readraw<-Readdata(file=F2data,fileFormat="GCIM",method="GCIM-QEI",filecov=NULL,MCIMmap=NULL,MultiEnv=TRUE)doda<-Dodata(fileFormat="GCIM",Population="F2",method="GCIM-QEI",Model="Random",readraw,MultiEnv=TRUE)F2data F2example data from2environmentsDescriptionGCIM format of F2dataset whith GCIM-QEI method.Usagedata(F2data)DetailsInputfile for ZhouF function.Author(s)Maintainer:Yuanming Zhang<******************>4markerinsert markerinsert To insert marker in genotype.Descriptiona method that can insert marker in genotype.Usagemarkerinsert(mp,geno,map,cl,gg1,gg2,gg0,flagRIL)Argumentsmp linkage map matrix after insert.geno genotype matrix.map linkage map matrix.cl walk speed.gg1raw covariate matrix.gg2code for type1.gg0code for missing.flagRIL RIL population or not.Author(s)Zhang Ya-Wen,Wen Yang-Jun,Wang Shi-Bo,and Zhang Yuan-MingMaintainer:Yuanming Zhang<******************>Examples##Not run:mp=matrix(c(197.9196,198.7536,199.5876,200.4216,201.2453,202.0691,202.8928,203.7521,204.6113,205.4706,206.3298,207.1891,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,2,2,2,3,3,3,3,3,3,1,1,1,2,2,2,3,3,3,3,3,3,1,2,3,4,5,6,7,8,9,10,11,12),12,5)map=matrix(c(1,1,1,1,197.9196,200.4216,202.8928,207.1891),4,2)geno=matrix(c(1,99,99,99),1,4)QTL.gCIMapping::markerinsert(mp,geno,map,cl=1,gg1=1,gg2=-1,gg0=99,flagRIL=1)##End(Not run)QTL.gCIMapping5 QTL.gCIMapping QTL Genome-Wide Composite Interval MappingDescriptionQTL Genome-Wide Composite Interval MappingUsageQTL.gCIMapping(file=NULL,fileFormat="GCIM",filecov=NULL,MCIMmap=NULL,Population=NULL,method="GCIM-QEI",MultiEnv=FALSE,Model="Random",WalkSpeed=1,CriLOD=3,CriDis=5,Likelihood="REML",SetSeed=11001,flagrqtl=FALSE,DrawPlot=TRUE,PlotFormat="jpeg",Resolution="Low",Trait=NULL,dir=NULL,CLO=NULL)Argumentsfile File path and name in your computer.fileFormat Format for inputfile:GCIM,ICIM,Cart,or MCIM.filecov Covariatefile of QTLIciMapping or QTLNetwork.MCIMmap Mapfile of QTLNetwork.Population Population type:F2,BC1,BC2,DH,RIL.method Method"GCIM"or method"GCIM-QEI".MultiEnv Whether to perform multi-environment analysis.Model Random orfixed model.WalkSpeed Walk speed for Genome-wide Scanning.CriLOD Critical LOD scores for significant QTL.6ReaddataCriDis The distance of optimization.Likelihood This parameter is only for F2population,including REML(restricted maximum likelihood)and ML(maximum likelihood).SetSeed In which the cross validation experiment is needed.Generally speaking,the random seed in the cross-validation experiment was set as11001.If some knowngenes are not identified by the seed,users may try to use some new randomseeds.At this case,one better result may be obtained.flagrqtl This parameter is only for F2population,flagrqtl="FALSE"in thefirst running.If the other software detects only one QTL in a neighborhood but this softwarefinds two linked QTLs(one with additive effect and another with dominant ef-fect)in the region,letflagrqtl="TRUE"DrawPlot This parameter is for all the populations,including FALSE and TRUE,Draw-Plot=FALSE indicates nofigure output,DrawPlot=TRUE indicates the outputof thefigure against genome position.PlotFormat This parameter is for all thefigurefiles,including*.jpeg,*.png,*.tiff and*.pdf.Resolution This parameter is for all thefigurefiles,including Low and High.Trait Trait=1:3indicates the analysis from thefirst trait to the third trait.dir This parameter is for the save path.CLO Number of CPUs.Examplesdata(F2data)QTL.gCIMapping(file=F2data,Population="F2",MultiEnv=TRUE,Model="Random",CriLOD=3,Trait=1,dir=tempdir(),CLO=2)Readdata Read raw dataDescriptionRead raw dataUsageReaddata(file=NULL,fileFormat=NULL,method=NULL,filecov=NULL,MCIMmap=NULL,MultiEnv=FALSE)WangF7 Argumentsfile Dataset inputfileFormat Format of dataset.method Method"GCIM"or method"GCIM-QEI"filecov Covariatefile of QTLIciMapping or QTLNetwork.MCIMmap Mapfile of QTLNetwork.MultiEnv Whether to perform multi-environment analysisValuea listExamplesdata(F2data)Readdata(file=F2data,fileFormat="GCIM",method="GCIM-QEI",filecov=NULL,MCIMmap=NULL,MultiEnv=TRUE)WangF To perform QTL mapping with wang methodDescriptionTo perform QTL mapping with wang methodUsageWangF(pheRaw=NULL,genRaw=NULL,mapRaw1=NULL,yygg1=NULL,flagRIL=NULL,cov_en=NULL,Population=NULL,WalkSpeed=NULL,CriLOD=NULL)ArgumentspheRaw phenotype matrix.genRaw genotype matrix.mapRaw1linkage map matrix.yygg1the transformed covariate matrix.flagRIL if RIL or not.cov_en raw covariate matrix.Population populationflag.WalkSpeed Walk speed for Genome-wide Scanning.CriLOD Critical LOD scores for significant QTL.Valuea listExamplesdata(DHdata)readraw<-Readdata(file=DHdata,fileFormat="GCIM",method="GCIM",filecov=NULL,MCIMmap=NULL,MultiEnv=FALSE) DoResult<-Dodata(fileFormat="GCIM",Population="DH",method="GCIM",Model="Random",readraw,MultiEnv=FALSE)ws<-WangF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw, mapRaw1=DoResult$mapRaw1,yygg1=DoResult$yygg1,flagRIL=DoResult$flagRIL,cov_en=DoResult$cov_en,Population="DH",WalkSpeed=1,CriLOD=2.5)WangS The second step of wang methodDescriptionThe second step of wang methodUsageWangS(flag=NULL,CriLOD=NULL,NUM=NULL,pheRaw=NULL,chrRaw_name=NULL,yygg=NULL,mx=NULL,phe=NULL,chr_name=NULL,gen=NULL,mapname=NULL,CLO=NULL)Argumentsflagfix or random model.CriLOD Critical LOD scores for significant QTL.NUM The number of trait.pheRaw Raw phenotype matrix.chrRaw_name raw chromosome name.yygg covariate matrix.mx raw genotype matrix.phe phenotype matrix.chr_name chromosome name.gen genotype matrix.mapname linkage map matrix.CLO Number of CPUs.Valuea listExamplesdata(DHdata)readraw<-Readdata(file=DHdata,fileFormat="GCIM",method="GCIM",filecov=NULL,MCIMmap=NULL,MultiEnv=FALSE)DoResult<-Dodata(fileFormat="GCIM",Population="DH",method="GCIM",Model="Random",readraw,MultiEnv=FALSE)W1re<-WangF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw, mapRaw1=DoResult$mapRaw1,yygg1=DoResult$yygg1,flagRIL=DoResult$flagRIL,cov_en=DoResult$cov_en,Population="DH",WalkSpeed=1,CriLOD=2.5)ws<-WangS(flag=DoResult$flag,CriLOD=2.5,NUM=1,pheRaw=DoResult$pheRaw,chrRaw_name=W1re$chrRaw_name,yygg=W1re$yygg,mx=W1re$mx,phe=W1re$phe,chr_name=W1re$chr_name,gen=W1re$gen,mapname=W1re$mapname,CLO=1)10WenF WenF To perform QTL mapping with Wen methodDescriptionTo perform QTL mapping with Wen methodUsageWenF(pheRaw=NULL,genRaw=NULL,mapRaw1=NULL,yygg1=NULL,cov_en=NULL,WalkSpeed=NULL,CriLOD=NULL,dir=NULL)ArgumentspheRaw phenotype matrix.genRaw genotype matrix.mapRaw1linkage map matrix.yygg1the transformed covariate matrix.cov_en raw covariate matrix.WalkSpeed Walk speed for Genome-wide Scanning.CriLOD Critical LOD scores for significant QTL.dirfile path in your computer.Valuea listExamplesdata(F2data)readraw<-Readdata(file=F2data,fileFormat="GCIM",method="GCIM",filecov=NULL,MCIMmap=NULL,MultiEnv=FALSE)DoResult<-Dodata(fileFormat="GCIM",Population="F2",method="GCIM",Model="Random",readraw,MultiEnv=FALSE)wf<-WenF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw1=DoResult$mapRaw1,yygg1=DoResult$yygg1,cov_en=DoResult$cov_en,WalkSpeed=1,CriLOD=2.5,dir=tempdir())WenS11 WenS The second step of Wen methodDescriptionThe second step of Wen methodUsageWenS(flag=NULL,CriLOD=NULL,NUM=NULL,pheRaw=NULL,Likelihood=NULL,SetSeed=NULL,flagrqtl=NULL,yygg=NULL,mx=NULL,phe=NULL,chr_name=NULL,v.map=NULL,gen.raw=NULL,a.gen.orig=NULL,d.gen.orig=NULL,n=NULL,names.insert2=NULL,X.ad.tran.data=NULL,X.ad.t4=NULL,dir=NULL)Argumentsflag random orfix model.CriLOD LOD score.NUM the number of trait.pheRaw raw phenotype matrix.Likelihood likelihood function.SetSeed random seed set in which,the cross validation is needed.flagrqtl do CIM or not.yygg covariate matrix.mx raw genotype matrix.phe phenotype matrix.12ZhouFchr_name chromosome name.v.map linkage map matrix.gen.raw raw genotype matrix.a.gen.orig additive genotype matrix.d.gen.orig dominant genotype matrix.n number of individual.names.insert2linkage map after insert.X.ad.tran.data genotype matrix after insert.X.ad.t4genotype matrix.dirfile storage path.Valuea listExamplesdata(F2data)readraw<-Readdata(file=F2data,fileFormat="GCIM",method="GCIM",filecov=NULL,MCIMmap=NULL,MultiEnv=FALSE)DoResult<-Dodata(fileFormat="GCIM",Population="F2",method="GCIM",Model="Random",readraw,MultiEnv=FALSE)WEN1re<-WenF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw1=DoResult$mapRaw1,yygg1=DoResult$yygg1,cov_en=DoResult$cov_en,WalkSpeed=1,CriLOD=2.5,dir=tempdir())ws<-WenS(flag=DoResult$flag,CriLOD=2.5,NUM=1,pheRaw=DoResult$pheRaw,Likelihood="REML",SetSeed=11001,flagrqtl=FALSE,yygg=WEN1re$yygg,mx=WEN1re$mx,phe=WEN1re$phe,chr_name=WEN1re$chr_name,v.map=WEN1re$v.map,gen.raw=WEN1re$gen.raw,a.gen.orig=WEN1re$a.gen.orig,d.gen.orig=WEN1re$d.gen.orig,n=WEN1re$n,names.insert2=WEN1re$names.insert2,X.ad.tran.data=WEN1re$X.ad.tran.data,X.ad.t4=WEN1re$X.ad.t4,dir=tempdir())ZhouF To perform QTL mapping with Wen methodDescriptionTo perform QTL mapping with Wen methodUsageZhouF(pheRaw=NULL,genRaw=NULL,mapRaw1=NULL,WalkSpeed=NULL,CriLOD=NULL,dir=NULL)ArgumentspheRaw phenotype matrix.genRaw genotype matrix.mapRaw1linkage map matrix.WalkSpeed Walk speed for Genome-wide Scanning.CriLOD Critical LOD scores for significant QTL.dirfile path in your computer.Valuea listExamplesdata(F2data)readraw<-Readdata(file=F2data,fileFormat="GCIM",method="GCIM-QEI",filecov=NULL,MCIMmap=NULL,MultiEnv=TRUE)DoResult<-Dodata(fileFormat="GCIM",Population="F2",method="GCIM-QEI",Model="Random",readraw,MultiEnv=TRUE)ZhouMatrices<-ZhouF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw1=DoResult$mapRaw1,WalkSpeed=1,CriLOD=3,dir=tempdir())ZhouMethod The second step of Zhou method for multiple environmentsDescriptionThe second step of Zhou method for multiple environmentsUsageZhouMethod(Model=NULL,pheRaw=NULL,genRaw=NULL,mapRaw=NULL,CriLOD=NULL,NUM=NULL,EnvNum=NULL,yygg=NULL,genoname=NULL,Ax0=NULL,Hx0=NULL,Bx0=NULL,Ax=NULL,Hx=NULL,Bx=NULL,dir=NULL,CriDis=NULL,CLO=NULL)ArgumentsModel Random orfixed model.pheRaw phenotype matrix.genRaw genotype matrix.mapRaw linkage map matrix.CriLOD Critical LOD scores for significant QTL.NUM The serial number of the trait to be analyzed.EnvNum The number of environments for each trait is a vector.yygg covariate matrix.genoname linkage map matrix with pseudo markers inserted.Ax0AA genotype matrix.Hx0Aa genotype matrix.Bx0aa genotype matrix.Ax AA genotype matrix with pseudo markers inserted.Hx Aa genotype matrix with pseudo markers inserted.Bx aa genotype matrix with pseudo markers inserted.dirfile storage path.CriDis The distance of optimization.CLO Number of CPUs.Valuea listExamplesdata(F2data)readraw<-Readdata(file=F2data,fileFormat="GCIM",method="GCIM-QEI",filecov=NULL,MCIMmap=NULL,MultiEnv=TRUE)DoResult<-Dodata(fileFormat="GCIM",Population="F2",method="GCIM-QEI",Model="Random",readraw,MultiEnv=TRUE)ZhouMatrices<-ZhouF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw1=DoResult$mapRaw1,WalkSpeed=1,CriLOD=3,dir=tempdir())OutputZhou<-ZhouMethod(Model="Random",pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw=ZhouMatrices$mapRaw,CriLOD=3,NUM=1,EnvNum=DoResult$EnvNum,yygg=DoResult$yygg1,genoname=ZhouMatrices$genoname,Ax0=ZhouMatrices$Ax0,Hx0=ZhouMatrices$Hx0,Bx0=ZhouMatrices$Bx0,Ax=ZhouMatrices$Ax,Hx=ZhouMatrices$Hx,Bx=ZhouMatrices$Bx,dir=tempdir(),CriDis=5,CLO=2)ZhouMethod_single_env The second step of Zhou method for single environmentDescriptionThe second step of Zhou method for single environmentUsageZhouMethod_single_env(Model=NULL,pheRaw=NULL,genRaw=NULL,mapRaw=NULL,CriLOD=NULL,NUM=NULL,yygg=NULL,genoname=NULL,Ax0=NULL,Hx0=NULL,Bx0=NULL,Ax=NULL,Hx=NULL,Bx=NULL,dir=NULL,CriDis=NULL,CLO=NULL)ArgumentsModel Random orfixed model.pheRaw phenotype matrix.genRaw genotype matrix.mapRaw linkage map matrix.CriLOD Critical LOD scores for significant QTL.NUM The serial number of the trait to be analyzed.yygg covariate matrix.genoname linkage map matrix with pseudo markers inserted.Ax0AA genotype matrix.Hx0Aa genotype matrix.Bx0aa genotype matrix.Ax AA genotype matrix with pseudo markers inserted.Hx Aa genotype matrix with pseudo markers inserted.Bx aa genotype matrix with pseudo markers inserted.dirfile storage path.CriDis The distance of optimization.CLO Number of CPUs.Valuea listExamplesdata(F2data)readraw<-Readdata(file=F2data,fileFormat="GCIM",method="GCIM-QEI",filecov=NULL,MCIMmap=NULL,MultiEnv=FALSE)DoResult<-Dodata(fileFormat="GCIM",Population="F2",method="GCIM-QEI",Model="Random",readraw,MultiEnv=FALSE)ZhouMatrices<-ZhouF(pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw1=DoResult$mapRaw1,WalkSpeed=1,CriLOD=3,dir=tempdir())OutputZhou<-ZhouMethod_single_env(Model="Random",pheRaw=DoResult$pheRaw,genRaw=DoResult$genRaw,mapRaw=ZhouMatrices$mapRaw,CriLOD=3,NUM=1,yygg=DoResult$yygg1,genoname=ZhouMatrices$genoname, Ax0=ZhouMatrices$Ax0,Hx0=ZhouMatrices$Hx0,Bx0=ZhouMatrices$Bx0,Ax=ZhouMatrices$Ax,Hx=ZhouMatrices$Hx,Bx=ZhouMatrices$Bx,dir=tempdir(),CriDis=5,CLO=2)Indexchr(DHdata),2DHdata,2Dodata,2F001(F2data),3F002(F2data),3F003(F2data),3F2data,3marker(DHdata),2markerinsert,4pos(DHdata),2QTL.gCIMapping,5Readdata,6WangF,7WangS,8WenF,10WenS,11ZhouF,12ZhouMethod,13ZhouMethod_single_env,1518。
QTL_IciMapping3.0_简单教程
QTL IciMapping3.0 定位简单应用教程张茜中国农科院2012.6.14主要步骤•数据准备•新建project•导入数据•构建图谱•QTL定位准备数据•.map格式将txt格式后缀名改成.map即可(表头信息不能动),一个map文件中包括General Information、Marker Types 、Information for Chromosomes and Markers三部分信息主要更改数据:7为F2群体;1一般不动;Marker space type 选1或2均可,只要保持数据对应Maker Types带型统计方法这些数据是标记在第几条染色体(group)上,未构建图谱侧全为0点File 选New Project新建一个工作项命名保存路径点File 选*map导入构建准备好的map格式图谱的数据打开,完成数据导入点击分组,在此处出现group群点可以看到一个group下所含标记,右键点击一个标记可以对其位置调动或者删除完成分组后,点击ordering,转换成染色体组再点此按钮完成沟通准备工作,工具栏上的map图标变蓝可以点击构图了点击map 按钮出现图谱(右)点击即可出现下一个染色体图谱点击出现整体图谱Save 可以保存各种格式的图QTL定位数据准备将构图所得结果F2bip(在project-map-result文件下)先复制一份,再用txt打开方式打开所复制文件。
Bip文件中包含5部分General Information、Information for Chromosomes andMarkers、Linkage map (Marker namefollowed by position or the interval length)、 Marker Type 、Phenotypic Data更改数据:0改成1选File-open file-*bip打开更改保存好的bip格式文件选ICIM-ADD添加的下框(一般都默认),此时start按钮从灰色变黑色,单击即可进行定位Start 完了点ADD即出现下图加性效应图显性效应图总染色体添加lod值线下一个染色体在Graph 下可以选择连锁图和lod (上)或者连锁图和QTL(下)图谱结果信息在map目录下QTL结果信息在BIP目录下信息栏补充•QTL ICIMapping是在*map(oppen file子菜单下)下完成构件图谱,在*bip(oppen file子菜单下)下完成QTL定位。
GlobalMapper软件操作教程

Global Mapper软件操作教程一软件简介Global Mapper软件可支持多类型,多数据读取,且支持不同格式的数据输出,对矢量数据的编辑和管理简便的特点,其作为Lidar数据的质检环节,所带来的和易操作性,更适应于高效生产中。
二软件工具介绍1 . 软件界面Global Mapper软件界面简洁、友好,提供多途径常用的选项,且软件的大部分菜单和工具项配置了快捷键,所以操作很方便。
Global Mapper软件界面2 . 常用工具Open Data File(s)Ctrl+O 可一次多文件读取打开数据文件Save WorspaceCtrl+S 存储个人工作环境,视图和软件的配置保存工作空间Open Control CenterAlt+C 用于数据显隐控制和层管理打开控制中心Configuration 配置和设置软件全局变量配置Zoom InPgDn 定比例放大视图放大Zoom OutPgUp 定比例缩小视图缩小Fill ViewHome 用于数据全屏显示操作,其依据所有填加的数据(矢量和三维数据)全屏Zool toolAlt+Z 感性区域放大区域放大工具Pan toolAlt+G 视图的平移操作平移工具Measure ToolAlt+M 用自定义的路径测量长度、角度、周长、面积以及其终点于所属数据高程信息测量工具Feature Info ToolAlt+P 对矢量数据的属性信息查看要素信息工具3D path Profile/Line of Sight ToolAlt+L 用于自定义的路径示意出各节点所属数据剖切高程趋势图3D路径剖面/瞄准线工具View shed ToolDigitizer ToolAlt+D 创建具有一定属性的点、线、面矢量数据,可供编辑,删除等操作矢量化工具Search by Attribute\Name\Description 快速检索类型名称区域位置及属性由属性\名称\描述查询Eanble\Disable Hill Shading 用于Tin高程的夸显示开启\关闭山丘阴影Show 3D ViewCtrl+3 数据三维视图查看,可于数据同步缩放,移动和旋转操作显示3D 视图3 . 菜单介绍Files(文件)菜单Eait (编辑)菜单View (查看)菜单Tools(工具)菜单Search (查询)菜单Digitizer Tool (矢量化工具)右键菜单三QC操作流程1.拷贝custom_shaders.txt (颜色表文件)和custom_area_types.txt (质检类型层文件)文件至GlobalMapper软件安装目录C:\Program Files\GlobalMapper10\下。
Mapmaker3.0winQTLCart2.0简单中文教程和举例

Mapmaker3.0winQTLCart2.0简单中文教程和举例内部使用Mapmaker3.0 winQTLCart2.0简单中文教程和举例胡磊(新疆农业大学农学院棉花遗传育种2006级研究生)本文是笔者自己的试验方法记录里得来的,尽量使用通俗易懂的语言,配合截图,基本上看过的人都可以学会使用。
但是由于时间紧促,难免存在疏漏和错误的地方,欢迎指正!此资料仅供参考,因此引起的后果概不负责,特此声明。
一、前期数据准备1 数据调查和采样数据调查:我们把群体中的每个材料种成行,比如3m 一行,在这一行里选取10株做调查,已果枝始节为例,调查10株,并记录在记录本或者电脑里备用。
采样:在上面说的3m 一行的里取指甲盖大小的叶片,回实验室提NDA ,跑电泳,和读带。
2 数据输入 2.1 大田数据先把你的田间调查数据输入excel ,以f2代果枝始节调查数据为例。
在最后一列算出平均数。
注意!我们需要用的就是这个平均数!数据输入的时候,材料编号那里不能出错。
2.2 实验室数据然后就是我们采回来的叶片跑的电泳记录,这里的叶片都是大田里取来的,全部需要提取DNA 和跑电泳。
最后在所跑出来的电泳条带上读取数据,读取数据的方法如下:如图所示,此图显示的是凝胶上的带型。
内用共显性标记:母本带型记为“B ”,父本带型记为“A ”,杂合带型记为“H ”。
显性标记记载有两种情况:若母本等位基因为显性,则父本带型记为“A ”,杂种带型记为“D ”;若父本等位基因为显性,则母本带型记为“B ”,杂种带型记为“C ”。
无论在显性标记还是共显性标记中,由于某些原因造成的带型不清或数据缺失均用“–”表示。
而我们在读取的时候一般用数字表示,即:1=A 2=B 3=H 4=C 5=D 。
在读带后的数据输入也要注意编号,编号一定不能出错。
3 数据处理 3.1 数据核对将DNA 的编号和大田数据进行对比,如果DNA 里有的编号,在材料编号里没有,就把那一行数据用“-”表示,单元格下移。
colmap操作流程

colmap操作流程colmap 操作流程1. 新建⼀个项⽬数据库⽂件,放在Project workplace⽂件夹下2. 点击 Processing > Feature Extraction 进⾏特征提取参数,默认即可3. 点击Processing > Feature matching 进⾏特征匹配,参数默认,时间会⽐特征提取长4. 点击reconstruction > start reconstruction 进⾏ SfM 与三⾓化建⽴稀疏点云,期间伴随着光束法平差(Bundle Adjustment)。
重建的结果是稀疏点云(就是刚刚提取的特征点三⾓化后的三维坐标)和相机位姿恢复的⽰意图。
可以把稀疏点云导出为.ply⽂件查看5. 点击reconstruction>dense reconstruction 进⼊稠密重建步骤(如果你电脑没cuda到这⼀步之后就可以结束了)6. 点击右上⾓select选择稠密重建项⽬保存的⽂件夹,可以在workplace下建⼀个dense⽂件夹来保存。
7. 点击Undistortion 进⾏图像的去畸变8. 点击Stereo 进⾏密集匹配(过程漫长)。
完成密集匹配后可以看到⽣成的深度图,colmap采⽤的是PatchMatch的倾斜窗⼝密集匹配算法。
9. 点击 Fusion 进⾏深度图融合⽣成稠密点云。
可以导出稠密点云结果将其保存。
10. 这⾥有两个选项,Possion是泊松表⾯重建,Delaunay是狄洛尼三⾓⽹重建。
11. 结果需要在Meshlab上看,打开dense⽂件夹下的meshed-possion.ply⽂件。
colmap官⽹对于流程步骤的作⽤解释Structure-from-MotionStructure-from-Motion (SfM) is the process of reconstructing 3D structure from its projections into a series of images. The input is a set of overlapping images of the same object, taken from different viewpoints. The output is a 3-D reconstruction of the object, and the reconstructed intrinsic and extrinsic camera parameters of all images. Typically, Structure-from-Motion systems divide this process into three stages:1.Feature detection and extraction2.Feature matching and geometric verification3.Structure and motion reconstructionMulti-View StereoMulti-View Stereo (MVS) takes the output of SfM to compute depth and/or normal information for every pixel in an image.Fusion of the depth and normal maps of multiple images in 3D then produces a dense point cloud of the scene.Using the depth and normal information of the fused point cloud, algorithms such as the (screened) Poisson surface reconstruction can then recover the 3D surface geometry of the scene.。
mrbase和 qtls使用方法

mrbase和 qtls使用方法以mrbase和qtls使用方法为标题,我们将分别介绍这两个工具的使用方法。
一、mrbase使用方法mrbase是一个强大的基因组和转录组数据库,提供了大量的基因组学和转录组学数据资源。
下面是mrbase使用的步骤:1. 打开mrbase官方网站,在搜索栏中输入感兴趣的基因或转录本的名称。
2. 筛选结果。
在搜索结果页面,可以根据不同的过滤选项,如物种、数据类型、样本类型等进行筛选,以找到最符合需求的数据。
3. 查看基因或转录本信息。
点击搜索结果中的基因或转录本名称,可以查看详细的信息,包括基因或转录本的序列、调控信息、表达谱图等。
4. 下载数据。
在基因或转录本信息页面,可以选择下载所需的数据文件,如基因组序列、表达谱数据等。
5. 分析数据。
将下载的数据文件导入到相应的分析软件中,进行后续的数据分析和挖掘。
二、qtls使用方法qtls是用于研究基因型和表型之间关联的一种工具,可以帮助研究人员发现基因型与表型之间的遗传关系。
下面是qtls使用的步骤:1. 准备数据。
首先需要准备基因型和表型数据,基因型数据可以是SNP、基因表达数据等,表型数据可以是某种性状的测量值。
2. 安装qtls软件。
根据操作系统的不同,选择合适的版本并下载安装qtls软件。
3. 导入数据。
打开qtls软件,通过菜单或命令行将准备好的基因型和表型数据导入到软件中。
4. 运行分析。
在qtls软件中选择适当的分析方法,如GWAS、eQTL等,设置参数并运行分析。
5. 结果解读。
分析完成后,软件会生成相应的结果文件,可以通过软件内置的工具进行结果可视化和解读,以发现基因型和表型之间的关联。
6. 结果验证。
为了验证结果的可靠性,可以进行重复实验或与其他研究结果进行比对。
以上是mrbase和qtls使用的基本方法。
使用这两个工具可以帮助研究人员更好地理解基因组和转录组的特征,并发现基因型和表型之间的关联。
希望这些信息对您有所帮助。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
QTL IciMapping3.0 定位简单应用教程
张茜
中国农科院
2012.6.14
主要步骤
•数据准备
•新建project
•导入数据
•构建图谱
•QTL定位
准备数据
•.map格式
将txt格式后缀名改成.map即可(表头信息不能动),一个map文件中包括General Information、Marker Types 、Information for Chromosomes and Markers三部分信息
主要更改数据:7为F2群体;1一般不动;Marker space type 选1或2均可,只要保持数据对应
Maker Types
带型统计方法
这些数据是标记在第几条染色体(group)上,未构建图谱侧全为0
点File 选New Project新建一个工作项
命名
保存路径
点File 选*map导入构建准备好的map格式图谱的数据打开,完成数据导入
点击分组,
在此处出现group群
点可以看到一个group下所含标记,右键点击一个标记可以对其位置调动或者删除
完成分组后,
点击ordering,
转换成染色体组再点此按钮完成沟通准备工作,
工具栏上的map图标变蓝可以点击构图了
点击map 按钮出现图谱
(右)
点击即可出现下一个染色体图谱
点击出现整体图谱
Save 可以保存各种格式的图
QTL定位
数据准备
将构图所得结果F2bip(在project-map-result文件下)先复制一份,再用txt打开方式打开所复制文件。
Bip文件中包含5部分General Information、Information for Chromosomes and
Markers、Linkage map (Marker name
followed by position or the interval length)、 Marker Type 、Phenotypic Data
更改数据:0改成1
选File-open file-*bip打开更改保存好的bip格式文件
选ICIM-ADD添加的下框
(一般都默认),此时start按钮从灰色变黑色,单击即可进行定位
Start 完了点ADD即出现下图
加性效应图显性效应图总染色体添加lod值线
下一个染色体
在Graph 下可以选择连锁图和lod (上)或者连锁图和QTL(下)
图谱结果信息在map目录下QTL结果信息在BIP目录下
信息栏
补充
•QTL ICIMapping是在*map(oppen file子菜单下)下完成构件图谱,在*bip(oppen file子菜单下)下完成QTL定位。
•前面是以map格式构建图谱,再在其结果中(bip文件)添加表型数据进行定位。
•也可以用excel数据构建图谱和定位,同样是在*map下完成构建图谱和在*bip下完成QTL定位(在导入数据时选中)
标记名标记名
•第一列为标记名称,
•第二列为第几条染色体,
•第三列为标记间距或位点,这一列数据要
与GeneralInfo相对应!
性状数据
和构建图谱一样
其他步骤不变
谢谢!。