《生物信息学》上机作业
生物数据上机实验报告

一、实验名称生物数据上机实验二、实验目的1. 熟悉生物数据处理的常用软件及其基本操作。
2. 学习生物数据的整理、分析和可视化方法。
3. 培养对生物数据的敏感性和分析能力。
三、实验原理生物数据是指生物科学研究中收集到的各种数据,包括基因组学、蛋白质组学、代谢组学等领域的实验数据。
本实验旨在通过上机操作,学习如何使用生物信息学软件对生物数据进行整理、分析和可视化,从而更好地理解生物学现象和规律。
四、实验器材1. 电脑2. 生物信息学软件(如R、Python、MATLAB等)3. 生物数据集五、实验步骤1. 数据整理- 下载并导入生物数据集。
- 检查数据完整性,包括数据类型、缺失值等。
- 对数据进行清洗,去除异常值和噪声。
2. 数据分析- 使用R或Python等软件进行数据分析。
- 根据实验目的,选择合适的统计方法进行分析,如相关性分析、差异分析等。
- 使用可视化工具(如ggplot2、Seaborn等)展示分析结果。
3. 结果可视化- 将分析结果以图表形式展示,如散点图、柱状图、热图等。
- 对图表进行美化,包括字体、颜色、标题等。
4. 结果讨论- 根据分析结果,对生物学现象进行解释和讨论。
- 提出进一步研究的方向和假设。
六、实验结果1. 数据整理- 导入数据集:成功导入基因组学数据集,数据包含基因表达水平、样本信息等。
- 数据检查:发现数据集中存在缺失值,已进行清洗处理。
2. 数据分析- 相关性分析:分析基因表达水平与样本信息之间的相关性,发现某些基因与样本类型之间存在显著相关性。
- 差异分析:分析不同样本类型之间的基因表达差异,发现某些基因在特定样本类型中表达水平显著升高或降低。
3. 结果可视化- 散点图:展示基因表达水平与样本信息之间的相关性。
- 柱状图:展示不同样本类型中基因表达水平的差异。
- 热图:展示基因表达水平的聚类情况。
4. 结果讨论- 根据分析结果,推测特定基因可能与特定样本类型相关,进一步研究该基因在生物学过程中的作用。
生物信息学分析上机实验教学大纲

生物信息学分析上机实验教学大纲一、制定本大纲的依据依据《生物信息学分析教学大纲》制定本上机实验大纲。
生物信息学是当今生命科学和自然科学的核心领域和最具活力的前沿领域之一,是一门新兴的交叉学科,是现代生物学研究的重要工具。
它所研究的材料是生物学的数据,而它进行研究所采用的方法,则是从各种计算技术衍生出来的。
随着Internet的广泛应用和基因组研究的深入进行,生物信息学也得到了飞速的发展。
只有通过系统的理论学习和实际的上机操作,才能使学生了解当今生物信息学网络资源,学会常用生物信息数据库查询、数据库搜索方法、生物大分子序列分析和分子进化分析软件等的使用方法,初步解决科研和实际工作中生物信息的存储、检索、分析和利用的问题。
二、本实验课程的具体安排实验项目的设置及学时分配三、本实验课在该课程体系中的地位与作用根据《生物信息学分析教学大纲》开设的上机实验,能够使学生掌握生物信息学的基础知识与概念,了解生物信息学网络资源,实践具体的操作方法。
培养学生具有生物信息学方面的理论基础和基本技能,并且能够运用所掌握的生物信息学理论、方法和技术,初步解决科研和实际工作中生物信息的存储、检索、分析和利用的问题。
四、学生应达到的实验能力与标准:通过上机实验的开设,学生应了解生物信息学的主要内容, 理解生物信息技术的原理和应用领域,掌握并能使用生物信息学的基本工具,提高分析和解决实际问题的能力,为今后开展相关研究打下基础。
通过上机实验具体的操作过程,学生应达到以下要求:1、熟悉并掌握各生物数据库的查询检索方法。
2、了解生物大分子结构生物信息学的内容与分析方法。
3、熟悉网上数据分析预测工具的使用。
4、培养学生进行生物绘图、生物计算、数据处理、分析结果的基本能力。
5、培养学生独立从事科研实验的技能和素养、与组员分工合作能力及对在上机实验过程中遇到问题的解决能力。
五、上机实验的基本理论与实验技术知识:实验一常用分子生物学数据库的使用基本要求:了解生物信息学的各大门户网站以及其中的主要资源,掌握主要数据库的内容及结构,理解各数据库注释的含义。
生物信息学作业(一)

生物信息学实验作业一1、了解NCBI、DDBJ、EMBL上网的方法自学各网站相关介绍。
答:(1)、NCBI: (National Center of Biotechnology Information,简称NCBI)美国国立生物技术信息中心。
其主页为:。
NCBI 是在NIH的国立医学图书馆(NLM)的一个分支。
NLM是因为它在创立和维护生物信息学数据库方面的经验被选择的,而且这可以建立一个内部的关于计算分子生物学的研究计划。
NCBI的任务是发展新的信息学技术来帮助对那些控制健康和疾病的基本分子和遗传过程的理解。
NCBI有一个多学科的研究小组包括计算机科学家,分子生物学家,数学家,生物化学家,实验物理学家,和结构生物学家,集中于计算分子生物学的基本的和应用的研究。
他们一起用数学和计算的方法研究在分子水平上的基本的生物医学问题。
这些问题包括基因的组织,序列的分析,和结构的预测。
在1992年10月,NCBI承担起对GenBank DNA序列数据库的责任。
NCBI 受过分子生物学高级训练的工作人员通过来自各个实验室递交的序列和同国际核酸序列数据库(EMBL和DDBJ)交换数据建立起数据库。
同美国专利和商标局的安排使得专利的序列信息也被整合。
BLAST是一个NCBI开发的序列相似搜索程序,还可作为鉴别基因和遗传特点的手段。
BLAST能够在小于15秒的时间内对整个DNA数据库执行序列搜索。
NCBI提供的附加的软件工具有:开放阅读框寻觅器(ORF Finder),电子PCR,和序列提交工具,Sequin和BankIt。
所有的NCBI数据库和软件工具可以从WWW 或FTP来获得。
NCBI还有E-mail服务器,提供用文本搜索或序列相似搜索访问数据库一种可选方法。
主要任务:(1)建立关于分子生物学,生物化学,和遗传学知识的存储和分析的自动系统(2)实行关于用于分析生物学重要分子和复合物的结构和功能的基于计算机的信息处理的,先进方法的研究(3)加速生物技术研究者和医药治疗人员对数据库和软件的使用。
2017 研究生 生物信息学 蛋白部分(上机)_2

分析蛋白质的跨膜区
直接输入http://embnet.vital-it.ch/software/TMPRED
采用的氨基酸标度 采用Tmbase作为跨膜蛋白数据库
最短和最长的跨膜螺旋疏水区长度 选择合适的输入格式
1、贴入蛋白质序列 2、选择合适的参数 3、运行计算
氨基酸数量 分子量 理论等电点
氨基酸组成
消光系数
预测半衰期 不稳定系数 脂肪氨基酸系数
GRAVY值
消光系数—反映了蛋白在特定波长下吸收可见光或 不可见光的能力,可用来测蛋白浓度。
不稳定系数—预测对应蛋白质在试验中稳定性。
小于40时,预测蛋白稳定
大于40时,预测蛋白不稳定
脂肪系数—计算球状蛋白脂肪族氨基酸侧链所占相 对体积,反映了蛋白质的热稳定性。
比对的数据库
该序列结构域信息 (PROSITE数据库信息)
图形化比对结果
可能匹配的序列列表
BLAST结果评价
Score:使用打分矩阵对匹配的片段进行打分,这是
对各对氨基酸残基(或碱基)打分求和的结果,一般来 说,匹配片段越长、 Score值越大,则相似性越高 。
E value:在相同长度的情况下,两个氨基酸残基(或
选择“TMHMM”分析软件 ( http://www.cbs.dtu.dk/services/TMHMM-2.0/)
在TMHMM主页粘贴序列进行分析
分析蛋白质的跨膜区
直接输入 http://embnet.vital-it.ch/software/TMPRED
1、输入序列 2、运行软件
结果输出
1、胞外区 2、跨膜区 3、胞内区
选择“protparam”分析软件 ( /protparam/)
生物信息学作业

生物信息学作业
一、Blast搜索
首先在NCBI的网页上打开Blast的网页,找到需要的数据库类型。
查询的序列直接粘贴到序列框中
1、可在该页面Algorithm parameters”栏目中更改相关的参数
2、点击BLAST以及Show results in a new window选择用新窗口展示分析结
果
3、点击“Formatting options”,在新网页选择变换格式
如:将其改变为Pairwise with dots for identities”格式
4、通过选择几个需要比较的序列,然后点击Distance tree of results”显示检索到的序列之间的同源关系
5、结果显示为:
6、保存:选择需要的序列,按Download保存
二、在记事本中可得到结果
比对法
一:ClustalW比对法
1、进入http://www.expasy.ch网页
2、在查找框中找到Find resources 以及ClustalW,得到页面
3、点击Clastw得
4、可在该页面上进行先关参数的设计,同时可在框中输入需要比对的序列,按下Run Clustalw可得比对结果
(由于网速问题只能进行到该阶段)
二、CLUSTAL X对比法
1、打开相应软件
将需要比对的序列从软件中导入
2、可对相关的参数进行设计:即按Alignment中Alignment Paramenter下的Multiple Alignment Paramenter即可进行
3、比对:按下Alignment中Do Complete Alignment即可得到比对结果
4、保存:。
研究生 生物信息学 蛋白部分(上机)_1[30页]
![研究生 生物信息学 蛋白部分(上机)_1[30页]](https://img.taocdn.com/s3/m/2c2e9d8f83c4bb4cf6ecd138.png)
蛋白质表达
蛋白质参与的相互作用
STRING数据库中 SOD1蛋白与其他蛋白相互作用信息
蛋白质结构
点击进入PDB数据库中该蛋白的链接
蛋白家族和结构域
Prosite数据库中的保守结构域
Prosite数据库中的人SOD1蛋白的保守结构域
蛋白质序列
FASTA格式序列
蛋白涉及5条通路 点击进入详细条目
SOD1参与的过氧化物酶途径
蛋白质在其他数据库中的链接
SOD1 相关的文献
课堂练习作业: 查询人类P53蛋白,说明其主要功能、主
要的结构域、主要的翻译后修饰、参与的代 谢途径、相互作用的蛋白、主要涉及的疾病
GO分析 分子功能、生物过程
Gene Ontology(GO分类)
Gene Ontology包含了基因参与的生物过程,所处的细 胞位置,发挥的分子功能三方面功能信息,并将概念 粗细不同的功能概念组织成DAG(有向无环图)的结 构。
Gene Ontology是一个使用有控制的词汇表和严格定义 的概念关系,以有向无环图的形式统一表示各物种的 基因功能分类体系,从而较全面地概括了基因的功能 信息。
UniProKB数据库
实例: 获取SOD1人超氧化物歧 化酶的功能及结构信息。
输入
以人类SOD1为例,介绍Uniprot数据库中贮存形式
选择目标数据库
输入目标蛋白
点击查找
最常见物种
选择物种为人的SOD1蛋白
快速导航栏 方便查找
蛋白主要的功能
特征序列注释
KEGG通路分析
KEGG日本京都基因和基因组百科全书
全球影响力最大的代谢数据库之一,它的生物学 途 径 ( pathway ) 数 据 库 有 细 分 成 代 谢 ( metabolism ) 、 遗 传 信 息 处 理 ( genetic information processing ) 、 环 境 信 息 处 理 (environmental information processing)细胞代谢 (cellular process)和人类疾病(human disease)5 个方面
生物信息学作业1.doc

生物信息学实验作业试验一一.找到编码拟南芥(arabidopsis)phyA(光敏色素A)基因的核酸序列编号, 并记录查找过程。
GI:224576211步骤1.进入NCBI主页2.搜索arabidopsis phyA3.Arabidopsis thaliana phytochrome A (PHYA) gene, partial cds4.VERSION:GI:224576211二.以phyA为检索词,在pubmed数据库中分别检索在题目和关键词字段中含有该检索词的文献,记录检索出的条目数目。
Results: 614三.仔细阅读所查询核酸序列在NCBI和EMBL数据库中格式的解释,理解各字段的含义,并比较NCBI 与EMBL中序列格式的异同。
实验二一.分析你感兴趣核酸序列的分子质量、碱基组成。
Composition 35 A; 25 C; 35 G; 15 T; 0 OTHERPercentage: 32% A; 23% C; 32% G; 14% T; 0%OTHERMolecular Weight (kDa): ssDNA: 34.26 dsDNA: 67.8二.列出你所分析核酸序列(或部分序列)的互补序列、反向序列、反向互补序列、DNA双链序列和RNA 序列。
R S1 ACTACTCGAG AAGCAGCGAC AGAGGCGTTA GCCCGCTCAG CAGACTGGCA GTTCTCTACC61 GACAAAAAAG AGGTAGGAGG CACAGTAATG ATACAGGCGT AGCAGGAGGGC S1 CCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 TGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTR C S1 TGATGAGCTC TTCGTCGCTG TCTCCGCAAT CGGGCGAGTC GTCTGACCGT CAAGAGATGG61 CTGTTTTTTC TCCATCCTCC GTGTCATTAC TATGTCCGCA TCGTCCTCCCD DNA S1 GGGAGGACGA TGCGGACATA GTAATGACAC GGAGGATGGA GAAAAAACAG CCATCTCTTGCCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 ACGGTCAGAC GACTCGCCCG ATTGCGGAGA CAGCGACGAA GAGCTCATCATGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTRNA S1 GGGAGGACGA UGCGGACAUA GUAAUGACAC GGAGGAUGGA GAAAAAACAG CCAUCUCUUG61 ACGGUCAGAC GACUCGCCCG AUUGCGGAGA CAGCGACGAA GAGCUCAUCA三.列出核酸序列的限制性酶切位点分析结果(酶及识别位点)。
生物信息学习题

GTATCACACG ACTCAGCGCA GCATTTGCCC
GTATCACATA GCTCAGCGCA GCATTTGCCC
6、对于下列距离矩阵,用 UPGMA 构建系统发生树。
ABCDE
A0
B3 0
C6 5 0
D 9 9 10 0
E 12 11 13 9 0 7、对下面距离矩阵,用 UPGMA 法构建系统发生树
1、蛋白质得分矩阵类型有 、
、、
和
等。
2、对位排列主要有局部比对和 三、运算题 1、画出下面两条序列的简单点阵图。将第一条序列放在 x 坐标轴上,将第二条序列放在 y
坐标轴上。 TGAACTCCCTCAGATATTA CGAACCCTCACATATTAGCG
2、对两个核酸序列 ACACACTA 和 AGCACACA 进行全局比对
第八章 后基因组时代的生物信息学(问题与练习)
1、 比较生物还原论与生物综合论的异同 2、 简述“后基因组生物信息学”的基本研究思路 3、 后基因组生物信息学的主要挑战是什么? 4、 功能基因组系统学的基本特征是什么? 5、 说明后基因组生物信息学对信息流动的最新理解 6、 列举几种预测蛋白质-蛋白质相互作用的理论方法 7、 解释从基因表达水平关联预测蛋白质-蛋白质相互作用的理论方法 8、 解释基因保守近邻法预测蛋白质-蛋白质相互作用的理论方法 9、 解释基因融合法预测蛋白质-蛋白质相互作用的理论方法 10、解释种系轮廓发生法预测蛋白质-蛋白质相互作用的理论方法
正确的树的可能性比前一种情况大还是小?
5、对于下列 5 条序列的比对构造一个距离矩阵,其中序列之间的距离值为比对中失配的碱
基数目,但是颠换的权值为转换的两倍。
GTGCTGCACG GCTCAGTATA GCATTTACCC
生物信息 上机
生物信息学 Bioinformatics
1
bioinformation2009@ 密码: 密码: bioinformation 进入以上公用邮箱下载幻灯片 2009-11- 上机1 “2009-11-8上机1” 到电脑桌面
2
第三章
生物信息学数据库资源
3
主要的数据库资源
核酸序列数据库主要有 核酸序列数据库主要有GenBank, EMBL, DDBJ等. 主要有 等 蛋白质序列数据库主要有 蛋白质序列数据库主要有SWISS-PROT, PIR, 主要有 TrEMBL等, 等 蛋白结构数据库有 蛋白结构数据库有PDB, MMDB等, 等 与基因组有关的数据库还有dbEST, OMIM等, 与基因组有关的数据库还有 等
15
如何获取GenBank中的序列 中的序列? 如何获取 中的序列
16
如何获取GenBank中的序列 中的序列? 如何获取 中的序列 同样存在限制字段: 同样存在限制字段 常用的有: Author: Zhang H[au] 常用的有 title: Zinc[ti] organism: rice[organism] 如: Zhang H[au] AND zinc[ti] AND rice[organism] 或者直接输入: 或者直接输入 Accession: AY077725[Accession] Gene Name: ZFP15[Gene Name] Protein Name: ZFP15[Protein Name]
12
复杂检索
2. 布尔逻辑运算: 布尔逻辑运算: AND、OR、NOT必须大写。 、 必须大写。 、 必须大写 逻辑符的运算次序是从左至右, 逻辑符的运算次序是从左至右,括号内的检索式可作为一个 左至右 单元,优先运行。 单元,优先运行。 布尔逻辑检索允许在检索词后面附加字段标识 布尔逻辑检索允许在检索词后面附加字段标识
生物信息学上机指南3

《生物信息学》上机指南(三)实验三、分子系统发育分析 2学时教学要求:1、了解系统发育分析原理、步骤、方法。
2、掌握phylip、Mega等软件的下载与使用。
3、学习进化树结果分析。
重点掌握phylip、Mega的使用。
实验步骤:1.基于细胞色素c氨基酸序列的真核生物系统发育分析细胞色素c(cytochrome c)是一种含血红素的电子转运蛋白,它存在于所有真核生物的线粒体中,参加呼吸作用。
细胞色素c的氨基酸顺序分析资料已经用来核对各个物种之间的分类学关系,以及绘制进化树。
本实验利用Mega软件,采用邻位相接法,构建43种真核生物细胞色素c系统进化树。
类群中文名称拉丁学名蛋白质登录号哺乳类人Homo sapiens P99999黑猩猩Pan troglodytes P99998恒河猴Macaca mulatta P00002大袋鼠Macropus giganteus P00014家兔Oryctolagus cuniculus P00008小家鼠Mus musculus CAA25899 马Equus caballus P00004绵羊Ovis aries P62896牛Bos taurus P62894野猪Sus scrofa P62895狗Canis familiaris P00011南象海豹Mirounga leonina P00012长翼蝠Miniopterus schreibersii P00013河马Hippopotamus amphibius P00007鸟类鸸鹋Dromaius novaehollandiae P00018 鸵鸟Struthio camelus P00019 原鸡Gallus gallus P67881 火鸡Meleagris gallopavo P67882企鹅Aptenodytes patagonicus P00017 家鸽Columba livia P00021绿头鸭Anas platyrhynchosP00020爬行类拟鳄龟Chelydra serpentina P00022 两栖类牛蛙Rana catesbeiana P00024硬骨鱼类长鳍金枪鱼Thunnus alalunga P81459 太平洋鲣鱼Katsuwonus pelamis P00025 斑马鱼Danio rerio Q6IQM2软骨鱼类角鲨Squalus sucklii P00027 圆口类七鳃鳗Entosphenus tridentatus P00028 棘皮动物红海星Asterias rubens P00029 环节动物赤子爱胜蚓Eisenia fetida P00030昆虫沙漠蝗Schistocerca gregaria P00040 烟草天蛾Manduca sexta P00039 眉纹天蚕蛾Samia cynthia P00037 铜绿蝇Lucilia cuppina P00036植物小麦Triticum aestivum P00068水稻Oryza sativa BAA02159 向日葵Helianthus annuus P00070菠菜Spinacia oleracea P00073银杏Ginkgo biloba P00074芝麻Sesamum indicum P00054真菌毕赤酵母Pichia anomala P00042 白色念珠菌Candida albicans P53698 粗糙脉胞菌Neurospora crassa P000481.1.序列获取(1) 用记事本将蛋白质登录号粘进去,每个登录号占一行,存为Sequence_ID.txt。
生物信息学作业题

生物信息学作业题生物信息学作业题绪论1.什么是生物信息学?2.生物信息学有哪些主要研究领域?第一章生物信息学的分子生物学基础1.DNA的双螺旋结构要点是什么?2.什么是基因组和蛋白质组?对它们的研究有何意义?第二章生物信息学的计算机基础1.简述网络操作系统的类型。
第三章核酸序列分析1.什么是全局比对?2.什么是局部比对?有哪些优点?第四章分子进化分析1.分子进化分析具有哪些优点?2. 简述分子进化的中性学说。
第五章基因组分析1. 什么是基因组学?其主要研究内容是什么?2.简述基因预测分析的一般步骤。
第六章蛋白质组分析1. 蛋白质组学的概念和主要研究的大致方向是什么?2. 蛋白质组功能预测的程序是怎样的?第七章生物芯片数据分析1. 什么是生物芯片?2. 生物芯片有哪些方面的应用?第八章核酸与蛋白质结构预测1. RNA二级结构典型的预测方法有哪些?2. 基于统计学的预测蛋白质二级结构的方法有哪些?第九章生物信息学平台与工具软件1. 请利用Clustal X软件对下列6条蛋白质序列进行多重比对(比对结果用BioEdit软件打开,用“截图”方式显示比对结果)。
>1mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>2mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl>3mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>4mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl>5mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>6mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl2. 现有一ZmPti1b蛋白质序列,请用DNAMAN软件分析其二级结构,给出分析结果。
生物信息上机作业

生物信息学上机作业上机一生物信息数据库信息检索上机内容:1、了解NCBI、DDBJ、EMBL上网的方法自学各网站相关介绍。
2、了解北大生物信息学中心等几大中文生物信息学网站。
3、了解一些生物论坛中有关生物信息学的部分。
如:Biooo和Bioon。
4、利用NCBI的Entrenz查询系统和EBI的SRS检索文献和核酸或蛋白质序列。
(phyA)并对照所学复习各字段的含义。
5、将所得记录的ID或Accession记录下来备用。
作业:1、记录相关网站及论坛网址(或如何查询到该网址的方法)。
(1)NCBI :/(2)DDBJ :http://www.ddbj.nig.ac.jp/(3)EMBL :/(4)北大生物信息学中心 /chinese/(5)中科院计算所智能信息处理重点上机室生物信息学:/index.php(6)北大生物信息中心:/chinese/documents/bioinfor/overview/web1/1.html (7)生物谷生物信息学:/bioinfo.htm(8)中国生物论坛:/(9)中国生物谷论坛:/(10)生物谷:/2、找到编码拟南芥(arabidopsis)phyA(光敏色素A)基因的核酸序列编号。
并记录查找过程。
上机二核酸及蛋白质序列的比对一、上机内容利用检索出的蛋白质和核酸序列进行序列比对并进行分子进化树分析。
二、作业1、绘制分子进化树,并标明各个物种phyA蛋白之间的序列相似性。
2、根据你所学生物分类的知识,试解释该分子进化树的合理性。
3、找出一条可能的保守序列(多条蛋白共同的氨基酸序列)。
上机三核酸序列分析(一)一、上机内容1、使用DNAstar进行核酸基本信息分析2、ORF分析二、作业1、记录拟南芥phyA NM_100828序列的序列组成2、记录拟南芥phyA NM_100828序列最长的ORF的起止区间。
上机四核酸序列分析(二)一、上机内容1、PCR引物设计2、核酸序列的电子基因定位二、作业1、记录拟南芥phyA NM_100828序列最长的ORF的起止区间。
生物信息学上机实验4 用DNAMAN软件进行引物设计

生物信息学上机实验四用DNAMAN软件设计PCR引物一、目的要求DNAMAN 是一种常用的核酸序列分析软件。
由于它功能强大,使用方便,已成为一种普遍使用的DNA序列分析工具。
通过本实验,使学生掌握PCR引物的设计方法。
二、实验准备DNAMAN的使用说明书(word文档)一份、DNAMAN软件5.2.2版本、实验分析所用的4个序列见下面。
三、实验内容1、将待分析4个序列装入4个Channel,熟悉Channel的使用方法2、显示“序列(2)”的反向互补序列、互补序列、反向序列3、分析“序列(3)”的限制性酶切位点4、设计一对引物扩增“序列(1)”中的微卫星重复区域四、作业将上述前5项操作所得结果保存到电脑桌面,发到xiaopingjia@(1)CCAGA TGAGCGTGCGTTCGTTCCACGTACGTGTGCTGTGTGAGACGACACA TCT GCACCTGCACGTCAGCACGTACGTGCACCCGGTA TGTGTGCGCGTGTACTTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTCTGAGA TGAGGCCGGA TTCAGGA GCTGCGAGCTCA TAGGCCACAGTCACAGAA TTGCAACGGTACTTCAGTTCAGTCA TCTCCTAGTCCTTGAGAG(2)GGAAAAAAGA TACGTA TGTACA TA TACGTGTACGTGTGTGTGTGTGTGTGTGTGT GTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGAAGCAAAGACA TTGA TA TTGTTGCTGGTGGCGAGGTTGA TGCGCACAGCTCACTCCCGCGCTGACTGACACG(3)GGTCAGCAGAAAGCA TGCCGTAGTCAAACGA TCGACCTAGCTAGTAGCAGTGTG TGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTTTTGCAAAAACCTAGACCTTAGCAGCCTAG(4)CCTGA TTTGGA TCCAACAAAA TGCA TTTGACCA TA TAGTGTGTGTGTGTGTGTGTG TGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTTCACAGTCACAGAA TTGCAACGGTACTTCAGTTCAGTCA TCTCCTAGTCCTTGAGAG(2)题 SEQ DNAMAN2: 172 bp;Composition 57 A; 62 C; 22 G; 31 T; 0 OTHERPercentage: 33.1% A; 36.0% C; 12.8% G; 18.0% T; 0.0%OTHERMolecular Weight (kDa): ssDNA: 52.42 dsDNA: 106.04COLOURSsequence = 1features = 0ORIGIN1 CGTGTCAGTC AGCGCGGGAG TGAGCTGTGC GCATCAACCT CGCCACCAGC AACAATATCA 61 ATGTCTTTGC TTCACACACA CACACACACA CACACACACA CACACACACA CACACACACA 121 CACACACACA CACACACACG TACACGTATA TGTACATACG TATCTTTTTT CCSEQ DNAMAN2: 172 bp;Composition 57 A; 62 C; 22 G; 31 T; 0 OTHERPercentage: 33.1% A; 36.0% C; 12.8% G; 18.0% T; 0.0%OTHERMolecular Weight (kDa): ssDNA: 52.42 dsDNA: 106.04COLOURSsequence = 1features = 0ORIGIN1 CCTTTTTTCT ATGCATACAT GTATATGCAC ATGCACACAC ACACACACAC ACACACACAC 61 ACACACACAC ACACACACAC ACACACACAC ACACACACAC TTCGTTTCTG TAACTATAAC 121 AACGACCACC GCTCCAACTA CGCGTGTCGA GTGAGGGCGC GACTGACTGT GCSEQ DNAMAN2: 172 bp;Composition 31 A; 22 C; 62 G; 57 T; 0 OTHERPercentage: 18.0% A; 12.8% C; 36.0% G; 33.1% T; 0.0%OTHERMolecular Weight (kDa): ssDNA: 53.79 dsDNA: 106.04COLOURSsequence = 1features = 0ORIGIN1 GCACAGTCAG TCGCGCCCTC ACTCGACACG CGTAGTTGGA GCGGTGGTCG TTGTTATAG T 61 TACAGAAACG AAGTGTGTGT GTGTGTGTGT GTGTGTGTGT GTGTGTGTGT GTGTGTGTGT 121 GTGTGTGTGT GTGTGTGTGC ATGTGCATAT ACATGTATGC ATAGAAAAAA GG(3)题 Restriction analysis on DNAMAN3Methylation: dam-No dcm-NoScreened with 180 enzymes, 19 sites foundAluI AG/CT 1: 40BbvI GCAGCNNNNNNNN/ 1: 151BsaOI CGRY/CG 1: 32Bst71I GCAGCNNNNNNNN/ 1: 151DdeI C/TNAG 1: 135DpnI GA/TC 1: 31Fnu4HI GC/NGC 1: 140MaeI C/TAG 4: 37, 41, 129, 144MboI /GATC 1: 29NlaIII CATG/ 1: 17NspI RCATG/Y 1: 17PvuI CGAT/CG 1: 32Sau3AI /GATC 1: 29SphI GCATG/C 1: 17TaqI T/CGA 1: 32XorII CGAT/CG 1: 32List by Site Order17 NlaIII 31 DpnI 37 MaeI 140 Fnu4HI17 SphI 32 TaqI 40 AluI 144 MaeI17 NspI 32 PvuI 41 MaeI 151 BbvI29 Sau3AI 32 BsaOI 129 MaeI 151 Bst71I 29 MboI 32 XorII 135 DdeINon Cut EnzymesAatII Acc65I AccI AccII AccIII AclIAcyI AflII AflIII AgeI AhaIII Alw26IAlw44I AlwNI ApaBI ApaI ApaLI AscIAsp718I AsuI AsuII AvaI AvaII AvrIIBalI BamHI BanI BanII BbeI BbvIIBclI BglI BglII Bpu1102I BsaHI Bsc91IBsiI BsmI Bsp1286I Bsp1407I BspHI BspMIBspMII BssHII BstD102I BstEII BstNI BstXIBsu36I Cfr10I CfrI ClaI Csp45I CspICvnI DraI DraII DraIII DrdI EagIEam1105I Ecl136II Eco31I Eco47III Eco52I Eco56IEco57I Eco72I EcoHI EcoICRI EcoNI EcoRIEcoRII EcoRV EheI EspI FnuDII FokIFseI HaeII HaeIII HgaI HgiAI HhaIHindII HindIII HinfI HinP1I HpaI HpaIIHphI I-PpoI KpnI MaeII MaeIII MboIIMfeI Mlu113I MluI MnlI MscI MseIMspA1I MspI MstI MstII NaeI NarINcoI NdeI NheI NlaIV NotI NruINsiI NspBII PacI PflMI PinAI PleIPmaCI PmeI PpuMI PssI PstI PvuIIRleAI RsaI SacI SacII SalI SapISauI ScaI SciI ScrFI SduI SfaNISfiI SgrAI SmaI SnaBI SpeI SplISpoI SrfI SspI SstI SstII StuIStyI SunI SwaI ThaI Tth111I Tth111IIVspI XbaI XcmI XhoI XhoII XmaIXmaIII XmnIRestriction sites on DNAMAN3MaeIAluIMaeIXorIIBsaOIPvuITaqINspI DpnISphI MboINlaIII Sau3AI1 GGTCAGCAGAAAGCATGCCGTAGTCAAACGATCGACCTAGCTAGTAGCAGTGTGTGTGTGCCAGTCGTCTTTCGTACGGCATCAGTTTGCTAGCTGGATCGATCATCGTCACACACACAC61 TGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTTTTACACACACACACACACACACACACACACACACACACACACACACACACACACACACAAAAMaeIMaeI DdeI Fnu4HI121 GCAAAAACCTAGACCTTAGCAGCCTAGCGTTTTTGGATCTGGAATCGTCGGATC(4)题Primer List29 CGTGTGCTGTGTGAGAC 51.9癈 and 224 GGAGATGACTGAACTGAAG 50.1癈30 GTGTGCTGTGTGAGACG 51.9癈 and 224 GGAGATGACTGAACTGAAG 50.1癈32 GTGCTGTGTGAGACGAC 50.7癈 and 224 GGAGATGACTGAACTGAAG 50.1癈。
生物信息学课堂操作练习

生物信息学课堂操作练习一、生物信息学科的发展和研究内容通过下列internet上的自教课程,初步了解不同的数据库和分析工具/2can/Education二、生物数据库1. 熟悉各种数据库。
2. 重点了解GenBank和SWISS-PROT所包含的各种功能和适用范围。
三、关键词或词组为基础的数据库检索1. 熟练掌握Entrez检索体系。
2. 查找与水稻抗病基因Xa21有关的资料(1) 由多少碱基构成?编码多少个氨基酸?(2) exon和intron的位置?(3) 是否有3-D structure数据?1) 由多少碱基构成?编码多少个氨基酸?4623b.p., 1025A.a.;2) exon和intron的位置?Exon: 24~2700,3543~3943 intron: remaining;3) 是否有3-D structure数据?没有.3. 查找C. elegans基因组的资料。
(1) chromosome I的测序是否已完成?(2) 已知的chromosome I的序列有多少碱基?序列发表在哪份杂志上?期号和页码?1) chromosome I的测序是否已完成?完成.2) 已知的chromosome I的序列有多少碱基? 序列发表在哪份杂志上? 期号和页码? 15.0724Mb.p.(15072421b.p.), Science 1999 Jan 1;283(5398):35.4. 查看人类基因组第1染色体上基因的分布。
/mapview/maps.cgi?ORG=hum&MAPS=ideogr,est,loc&LINKS= ON&VERBOSE=ON&CHR=15. 查看Arabidopsis的系谱树,以及Arabidopsis第1染色体上的序列。
比较Arabidopsis基因组的资料提供形式与人类基因组有什么不同(/Taxonomy/Browser/wwwtax.cgi?id=3701,/mapview/maps.cgi?taxid=3702&chr=1)貌似没什么区别……比较Arabidopsis基因组的资料提供形式与人类基因组有什么不同。
生物信息学作业1---开放读码框与预测序列启动子(何婷,学号;1302008)

何婷学号:1302008 专业:病理学与病理生理学
1.使用Entrez信息查询系统检索与自己课题相关的基因核酸序列,预测开放读码框,并使用PromoterScan预测该序列中的启动子。
①查询HPV的核酸序列:输入网址:/,打开NCBI主页,在
检索窗口的选择数据库的下拉菜单选中Nucleotide项,在它右侧的文本输入栏输入检索词“HPV”,再点击“Search”按钮。
如下图所示:
搜索结果,如下图所示:
显示结果,如下图所示:
② 预测开放读码框:输入网址:/gorf/gorf.html ,打开NCBI
的ORF Finder 软件,输入HPV 的核酸序列的GI 号,最后点击“OrfFind ”按钮,如下图所示:
结果如下图所示:
点击“正链+1”,显示结果如下:
③使用PromoterScan预测该序列中的启动子:输入网址为
/molbio/proscan/,打开PromoterScan的在线操作页面,复制粘贴上述的HPV的核酸序列到指定的框中,点击submit按钮提交序列后,注意使用
软件时不需要设置任何参数,如下图所示:
输出结果为:
何婷学号:1302008 专业:病理学与病理生理学。
生物信息学作业
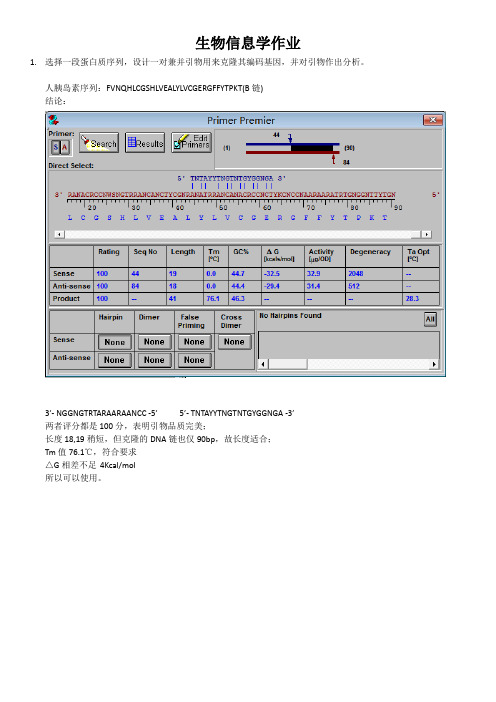
生物信息学作业1.选择一段蛋白质序列,设计一对兼并引物用来克隆其编码基因,并对引物作出分析。
人胰岛素序列:FVNQHLCGSHLVEALYLVCGERGFFYTPKT(B链)结论:3’- NGGNGTRTARAARAANCC -5’ 5’- TNTAYYTNGTNTGYGGNGA -3’两者评分都是100分,表明引物品质完美;长度18,19稍短,但克隆的DNA链也仅90bp,故长度适合;Tm值76.1℃,符合要求△G相差不足4Kcal/mol所以可以使用。
Step1:打开primer premier 5.0 输入蛋白质链,转化为DNA链。
获得DNA链。
2.选择一段基因,预测期编码RNA的二级结构,并分析功能。
取一段基因:ACGCG GGCGG GCATG TGGGC AGCTT TACCC AGTGC TACTG TGCTG GCCAGCACTG AAACA GGGGC ACTGG TTTGG GGTGG ATGAA GGGTA GAAGT GCAAGTTCCA TTGCC TGTGC AATCC CTGCC TTGCT CAGAC CCTGC TCACT CCTCAGGCCC CATCA GCCCC TCAAC TCTGC TAACC ATGGT GGTAG AAATC AGCTACAATA AACCC TGGAG CCAGT AAAAA AAAAA AAAAA AAAAA AAAAA AAAGT点击Fold as RNA点击START点击Draw Stuclture得到RNA二级结构RNA功能预测打开网址http://sidirect2.rnai.jp/输入DNA序列得出结论:。
生物信息学上机

实习一1、根据课件(或教材)提供的地址,访问NCBI、EBI主页,了解其结构、内容。
@整理记录以下信息:1)、NCBI和EBI的英文及中文全名;2)、在NCBI和EBI所管理的生物学数据库或所提供的服务(工具)里,各选出10个(NCBI 10,EBI 10),列表整理出这些数据库或服务的简称、全称、中文名。
2、分别进入三大核酸序列数据库,Genbank、ENA(EMBL bank)、DDBJ,了解其结构,@记录以下内容:1)、访问地址2)、数据库全称3)、最新发布的版本(日期)、目前可获得的核酸记录条数等信息。
3、分别进入三个核酸数据库的序列提交界面(如Genbank的bankit),@记录其地址。
了解序列提交方法。
4、学习使用Pubmed:选择一关键词,查询文献。
5、访问某一核酸序列数据库(如 Genbank),进入其查询系统,在下表中选择序号和你的座位号相同的基因名(每组一个)作为检索词,检索核苷酸数据库。
浏览查询结果;选择、@保存来自物种为小鼠的mRNA or cDNA or complete CDS (即编码一条蛋白质的完整序列)的记录。
请分别保存两种格式:GBFF 和FASTA。
1) 1700019D03Rik;2) 5730528L13Rik;3) Cnot10;4) Gid8;5) Lrrc2;7) 4933403G14Rik;8) 8430410A17Rik;9) Bend3;10) Prrc2a;11) Rmnd5b;12) Tmem131;13) Tmem170;14) Tmem2;15) Tmem8;16) Vrtn;17) 1110001J03RIK;18) 1110059E24Rik;19) 2410137M14RIK;20) 2610042L04Rik;21) 2900010M23RIK;22) 2900011O08RIK;22) 9130011E15Rik;23) Ankrd10;24) BC055324;25) Cdc37l1;26) Commd3;27) Fam102a;28) Fam43a;29) Fam98a;30) Gm3696;32) Hist1h2an;33) Ier2;34) Ifitm7;35) Igsf21;36) Igsf3;Lrrc34; Ng23; Nucks1; Nudcd3; Rbmxl2; Reep3; Sdf2; Ssr2; Tmem60; Zfp 280b; Zfp296; Znrd1as;实习二1、打开上次保存的Genbank文件。
2011春生物信息学上机考核题及要求

考查题从生物信息数据库中检索近缘物种某一种基因或蛋白质近年来的相关记录,从中选取6条以上序列进行多序列比对,并在此基础上构建系统发育树。
要求:①用A4纸打印;页眉为2.5,页脚为2,左边距为2.5,右边距为2。
②第一页第一行左上角为“生物信息学课程考核”,左对齐,黑体5号字体;第三行为班级、姓名和学号,楷体小4号,右对齐。
③题目“×××基因(或蛋白质)序列系统发育分析”居中,宋体4号字体,加粗。
④简要描述步骤。
如序列获得、序列GenBank登录号、多序列比对、系统发育树构建等。
生物信息学课程考核自噬基因序列系统发育分析学院:生命科学与技术学院班级:08(2)姓名:杨彬学号:08243147 步骤:1、序列获得:①进入NCBI主页,点击“Entrez”按钮进入Entrez查询系统,点击“Nucleotide”按钮选择核酸序列数据库②点击“Limits”按钮,在检索栏中填入“Autophagy”,选择“Title word”,点击“Search”按钮2、序列GenBank登录号:HQ447598.1,HQ447703.1,HQ447996.1,NM121735.4,XM003341563.1,XM003341564.13、多序列比对:①将获取的六条基因序列以纯文本文档保存②点击Clustal x.exe图标进入界面③点击File下拉菜单下的Load sequences按钮,导入纯文本文档④点击Alignment下拉菜单下的Do complete Alignment按钮,弹出对话框,点击ALIGN 按钮,执行多序列比对,保存生成的结果到指定的路径目录下⑤点击Mega2图标进入界面⑥点击File下拉菜单下的Convert To MEGA Format按钮,导入经Clustal X软件比对好的文档,将其转换为MEGA格式文档并保存⑦关闭其他窗口,返回主界面;点击主界面上的Click To activate a data file按钮,打开转换好的MEGA格式文档,选择序列类型⑧点击页眉上的下拉菜单,执行相关分析内容保守位点:变异位点:简约信息位点:(在所有群体中>=2个变异的位点)在所有群体中只有1个群体中发生变异的位点:四种碱基及密码子第1、2、3位在各群体中的使用概率Transitions only:转换的百分率Transvertions only:颠换的百分率、4、系统发育树构建:5、系统发育树可靠性检验:。
《生物信息学》上机作业

《生物信息学》上机作业题目:对人血红蛋白(HBA1)编码基因序列的生物信息分析目录引言 .............................................................................................................................................. - 1 -1 正文......................................................................................................................................... - 2 -1.1 NCBI上对相关核苷酸序列的查找............................................................................ - 2 -1.2 BLAST运行及其结果.................................................................................................. - 2 -1.3 BLASTX运行及其结果................................................................................................ - 6 -2 其他软件的运行及其结果..................................................................................................... - 8 -2.1 Clustal W运行及其结果 ............................................................................................. - 9 -2.2 MEGA4.0运行及其结果............................................................................................. - 10 -结论 ............................................................................................................................................ - 10 -引言血红蛋白又称血色素,是红细胞的主要组成部分,能与氧结合,运输氧和二氧化碳。
生物信息学

多序列比对
• 多序列比对完成
• Dateexport alignment, 导出MEGE format和 Fasta format两份结果, 得到一个*.meg文件 和一个*.fas文件
进化树构建
• 关闭Alignment窗口,回到MEGA软件主窗口, File -> Open A File/Session,打开之前 保存的*.meg文件
• 选择Protein
MEGA 5软件使用
• 在新弹出的窗口中,选择Data->Open>Retrieve Sequences from File,然后导 入刚才保存的fasta文件
多序列比对
• Ctrl+A选择全部序列,Aligment->Align by ClustalW
多序列比对
• 可以修改各补偿值等参数,点OK
• 每个序列的Title仅保留蛋白/基因名称+种 属来源,如:CY1_YEAST
• 序列名称中不含有 ‘=’ 字符
• 氨基酸序列可以分成多行,但内部不要有 空格
MEGA 5软件使用
• 打开MEGA 5,拉开Align菜单,选择 Edit/Build Alignment
MEGA 5软件使用
• Creat a new Alignment
创建Fasta
可直接下载或复制粘贴创建Fasta文件: 以>为开头,后接序列名称,重启一行,输入序列
>CY1_BOVIN MAAAAATLRGAMVGPRG… >CY1_YEAST MFSNLSKRWAQRTLSKS… >CY1_HUMAN MAAAAASLRGVVLGPRG… >…
Fasta文件要求
问题
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
《生物信息学》上机作业
题目:对人血红蛋白(HBA1)编码基因序列的生物信息分析
目录
引言 .............................................................................................................................................. - 1 -1 正文......................................................................................................................................... - 2 -
1.1 NCBI上对相关核苷酸序列的查找............................................................................ - 2 -
1.2 BLAST运行及其结果.................................................................................................. - 2 -
1.3 BLASTX运行及其结果................................................................................................ - 6 -
2 其他软件的运行及其结果..................................................................................................... - 8 -
2.1 Clustal W运行及其结果 ............................................................................................. - 9 -
2.2 MEGA4.0运行及其结果............................................................................................. - 10 -结论 ............................................................................................................................................ - 10 -
引言
血红蛋白又称血色素,是红细胞的主要组成部分,能与氧结合,运输氧和二氧化碳。
血红蛋白含量能很好地反映贫血程度。
血红蛋白是高等生物体内负责运载氧的一种蛋白质,具有四聚体蛋白质四级空间结构。
人体内的血红蛋白由四个亚基构成,分别为两个α亚基和两个β亚基,每个亚基由一条肽链和一个血红素分子构成。
肽链在生理条件下会盘绕折叠成球形,把血红素分子抱在里面,这条肽链盘绕成的球形结构又被称为珠蛋白。
每1Hb分子由1个珠蛋白和4个血红素(又称亚铁原卟啉)组成。
每个血红素又由4个吡咯基组成一个环,中心为一铁原子。
每个珠蛋白有4条多肽链,每条多肽链与1个血红至少连接构成Hb的单体或亚单位。
Hb是由4个单体构成的四聚体。
不同Hb分子的珠蛋白的多肽链的组成不同。
成年人Hb(HbA)的多肽链是2条α链和2条β链,为α2β2结构。
胎儿Hb(HbF)是2条α链和2条γ链,为α2γ2结构。
出生后不久HbF即为HbFA所取代。
多肽链中氨基酸的排列顺序已经清楚。
血红素的Fe2+均连接在多肽链的组氨基酸残基上,这个组氨酸残基若被其它氨基酸取代,或其邻近的氨基酸有所改变,都会影响Hb的功能。
可见蛋白质结构和功能密切相关。
1 正文
1.1 NCBI上对相关核苷酸序列的查找
首先运用NCBI数据库对相关的核弹酸序列进行查找,得到如下FASTA格式结果序列:
图1.1人体血红蛋白亚基1(HBA1),mRNA,NM_000558.5序列
1.2 BLAST运行及其结果
图1.2 与已知序列具有一定相似度的序列
图1.3 基因对比结果
从上述两张图可以看出:前三个对比序列与第7个对比序列到第17个对比序列所引用的核苷酸序列相似度高达100%,第4个对比序列到第6个对比序列与所引核苷酸序列相似度为99%。
在诸多相似的其他序列之中,选取以下三个相似度不同的序列做细致的比对,结果如下:
图1.4黑猩猩血红蛋白亚基1(HBA1) mRNA
此为与所引序列比对相似度高达99%的——黑猩猩血红蛋白亚基1(HBA1)mRNA:此序列来源于黑猩猩(猩猩)生物。
编号NM_001042626。
图1.5人类血红蛋白亚基2(HBA2), mRNA
此为与所引序列比对相似度为100%的——人类血红蛋白亚基2(HBA2),mRNA序列:此序列来源于智人(人类)生物编号为NM_000517。
图1.6东非狒狒血红蛋白,亚基1(HBA1), mRNA
此为与所引序列比对相似度为98%的——东非狒狒血红蛋白,亚基1(HBA1), mRNA序列:此序列来源于东非狒狒(狒狒)生物。
编号
NM_001168816.
1.3 BLASTX运行及其结果
通过BLASTX,将由NCBI获得的基因:人体血红蛋白亚基1 HBA1 基因,
NM_000558.5,序列进行翻译为蛋白质后再次进行比对,结果如下:
图1.7 BLASTX运行结果
图1.8 蛋白质比对结果
由运行结果可知,前7个序列与已知人体血红蛋白亚基1 HBA1 基因,部分cds基因序列翻译得到的蛋白质有高度相似性。
其得分均在300以上,这些蛋白具有高度同源性。
2 其他软件的运行及其结果
- 9 -
2.1 Clustal W 运行及其结果
为了比较不同物种间的亲缘关系,以及不同血红蛋白亚基之间的相似度,我选取了BLASTN 运行结果前十项与已知人体血红蛋白亚基1 HBA1 基因序列进行比对,运行了ClustalW 运行,得到其结果如下:
图2.1 Clustal W 运行结果phlogenetic Tree 图谱
由phlogenetic Tree 图谱可知:
AK223392.1序列与BC005931.1序列、BC032122.2序列与XM_011960067.1序列、XM_024233299.1序列与 XM_030924410.1序列、XM_010381857.2 序列与NM_000517.6序列之间的亲缘关系最近;
而NM_001042626.1序列与 BC050661.1序列之间亲缘关系较近,其余各序列之间亲缘关系较远。
图2.2 Clustal W 运行结果Guide Tree 图谱
由Guide Tree 图谱可知:
XM_011960067.1序列与NM_001042626.1序列亲缘性最高;同理可知:XM_010381857.2序列与AK223392.1序列、XM_030924410.1序列与
BC005931.1
- 10 - 序列亲缘关系最近;
NM_000558.5序列与BC005931.1序列间、XM_024233299.1序列与
BC032122.2序列之间亲缘关系较近;其余序列之间亲缘关系较远。
图2.3 Jalview 运行结果
2.2 MEGA4.0运行及其结果
利用邻域加入法来推断其进化历史,通过对生物序列的研究来推测物种的进化历史。
通过DNA 序列,蛋白质序列,蛋白质结构等来构建系统发育树建立结构进化树。
使用邻接法构建系统发生树。
通过MEGA4.0的运行得到如下进化树:
图2.4 MEGA4.0运行结果
结论
由人血红蛋白(HBA1)编码基因序列与其相似序列比对分析可知:人血红蛋白亚基1与血红蛋白亚基2具有高度同源性,可推知血红蛋白可能最初就是从同一种蛋白质发生基因突变,产生的两种不同亚基的血红的蛋白,所以人体的血红蛋白由四个亚基组成,两个α亚基,两个β亚基。
并且人体血红蛋白基因序列和人亲缘关系较近的黑猩猩、狒狒等灵长类动物体内的部分基因具有高度的相似性。