味精检测标准
味精的执行标准哪个是最好的

味精的执行标准哪个是最好的味精是一种常用的调味品,它能够增强食物的鲜味,提升食物的口感,因此备受人们喜爱。
然而,随着市场上味精产品的不断增加,消费者往往难以选择到最适合自己需求的产品。
因此,味精的执行标准成为了消费者们关注的焦点之一。
那么,味精的执行标准中哪个是最好的呢?本文将就此问题展开讨论。
首先,我们需要了解味精的执行标准是什么。
味精的执行标准主要包括对原料、生产工艺、质量指标等方面的规定。
在我国,味精的执行标准由国家标准化管理委员会制定和管理,其中包括GB/T8967-2007《味精》国家标准。
这一标准对味精的原料、生产工艺、质量指标等方面进行了详细的规定,以确保生产的味精产品符合国家的安全和质量要求。
其次,我们需要了解不同执行标准的差异。
在市场上,我们可以看到不同企业生产的味精产品标注着不同的执行标准,如GB/T8967-2007、企业标准、行业标准等。
这些不同的执行标准可能会对产品的原料选用、生产工艺、质量指标等方面有所不同,因此也会对产品的品质产生影响。
消费者在选择味精产品时,需要对不同执行标准进行了解,以便选择到符合自己需求的产品。
接下来,我们需要考虑如何选择最适合的执行标准。
在选择味精产品时,消费者可以从以下几个方面考虑,首先,了解产品的执行标准,选择符合国家标准的产品,以确保产品的安全和质量;其次,了解产品的生产企业,选择信誉良好、生产工艺先进的企业生产的产品;最后,可以参考他人的使用经验和口碑评价,以便选择到口感好、效果显著的产品。
总之,味精的执行标准对产品的质量和安全有着重要的影响,消费者在选择产品时需要对不同执行标准进行了解,并综合考虑产品的质量、生产企业、口碑评价等方面,以选择到最适合自己需求的产品。
希望本文能够帮助消费者更好地选择味精产品,享受到美味佳肴带来的愉悦体验。
gb2720-2003味精卫生标准

gb2720-2003味精卫生标准该标准是为了保障味精制品的质量与安全,对味精产品中有害物质的含量和微生物指标进行了规定。
首先,该标准针对味精产品的有害物质进行了规定。
有害物质是指可以对人体健康产生不良影响的物质。
该标准要求味精产品中的苯甲酸钠含量不得超过0.01%。
苯甲酸钠是一种食品防腐剂,过量摄入可能对人体肝脏和肾脏造成损害。
味精制品中的铅含量不得超过2mg/kg,这是因为铅是一种有害重金属,对人体神经系统、肾脏和骨骼造成损害。
标准还规定了味精制品中其他重金属如砷、镉、锑、锡等的限量要求,以保障味精产品的安全性。
其次,该标准还对味精产品的微生物指标进行了规定。
微生物是指可以对人体产生致病作用的微生物菌群。
标准规定了味精产品中的大肠菌群、沙门氏菌和金黄色葡萄球菌的检验方法及其限量要求。
这些微生物的存在和繁殖可能导致食品中毒,因此该标准规定了味精产品中这些致病菌的可容许范围。
此外,该标准还包含了味精产品的质量指标和感官要求。
标准规定了味精产品的外观、色泽、味道和杂质等方面的要求,以确保味精的品质。
标准还对味精产品的干燥失重、还原能力和pH值等指标进行了规定,这些指标可以客观反映味精产品的含水量、味道和稳定性等特性。
综上所述,GB2720-2003《味精卫生标准》通过限制有害物质的含量和微生物指标,确保味精产品的质量与安全。
这些规定可以帮助生产者控制产品的质量,并保证消费者的健康。
同时,该标准还规定了产品的质量指标和包装要求,为味精制品行业提供了统一的标准和依据。
味精检测SOP

味精检测SOP1. 目的规范味精的检测操作,确保检测的准确性。
2. 范围本标准适用于味精的测定。
3. 定义无4. 职责4.1.QC负责本规程的起草、修订、培训及执行。
4.2.QA、QC组长、质量管理部经理负责本规程的审核。
4.3. 质量总监负责批准本规程。
4.4.QA负责本规程执行的监督。
5. 引用标准《中国生物制品主要原辅材料质控标准》6. 材料见程序7. 流程图无8. 程序8.1.性状本品是以粮食为原料发酵提纯的谷氨酸钠结晶或粉末。
8.2.谷氨酸钠含量(旋光计法)8.2.1.原理谷氨酸钠分子结构中含有一个不对称碳原子,具有光学活性,能使偏振光面旋转一定角度,可用旋光仪测定其旋光度。
8.2.2.仪器及设备旋光仪(备钠光灯钠光谱 D线 589.3nm)、电子天平(万分之一)、100ml容量瓶等。
8.2.3.试剂及配制盐酸溶液(6mol/L):取54ml盐酸,加水稀释至100ml,摇匀,即得。
8.2.4.操作步骤取样品5g于烧杯中,精密称定,加水20~30ml,加盐酸溶液(6mol/L )16ml,使溶解,然后移入50ml量瓶中,加水至刻度,摇匀,测定旋光度及溶液的温度。
按《目视旋光仪使用、清洁保养SOP》进行检测。
8.2.5.结果计算:d0=25.16+0.047(20-t) =26.1-0.047 t式中:X2—样品中谷氨酸钠含量,%;d—实测样液的旋光度;d0—纯谷氨酸的旋光度;L—旋光管长度(即液层厚度), dm;25.16—谷氨酸钠的比旋光度[a];t—测定时样液温度, ℃;0.047—温度校系正数。
同一样品进行两次测定,取平均值,结果修约到保留一位小数。
8.2.6.结果判断:含量应不小于95%。
8.3.锌8.3.1.仪器及设备UV-2550分光光度计、125ml分液漏斗、电子天平(千分之一)、100ml烧杯、100ml容量瓶等。
8.3.2.试剂及配制甲基橙指示液:取甲基橙0.1g,加水100ml使溶解,即得。
味精掺假检测

实验二味精掺伪实验
一、味精中掺入食盐的检验
(1)感官鉴别: ①色泽:掺入食盐的味精,色泽灰白,不透明,无光泽。
②形状:食盐颗粒子,呈长形。
③口尝:入嘴尝之,舌头发咸。
(2)化学检验:每一种规格味精中的氯化钠的含量,国家标准中都有规定,如99%味精,氯化钠含量应小于0.5%,如果大于国家规定要求,说明味精中多掺入了食盐。
取5毫升配制成浓度为5%的味精溶液,放在玻璃试管中,先加1滴5%的铬酸钾溶液,再加1毫升0.73%的硝酸银溶液,轻轻摇匀,观察溶液变色情况。
如果溶液显示桔红色,说明味精中氯化钠含量小于1%;如果溶液显示出黄色,说明味精中氯化钠含量大于1%。
(3)实验结果:实验组?对照组?
二、味精中掺入碳酸盐的检验
(1)原理:碳酸盐与盐酸作用即产生二氧化碳气体,可据此现象判断。
(2)测定:取样品少许,加少量的水溶解后,加数滴稀盐酸,如有碳酸盐存在,即有气泡产生。
(3)实验结果:实验组?对照组?
三、味精中掺入硼酸盐的检验
(1)原理:硼酸盐在浓硫酸中和乙醇反应生成极易挥发的硼酸乙酯使火焰呈绿色。
(2)测定:取少许样品于瓷皿中,加数滴浓硫酸和2ml乙醇,混匀后点燃,若火焰呈绿色说明掺有硼酸盐。
(3)实验结果:实验组?对照组?
四、味精中掺入蔗糖的检验
(1)原理:蔗糖在强酸条件下加热可分解生成生成葡萄糖和果糖,果糖可与间苯二酚反应生成玫瑰红色的糖醛衍生物。
(2)测定:称1g样品于烧杯中,加0.1g间苯二酚和3-5滴浓盐酸煮沸5min,若溶液呈现玫瑰红色说明掺有蔗糖。
(3)实验结果:实验组?对照组?。
gb2720-2003味精卫生标准

gb2720-2003味精卫生标准
GB2720-2003味精卫生标准是国家新版的有关味精安全卫生的重要规范性文件。
它主要针对我国食品工业用味精的质量指标及其监测方法进行了详细的规定,保证了我国食品安全健康的生产和使用。
GB2720-2003味精卫生标准的主要内容包括:
(一)定义一、类别。
根据主要成分的不同,味精可分为氨基酸类、氯代烃类、烯醇类和其他特殊类;
(二)指标要求二、味精指标要求。
包括使用指标、性能指标、无害成分指标、监测指标等;
(三)三、监测检测方法。
根据GB2720-2003的规定,味精
的检测方法大体上分为六大类:尺寸分析、物理检测、化学检测、鉴定检测、微生物检测和放射性检测;
(四)四、其他特殊要求。
除上述指标要求外,还要求味精应符合《食品添加剂标准》(GB2760-2011)的相关规定,并需
依据《食品安全法》、《食品安全法实施条例》、《食品添加剂监管管理办法》( GB 5574-2010)等要求进行管理,以确
保味精安全健康。
总之,GB2720-2003 味精卫生标准的作用是为了提高我国食品工业使用的味精的质量,保障我国食品都是安全健康的,所以相关部门应严格遵守这一标准,以实现食品安全的绿色生产。
总而言之,GB2720-2003 味精卫生标准对于促进我国食品安全的质量有着重要的作用,相关部门应该加强把控和管理,严格执行国家有关规定和相关法律法规,以确保我国食品的安全健康。
只有贯彻落实这一标准,我国食品才能真正健康安全,给大家带来更多的安心和安全。
gb 2720-1996味精卫生标准.doc

GB 2720-1996前言本标准代替了GB 2720-81味精卫生标准。
在对GB2720-81进行修订时,保留了GB2720-81中实践证明适合我国情况而又不妨碍国际通用内容。
在卫生要求中对味精中谷氨酸钠的含量进行了限定。
本标准在编写和规则上则依据GB/T1.1-1993和GB/T22-1993的要求。
本标准于1982年6月首次发布,1994年11月进行第一次修订。
本标准从实施之日起,同时代替GB2720-81。
本标准由卫生部卫生监督司提出。
本标准主要起草单位:北京市食品卫生监督检验所、国内贸易部北京食品酿造研究所、山西省卫生防疫站。
本标准主要起草人:丁秀英、胡克强、钟冠山、朱荭、孟海鹰。
本标准由卫生部委托技术归口单位卫生部食品卫生监督检验所负责解释。
中华人民共和国国家标准GB 2720-1996味精卫生标准代替GB 2720-81Hygienic standard of sodium glutamate──────────────────────────────────1 范围本标准规定了味精的卫生要求、检验方法。
本标准适用于味精。
2 引用标准下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文。
本标准出版时,所示版本均为有效,所有标准都会被修订,使用本标准的各方应探讨使用下列标准最新版本的可能性。
GB 2760-86 食品添加剂使用卫生标准GB/T 5009.43-1996 味精卫生标准得分析方法3 定义本标准采用下列定义。
3.1 味精:以粮食及其制品为原料,经发酵提纯谷氨酸钠产品。
4 卫生要求4.1 感官指标具有正常味精的色泽、滋味,不得有异味及夹杂物。
4.2 理化指标理化指标应符合表1的规定。
表1───────────────────┬─────────────项目│指标───────────────────┼─────────────谷氨酸钠(%) ≥│80砷(以As计,mg/kg) ≤│0.5铅(以pb计,mg/kg) ≤│ 1锌(以pb计,mg/kg) ≤│ 5───────────────────┴─────────────5 检验方法5.1 感官指标按本标准4.1规定执行。
谷氨酸钠(味精)标准
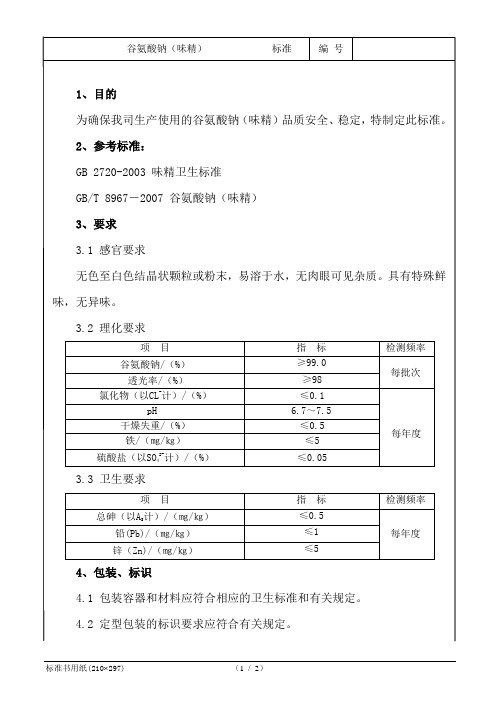
4、包装、标识
4.1包装容器和材料应符合相应的卫生标准和有关规定。
4.2定型包装的标识要求应符合有关规定。
5、贮存及运输
5.1产品应贮存在干燥、通风良好的场所。不得与有毒、有害、有异味、易挥发、易腐蚀的物品同处贮存。
5.2运输产品时应避免日晒、雨淋。不得与有毒、有害、有异味或影响产品质量的物品混装运输。
≥99.0
每批次
透光率/(%)
≥98
氯化物(以CL-计)/(%)
≤0.1
每年度
pH
6.7~7.5
干燥失重/(%)
≤0.5
铁/(㎎/㎏)
≤5
硫酸盐(以SO42-计)/(%)
≤0.05
3.3卫生要求
项目
指标
检测频率
总砷(以AS计)/(㎎/㎏)
≤0.5
每年度
铅n)/(㎎/㎏)
1、目的
为确保我司生产使用的谷氨酸钠(味精)品质安全、稳定,特制定此标准。
2、参考标准:
GB 2720-2003味精卫生标准
GB/T 8967-2007谷氨酸钠(味精)
3、要求
3.1感官要求
无色至白色结晶状颗粒或粉末,易溶于水,无肉眼可见杂质。具有特殊鲜味,无异味。
3.2理化要求
项目
指标
检测频率
谷氨酸钠/(%)
谷氨酸钠(味精)-最新国标

谷氨酸钠(味精)1 范围本文件界定了谷氨酸钠(味精)的术语和定义,规定了谷氨酸钠(味精)的原辅材料、感官要求、理化指标等要求,描述了相应的试验方法,规定了检验规则和标志、包装、运输和贮存的内容。
本文件适用于谷氨酸钠(味精)的生产、检验和销售。
2 规范性引用文件下列文件中的内容通过文中的规范性引用而构成本文件必不可少的条款。
其中,注日期的引用文件,仅该日期对应的版本适用于本文件;不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T 191 包装储运图示标志GB/T 601 化学试剂标准滴定溶液的制备GB/T 602 化学试剂杂质测定用标准溶液的制备GB/T 603 化学试剂试验方法中所用制剂及制品的制备GB 1886.97 食品安全国家标准食品添加剂5’-肌苷酸二钠GB 1886.170 食品安全国家标准食品添加剂5’-鸟苷酸二钠GB 1886.306 食品安全国际标准食品添加剂谷氨酸钠GB 5009.43 食品安全国家标准味精中麸氨酸钠(谷氨酸钠)的测定GB/T 6682 分析实验室用水规格和试验方法GB 7718 食品安全国家标准预包装食品标签通则SB/T 10731 鸡精调味料JJF 1070 定量包装商品净含量计量检验规则国家市场监督管理总局令第70号定量包装商品计量监督管理办法3 化学名称、分子式、结构式、相对分子质量化学名称L-谷氨酸一钠一水化物(L-α-氨基戊二酸一钠一水化物)分子式C5H8NNaO4·H2O相对分子质量187.13(按照2022年国际相对原子质量)结构式HNaOOC-CH2-CH2-C-COOH·H2ONH24 术语和定义下列术语和定义适用于本文件。
谷氨酸钠 monosodium L-glutamate(MSG)味精以碳水化合物(如淀粉、玉米、糖蜜等糖质)为原料,经微生物(谷氨酸棒杆菌等)发酵、提取、中和、结晶、分离、干燥而制成的具有特殊鲜味的白色结晶或粉末状调味品。
味精标准

a 以 发酵制品为原辅料的 油辣椒除外。
指标 50 0 0
30 不得检出
四、 试 验 方法
取样 品 一瓶 (袋 ), 将 内容 物 置于 清 洁 的白 瓷 盘中 ,用 视 觉法 鉴 别外 观 , 用嗅觉鉴别气味,用味觉法鉴别滋味。
五、 标 签 要求
1. 预包装油辣椒产品标签应符合 GB 77 18 的规定。 2. 预包装油辣椒产品标签上应标明所使用食用油的具体产品名称,如:“菜 籽油”、“大豆油”、“食用猪油”等。 3 . 预 包装油辣椒产品中 使用转基因原料的 ,应在标签上标明。
1. 什 么 是绿 色食 品 ?
绿色食 品是指遵循可持续发展原 则,按照特 定生产方式生产,经专门机构认
定,许可使用绿色食品标志,无污染的安全、优质、营养类食品。合理使用肥料、
农药食品添加剂等生产资料是生产绿色食品的重要环节。
2 . 什么 是 天然 食品 添 加剂 ?
以物理 方法从天然物中分离出来 ,经过毒理 学评价确认其食用安全的 食品添
品的成分、品质和感观,提高加工性能。
(中国绿色食品发展中心)
【GB/ T 20293- 2006】
由中华人民共和 国国家质量监督检验检 疫总局、中 国国家标准化管理委员会 于 2 00 6 年 7 月 1 1 日发布,自 2 0 06 年 12 月 1 日起实施。
一、范围
本标 准适 用于 油辣椒 的生 产、销 售和监 督检验 。
表2 含盐味精理化指标
项目
谷 氨 酸 钠 含 量 (%) 透 光 率 (% ) 食用盐(以 Na CI 计)(%) 干 燥 失 重 (%)
9 5 %味精 ≥ 95. 0 ≥ 95 <5. 0 ≤0. 5
指标 9 0 % 味精 ≥ 90. 0 ≥ 92 <10. 0 ≤ 0. 7
食品原材料 味精 验收方案

味精验收方案
一、目的
明确检验标准,快速准确地对味精进行检验,特制定本检验方案。
二、抽样方法
按照兼前顾后的原则,随机多点取样。
五、拒收条件
1. 品种不符,整批拒收;
2. 在抽检时发现有明显的色泽差异,整批拒收;
3. 抽检时发现肉眼可见异物,整批拒收;
4. 如在抽检时未发现有异物或异味,而在生产使用时发现有异物或异味的现
象,经品控确认后,未使用的原料作退货处理,已使用的原料全部作报废处理;
5. 抽检时发现包装被有毒有害物污染,整批拒收;
6. 运输车辆运输过有毒有害物品的,整批拒收。
味精中谷氨酸钠的测定标准2023年

味精中谷氨酸钠的测定1 范围本标准规定了味精中谷氨酸钠的测定方法。
本标准适用于味精中谷氨酸钠的测定。
第一法高氯酸非水溶液滴定法2 原理谷氨酸钠的碱度在乙酸溶液中显著增强,用高氯酸标准溶液滴定样品中的谷氨酸钠,采用电位滴定法测定时,以电位突跃为依据判定滴定终点;采用化学指示剂法测定时,以d-葵酪術基甲醇为指示剂,滴定样品溶液至浅绿色为其终点,通过消耗高氯酸标准滴定溶液的量计算味精中谷氨酸钠的含量。
3 试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为GB/T6682规定的二级水。
3. 1 试剂3. 1. 1 甲酸(CH2 O2)>99% 。
3. 1. 2 乙酸(C2 H4 O2)>99% 。
3. 1. 3 高氯酸(HClO4)。
3. 1. 4 d-葵酪術基甲醇(C27H18O2)。
3. 2 试剂配制d-葵酪術基甲醇- 乙酸指示液(2g/L):称取0. 1gd-葵酪術基甲醇,用乙酸溶解并稀释至50mL,避光保存,有效期为60天。
3. 3 标准溶液高氯酸标准滴定溶液[C(HClO4)—0. 1mol/L]:按GB/T601配制与标定或购买有证书标准滴定溶液。
4 仪器和设备4. 1 电位滴定仪(精度三0. 2mv),具备动态滴定模式或等量滴定模式。
4.2非水相PH 电极,采用Ag/AgCl为内参比电极;内参比电解液为2mol/L氯化钮乙醇溶液或0. 4mol/L四乙基澳化锁乙二醇溶液。
4. 3 磁力搅拌器。
4. 4 分析天平:感量 0. 1mg 。
4. 5 酸式滴定管。
4. 6 超声清洗器。
4. 7 粉碎机。
4. 8 温度计。
5 分析步骤5. 1 试样制备取100g的味精样品用粉碎机磨碎均匀。
5. 2 试样前处理称取试样 0. 15g(精确至 0. 0001g)至100mL烧杯中,加甲酸3mL,超声至完全溶解,再加乙酸40mL,摇匀,制备成待测试液,可采用电位滴定法或化学指示剂法测定。
5. 2. 1 电位滴定法测定5. 2. 1. 1 参考条件参考条件如下:a)样品测定:采用动态或等量滴定模式;b)试剂空白测定:采用动态或等量滴定模式。
谷氨酸钠(味精)标准

标准
编 号
1、目的 为确保我司生产使用的谷氨酸钠(味精)品质安全、稳定,特制定此标准。 2、参考标准: GB 2720-2003 味精卫生标准 GB/T 8967-2007 谷氨酸钠(味精) 3、要求 3.1 感官要求 无色至白色结晶状颗粒或粉末,易溶于水,无肉眼可见杂质。具有特殊鲜 味,无异味。 3.2 理化要求
标准书用纸(210297)
(2 / 3)
谷氨酸钠(味精)
标准
编 号
项
目Item
指
标Limit ≥99.0 ≥98 ≤0.1
检测频率 每批次
谷氨酸钠Assay /(%) 透光率 state of solution (transmittance)/(%) 氯化物chloride (cl)/(%) pH 干燥失重/(%) 铁Iron/(㎎/㎏) 硫酸盐Sulfate(以SO4 计)/(%)
≤0.5 ≤1 ≤5
3.3 卫生要求 4、包装、标识 4.1 包装容器和材料应符合相应的卫生标准和有关规定。
标准书用纸(210297)
(1 / 3)
谷氨酸钠(味精)
标准编 号Fra bibliotek4.2 定型包装的标识要求应符合有关规定。 5、贮存及运输 5.1 产品应贮存在干燥、通风良好的场所。不得与有毒、有害、有异味、 易挥发、易腐蚀的物品同处贮存。 5.2 运输产品时应避免日晒、雨淋。不得与有毒、有害、有异味或影响产 品质量的物品混装运输。
项 目 指 标 检测频率 每批次 谷氨酸钠/(%) 透光率/(%) 氯化物(以CL-计)/(%) pH 干燥失重/(%) 铁/(㎎/㎏) 硫酸盐(以SO42-计)/(%) ≥99.0 ≥98 ≤0.1 6.7~7.5 ≤0.5 ≤5 ≤0.05
DB31 2021-2013 食品安全地方标准 味精中硫化钠的测定

7.1
X
式中:
(c c0 ) V 1000 m 1000
…………………………………………(1)
X —试样中硫化钠(以S2-计)含量,单位为微克每千克(g/kg); c —从标准曲线上得到的试样中硫化钠(以S2-计)溶液浓度,单位为纳克每毫升(ng/mL);
2
DB 31/ 2021—2013
0.2 µm 尼龙滤膜过滤后,加入 5.25 mL 50 % 氢氧化钠溶液(4.3)。淋洗液配好后立即置于惰性气体条 件下保护,以防污染。 4.6 4.7 硫化钠标准物质(Na2S·9H2O, CAS 号:1313-84-4):分子量 240.17,纯度大于等于 98%。 硫化钠标准储备液:量取 10 mL 250 mmol/L 氢氧化钠溶液(4.4)加入到放在天平上的聚丙烯材
注:所用玻璃仪器均用磷酸-丙酮溶液(11.12)涂渍后,烘干,备用。 按图 1 组装。允许使用功能相同的仪器取代 上述仪器。所有的磨口接口都要遵从 ISO 383 中的要求,并在组装前用甘油(11.11)密封。
图1 13 测定步骤
吹气吸收装置连接图
13.1 试样制备:取相当于试样 10.0 g 的样品制备液(6)于抽提瓶中,加入 50 mL 水。 13.2 试样消解:将试样溶液置于抽提烧瓶(12.4)中,将 10 mL 吸收液加入吸收管(12.7),组装实 验装置,将空气采样器流速调节为 1 L/min,从分液漏斗(12.6)中向抽提烧瓶(12.4)中缓慢加入盐 酸溶液(11.1)20 mL,吹气保持 30~40min,将吸收管(12.7)内吸收液转移至具塞比色管(12.2)中。
3
DB 31/ 2021—2013 12 仪器和设备
12.1 分析天平:感量为 0.01 g。 12.2 具塞比色管:10 mL。 12.3 紫外分光光度计。 12.4 抽提烧瓶(A):容积 150 mL,有 34/35 锥形磨口接口。 12.5 减速调节器(B)。 12.6 分液漏斗(C):容积 100 mL,有 14/23 锥形插口,活塞及 14/23 瓶塞。 12.7 吸收管(D):有 10 mL 刻度线。 12.8 流量计(E):流量范围 0.2~2 L/min,流量稳定。使用时,用皂膜流量计校准采样系列在采样 前和采样后的流量,流量误差应小于 5%。
味精标准解读及谷氨酸钠含量测定

TECHNOLOGY AND INFORMATION120 科学与信息化2022年5月下味精标准解读及谷氨酸钠含量测定吴珍珍 李静 杨振东(通讯作者)山东省食品药品检验研究院 山东 济南 250101摘 要 本文介绍了味精产品相关标准和谷氨酸钠含量测定的3种方法。
3种方法比较分析得出:味精中谷氨酸钠含量的测定不宜采用酸度计法,旋光法适用于不含有旋光性杂质的味精,高氯酸非水溶液滴定法适用于含有旋光性杂质的味精。
关键词 味精;谷氨酸钠;检验Interpretation on Monosodium L-glutamate Standards and Determination of Sodium Glutamate Content Wu Zhen-zhen, Li Jing, Yang Zhen-dong (corresponding author)Shandong Institute for Food and Drug Control, Jinan 250101, Shandong Province, ChinaAbstract This article introduces the relevant standards of monosodium L-glutamate and three methods for the determination of sodium glutamate content. The comparative analysis of three methods shows that: pH meter method is not suitable for the determination of sodium glutamate content in monosodium L-glutamate, polarimetry is suitable for the monosodium L-glutamate which does not contain other impurities with optical activity, and perchloric acid non-aqueous solvent titration is suitable for the monosodium L-glutamate which contains other impurities with optical activity.Key words mon-sodium L-glutamate; sodium glutamate; inspection引言味精是由德国化学家里德豪森1866年从植物蛋白中发现的[1]。
味精的质检报告

味精的质检报告1. 引言味精(化学名:谷氨酸钠)是一种常用的食品添加剂,广泛应用于餐饮业和家庭烹饪中,为食物增添鲜味。
本质检报告旨在对味精进行质量检测,评估其符合安全和卫生标准的程度。
2. 检测方法味精的质检方法主要包括以下几个方面:2.1 外观检查外观检查主要观察味精颗粒的形状、颜色和纯净度。
合格的味精应具有白色结晶状颗粒,无杂质和异味。
2.2 粒度分析粒度分析用于测量味精颗粒的大小分布。
常用的测试方法包括激光粒度仪和筛分法。
2.3 水分含量测试水分含量对于味精的质量非常重要。
一般采用烘干法或卤素酸法进行水分含量测试。
2.4 谷氨酸钠含量测定味精的主要成分是谷氨酸钠,因此检测谷氨酸钠的含量可以评估味精的质量。
常用的测定方法包括酸碱滴定法和高效液相色谱法。
2.5 重金属和有害物质检测重金属和有害物质对人体健康会产生潜在风险。
常见的检测项目包括铅、汞、砷等重金属离子的含量以及亚硝酸盐、致癌物质等的检测。
3. 实验结果3.1 外观检查经过外观检查,味精样品呈现出白色颗粒状,无明显杂质和异味。
3.2 粒度分析采用激光粒度仪进行粒度分析,测试结果显示,味精颗粒平均粒径为35微米。
3.3 水分含量测试采用烘干法进行水分含量测试,结果显示味精的水分含量为0.5%。
3.4 谷氨酸钠含量测定采用高效液相色谱法进行谷氨酸钠含量测定,结果显示味精样品中含量为99.8%。
3.5 重金属和有害物质检测经过重金属和有害物质检测,味精样品中未检出铅、汞、砷等重金属离子,亚硝酸盐和致癌物质的含量也低于卫生标准限值。
4. 结论根据实验结果分析,本次味精样品经过质量检测,外观正常,粒度均匀,水分含量和谷氨酸钠含量符合相关标准要求。
重金属和有害物质检测结果显示,味精样品符合食品卫生安全标准,可以放心使用。
5. 后续建议为了确保味精的质量和安全性,建议生产厂商和相关监管部门加强生产工艺控制和质量监测,并定期进行有害物质检测,确保产品的安全性和合规性。
味精检测标准

味精检测标准GB/T 8967—2000前⾔本标准⾮等效采⽤了 1994 年⽇本《⾷品添加物公定书》第六版中的“⾕氨酸钠”标准。
本标准是对 GB/T 8967—1988《⾕氨酸钠》的修订。
本标准与 GB/T 8967—1988 的主要差异如下:——标准名称修改为“⾕氨酸钠(99%味精)”;——取消了对锌的限量规定;——将“重⾦属(以 Pb 计)”指标改为“铅”,最⾼限量不得超过 1 mg/kg;——将硫酸盐指标由⼩于(或等于)0.03%修改为⼩于(或等于)0.05%;——增加了测定⼲燥失重的快速法(第⼆法);对其他有关试验⽅法也做了相应的调整;——将半成品 L-⾕氨酸(麸酸)质量要求列⼊附录 A。
本标准的附录 A 和附录 B 都是提⽰的附录。
本标准⾃实施之⽇起,同时代替 GB/T 8967—1988。
本标准由国家轻⼯业局提出。
本标准由全国⾷品发酵标准化中⼼归⼝。
本标准起草单位:中国⾷品发酵⼯业研究所、沈阳红梅企业集团有限责任公司、上海冠⽣园天厨⾷品有限公司、⼴州奥桑味精⾷品有限公司。
本标准主要起草⼈:⽥栖静、张世根、俞儒钧、苏振⽟、花惠颖、陈继⾼、徐爱菌。
本标准由全国⾷品发酵标准化中⼼负责解释。
GB/T 8967—2000中华⼈民共和国国家标准GB/T8967-2000⾕氨酸钠(99%味精)代替 GB/T8967-1988Monosodium L-glutamate(99%Wei Jing)1 范围本标准规定了⾕氨酸钠(99%味精)的定义、技术要求、试验⽅法、检验规则和标志、包装、运输、贮存要求。
本标准适⽤于⾕氨酸钠含量不低于 99%的产品。
2 引⽤标准下列标准所包含的条⽂,通过在本标准中引⽤⽽构成为本标准的条⽂。
本标准出版时,所⽰版本均为有效。
所有标准都会被修订,使⽤本标准的各⽅应探讨使⽤下列标准最新版本的可能性。
GB 191—1990 包装储运图⽰标志GB/T 601—1988 化学试剂滴定分析(容量分析)⽤标准溶液的制备GB/T 602—1988 化学试剂杂质测定⽤标准溶液的制备GB/T 603—1988 化学试剂试验⽅法中所⽤制剂及制品的制备GB 1354—1986 ⼤⽶GB/T 5009.11—1996 ⾷品中总砷的测定⽅法GB/T 5009.12—1996 ⾷品中铅的测定⽅法GB/T 6543—1986 ⽡楞纸箱GB/T 6682—1992 分析实验室⽤⽔规格和试验⽅法GB 7718—1994 ⾷品标签通⽤标准GB 9687—1988 ⾷品包装⽤聚⼄烯成型品卫⽣标准GB/T 12309—1990 ⼯业⽟⽶淀粉QB/T 1840—1993 ⼯业薯类淀粉国家技术监督局令[1995]第 43 号定量包装商品计算监督规定3 定义本标准采⽤下列定义.⾕氨酸钠( 99% 味精) sodium L-glutamate ; L- ⾕氨酸单钠—⽔化物monosodium L-glutamate monohydrate;缩写式:MSG(99%Wei Jing)以碳⽔化合物(淀粉、⼤⽶、糖蜜等糖质)为原料,经微⽣物(⾕氨酸棒杆菌等)发酵,提取,中和,结晶,制成的具有特殊鲜味的⽩⾊结晶或粉末。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
前言本标准非等效采用了 1994 年日本《食品添加物公定书》第六版中的“谷氨酸钠”标准。
本标准是对 GB/T 8967—1988《谷氨酸钠》的修订。
本标准与 GB/T 8967—1988 的主要差异如下:——标准名称修改为“谷氨酸钠(99%味精)”;——取消了对锌的限量规定;——将“重金属(以 Pb 计)”指标改为“铅”,最高限量不得超过 1 mg/kg;——将硫酸盐指标由小于(或等于)%修改为小于(或等于)%;——增加了测定干燥失重的快速法(第二法);对其他有关试验方法也做了相应的调整;——将半成品 L-谷氨酸(麸酸)质量要求列入附录 A。
本标准的附录 A 和附录 B 都是提示的附录。
本标准自实施之日起,同时代替 GB/T 8967—1988。
本标准由国家轻工业局提出。
本标准由全国食品发酵标准化中心归口。
本标准起草单位:中国食品发酵工业研究所、沈阳红梅企业集团有限责任公司、上海冠生园天厨食品有限公司、广州奥桑味精食品有限公司。
本标准主要起草人:田栖静、张世根、俞儒钧、苏振玉、花惠颖、陈继高、徐爱菌。
本标准由全国食品发酵标准化中心负责解释。
国家质量技术监督局 2000—04—05 批准 2000—09—01 实施GB/T 8967—2000中华人民共和国国家标准GB/T8967-2000谷氨酸钠(99%味精)代替 GB/T8967-1988Monosodium L-glutamate(99%Wei Jing)1 范围本标准规定了谷氨酸钠(99%味精)的定义、技术要求、试验方法、检验规则和标志、包装、运输、贮存要求。
本标准适用于谷氨酸钠含量不低于 99%的产品。
2 引用标准下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文。
本标准出版时,所示版本均为有效。
所有标准都会被修订,使用本标准的各方应探讨使用下列标准最新版本的可能性。
GB 191—1990 包装储运图示标志GB/T 601—1988 化学试剂滴定分析(容量分析)用标准溶液的制备GB/T 602—1988 化学试剂杂质测定用标准溶液的制备GB/T 603—1988 化学试剂试验方法中所用制剂及制品的制备GB 1354—1986 大米GB/T —1996 食品中总砷的测定方法GB/T —1996 食品中铅的测定方法GB/T 6543—1986 瓦楞纸箱GB/T 6682—1992 分析实验室用水规格和试验方法GB 7718—1994 食品标签通用标准GB 9687—1988 食品包装用聚乙烯成型品卫生标准GB/T 12309—1990 工业玉米淀粉QB/T 1840—1993 工业薯类淀粉国家技术监督局令[1995]第 43 号定量包装商品计算监督规定3 定义本标准采用下列定义.谷氨酸钠( 99% 味精) sodium L-glutamate ; L- 谷氨酸单钠—水化物monosodium L-glutamate monohydrate;缩写式:MSG(99%Wei Jing)以碳水化合物(淀粉、大米、糖蜜等糖质)为原料,经微生物(谷氨酸棒杆菌等)发酵,提取,中和,结晶,制成的具有特殊鲜味的白色结晶或粉末。
4 化学名称、分子式、结构式、分子量化学名称:L-谷氨酸单钠一水化物(或 L-α-氨基戊二酸单钠一水化物)分子式:C5H8NNaO4·H2O国家质量技术监督局 2000—04—05 批准 2000—09—01 实施GB/T 8967—2000结构式:NaOOC—CH2—CH2—CH—COOH·H2ONH2相对分子质量:(按 1995 年相对原子质量)5 技术要求5.1 原料要求玉米淀粉:应符合 GB/T 12309 的规定。
薯类淀粉:应符合 QB/T 1840 的规定。
大米:应符合 GB 1354 的规定。
半成品 L-谷氨酸(麸酸):应符合附录 A 的要求。
5.2 外观及感官要求本品为无色至白色结晶或粉末,无明显杂质,具有特殊的鲜味,无异味。
42−砷,mg/kg ≤铅,mg/kg ≤6 试验方法1本试验方法中实验室用水,应符合 GB/T 6682 三级或三级以上水规格。
所用试剂除另有注明外,均为分析纯。
6.1 外观和感官检查将样品撒在白色滤纸上,目视法检查其颜色和杂质品尝其滋味。
同一单位包装,100 g 样品中不得超过 1 个( mm 以上)肉眼可见杂质。
6.2 谷氨酸钠的鉴别必要时,可按附录 B 鉴别。
6.3 谷氨酸钠含量6.3.1 第一法高氯酸非水溶液滴定法6.3.1.1 方法提要在乙酸存在下,用高氯酸滴定样品中的谷氨酸钠,以电位滴定法确定其终点,或以α-萘酚苯基甲醇为指示剂,滴定溶液至绿色为其终点。
6.3.1.2 试剂和溶液国家质量技术监督局 2000—04—05 批准 2000—09—01 实施GB/T 8967—2000a) 高氯酸标准溶液[c(HClO4)= mol/L]:按 GB/T 601—1988 中配制和标定;b) 乙酸(GB/T 676);c) 甲酸(HG/T 3-1296);d) 2 g/Lα-苯基甲醇-乙酸指示液:称取α-萘酚基甲醇 g,用乙酸[b)]溶解并稀释至 50 mL。
6.3.1.3 仪器自动电位滴定仪(精度±5 Mv);或酸度计:以玻璃电极为指示电极,饱和甘汞电极为参比电极(或采用复合电极),并备用电磁搅拌器。
6.3.1.4 分析步骤a)第一法电位滴定先按仪器使用说明书处理电极和校正电位滴定仪。
用小烧杯称取样品 g,精确至 g,加甲酸[c)]3 mL,搅拌直至完全溶解,再加乙酸[b)]30 mL,摇匀。
将盛有试液的小烧杯置于电磁搅拌器上,插入电极,开启搅拌,从滴定管中分次滴加 mL 高氯酸标准溶液[a)],同时记录电位 E(或 pH 值)和消耗高氯酸标准溶液的体积 V;当滴定将至终点前,则每次滴加 mL 高氯酸标准溶液,记录一次电位 E(或 pH 值)和消耗高氯酸标准溶液体积 V,突跃点过去,仍继续滴加高氯酸标准溶液,直至电位 E (或 pH 值)无明显变化为止。
以电位 E(或 pH 值)为纵坐标,以滴定时消耗高氯酸标准溶液的体积 V 为横坐标,绘制 E-V 滴定曲线,以该曲线的转折点(突跃点)为其滴定终点。
b) 第二法指示剂法称取样品 g,精确至 g,加甲酸[c)]3 mL,搅拌直至完全溶解,再加乙酸[b)]30 mL、α-萘酚苯基甲醇-乙酸指示液[d)]10 滴,用高氯酸标准溶液[a)]滴定试液,直至颜色变绿即为终点,记录消耗高氯酸标准溶液的体积(V)。
同时做空白试验,记录消耗高氯酸标准溶液的体积(V0)。
c) 高氯酸溶液浓度的校正若滴定样品与标定高氯酸溶液时温度之差超过10℃,则须重新标定高氯酸溶液的浓度;若不超过10℃,则按式(1)加以校正。
cc=0(10) (1)11 + ×−t t式中:c1——滴定试液时高氯酸溶液的浓度,mol/L;c0——标定时高氯酸溶液的浓度,mol/L;t1——滴定试液时高氯酸溶液的温度,℃;t0——标定时高氯酸溶液的温度,℃;——乙酸的膨胀系数。
6.3.1.5 分析结果的表述样品中谷氨酸钠含量按式(2)计算:()X1=× V1−V0×100 (2)c×m式中:X1——样品中谷氨酸钠含量,%;国家质量技术监督局 2000—04—05 批准 2000—09—01 实施GB/T 8967—2000V1——试液消耗高氯酸标准溶液的体积,mL;V0——空白消耗高氯酸标准溶液的体积,mL;c——高氯酸标准溶液的浓度,mol/L;—— mL 高氯酸标准溶液[c(HClO4)= mol/L]相当于谷氨酸钠(C5H8NNaO4·H2O)的质量,g;m——样品质量,g。
计算结果精确至小数点后第一位。
6.3.1.6 允许差同一样品测定结果,相对平均偏差不得超过 %。
6.3.2 第二法旋光法6.3.2.1 方法提要谷氨酸钠分子结构中含有一个不对称碳原子,具有光学活性,能使偏振光面旋转一定角度,所以,可用旋光仪测定其旋光度。
根据旋光度换算成谷氨酸钠的含量。
6.3.2.2 试剂盐酸(GB/T 622)6.3.2.3 仪器旋光仪(精度±)备有钠光灯(钠光谱 D 线 nm)。
6.3.2.4 分析步骤a) 称取样品 10 g,精确至 g,加少量水溶解并全部移入 100 mL 容量瓶中,加盐酸 20 mL,混匀,待冷却至20℃,补加水至刻度,摇匀。
b) 在恒温室(20℃)里,先用标准旋光角校正仪器。
然后,将上述试液置于旋光管中(不得有气泡),观测其旋光度,同时记录旋光管中试液的温度。
6.3.2.5 分析结果的表述样品中谷氨酸钠含量按式(3)计算:aX2=×+ (20 −t)×100 (3)式中:X2——样品中谷氨酸钠含量,%;α——实测试液的旋光度;L——旋光管长度(即液层厚度),dm;c——1 mL 试液中含谷氨酸钠的质量,g/mL;——谷氨酸钠的比旋光度,[ ]20D;t——测定时试液的温度,℃;——温度校正系数。
计算结果精确至小数点后第一位。
6.3.2.6 允许差同一样品测定结果,相对平均偏差不得超过 %。
6.4 透光率6.4.1 仪器分光光度计(精度±%)。
6.4.2 分析步骤国家质量技术监督局 2000—04—05 批准 2000—09—01 实施GB/T 8967—2000称取样品 10 g ,精确至 g ,加水溶解并定容至 100 mL ,摇匀,作为试液。
用试液冲洗并注入 10 mm 比色皿中,以溶解样品的同批水调仪器零点,于波长 430 nm 处,测定其透光率。
测定结果准确至整数。
6.4.3 允许差同一样品两次测定,绝对值之差不得超过 1%。
6.5 比旋光度 6.5.1 方法提要同 。
6.5.2 试剂 同 。
6.5.3 仪器 同 。
6.5.4 分析步骤 同 。
6.5.5 分析结果的表述若采用钠光谱 D 线,1 dm 旋光管,在 20℃测定(液温为 20℃)时,可以直接读数;若试液温度为 t℃时,则须按式(4)换算:X 3= [ ] D−( 20− t ) (4)式中:X 3——样品的比旋光度, [ ]20D;[ ]tD ——在 t℃时试液的比旋光度;t ——测定时试液的温度,℃;——温度校正系数。
计算结果精确至小数点后第一位。
6.5.6 允许差同一样品两次测定,绝对值之差不得超过 。
6.6 氯化物 6.6.1 方法提要试液中含有的微量氯离子与硝酸银生成氯化银沉淀,其浊度与标准氯离子 产生的氯化银比较,进行目视比浊定量。
6.6.2 试剂和溶液a) 硝酸(GB/T 626);b) 氯化物标准溶液(1 mL 溶液含有 mg 氯):按 GB/T 602—1988 中配制。