FDA检查相关要求
fda对于润滑油的要求

美国食品和药物管理局(FDA)对食品级润滑油有严格的要求,以确保其安全性和合规性。
以下是一些关键要求:
1. 成分:食品级润滑油必须使用符合 FDA 规定的成分。
这些成分必须是无毒、无害、无刺激性的,并且不能对食品造成污染。
2. 生产过程:食品级润滑油的生产过程必须符合 GMP(良好生产规范)要求,以确保产品的质量和安全性。
3. 标识和标签:食品级润滑油必须在包装上明确标识为“食品级”或“食品用”,并提供相关的使用说明和安全信息。
4. 测试和验证:食品级润滑油必须经过严格的测试和验证,以确保其符合 FDA 的要求。
这些测试包括化学分析、毒性测试、微生物测试等。
需要注意的是,FDA 对食品级润滑油的要求可能会因产品类型和用途而有所不同。
因此,在选择和使用食品级润滑油时,建议咨询相关专业人士或参考 FDA 的相关规定和指南。
FDA oos调查指南

药品检验结果超标调查工业指南草案本指南颁布的目的只是为了提供建议。
此指南的草案将在联邦注册通知上宣布生效,若有关于此草案的评论和提议,应在宣布生效后60天内提交。
提议应提交到……地址(5230 Fishers Lane., rm. 1061. Rockville, MD 20852)的FDA 诉讼事件管理部。
所有的评论均应与列于联邦注册发布的生效通知上的事件编号相一致。
此文件在HFD-210,5600 Fishers Lane. Rockville, MD 20857,(Tel)301-827-4573通信管理司的药物信息部和网址/cder/guidance/index.htm.上均有副本。
对草案内容如有疑问,请联系C. Russ Rutledge (301)594-2455美国健康与人服务部食品药品监督管理局药物评审中心1998年9月CP号目录表药品检验结果超标调查工业指南 (1)目录表 (2)1.前言 (3)2.背景 (3)3.检验结果鉴定与评估 (4)4.OOS 检验结果调查 (7)5.调查总结 (14)草案-不用于执行药品检验结果超标调查工业指南1.前言此工业指南提供评审部门目前在怎样评估可疑和超标检验结果上的看法,从此文档的目的出发,OOS 结果条款包含了所有的不符合新药申请、官方说明或者生产企业中制定的规范或可接收标准的可疑结果。
此指南适用于适应CGMP规范的原料药、赋形剂和其他成分生产期间的实验室检验和成品检验,特别是,指南论述了怎样调查可疑或OOS检验结果,包括实验室人员职责、调查的实验室阶段、必要的额外的检验,什么时候进行实验室外的扩展调查,和所有检验结果的最终评估。
2.背景FDA 关心在药品生产期间非常重要的实验室检验和文件记录的完整性。
CGMP法规必需的实验室检验对于确定成分、容器和密封件、进程物料和成品是否符合要求是非常重要的,包括稳定性试验在内。
检验也支持分析和工艺验证结果。
FDA检查准备工作及注意事项

补充资料——FDA查厂依据
法律依据 - 食品药品及化妆品法案 法规 21 CFR PART 820, 即QSR (以前叫做cGMP) 21 CFR PART 803, 即MDR (医疗器械不良反应报告) 21 CFR PART 801, 有关标签的规定 21 CFR PART 807, 厂商注册,510(K)等方面的相关规定 审核指南 QSIT Quality System Inspection Technique (质量体系检查技术) QSIT 的四个部分: 管理职责 (management control), 设计控制 (design control), 生产和过程控制 (p&pc), 要想办法找到这些法规, 纠正预防措施 (CAPA)
法规体系 FD&C Act Medical Device Amendment (1976-5-21大章800-898部分 法规主要关注医疗器械的安全性和有效性 医疗器械分为3类 CLASS I (general control) 例如:检查手套, 洗耳球 CLASS II (special control) 例如:注射器,静脉留置针 CLASS III(PMA) PS:FDA不喜欢QMS,不要只有ISO13485和CE等法规,也要有QSR820呢!
QSR820 & FDA工厂检查
美国对医疗器械企业的质量体系要求
Wendy Zou 2011.09.16
说明
因应将来FDA查厂,建议以此逐条查检公司的执行是不是符合要求? 再以ISO13485标准审核。(以补不足) 坐而言不如起而行,各公司就自己所负责的工作内容,逐一自审,就从 现在开始做都不嫌晚,最怕是没有要做的意念,消极被动,都不足取, FDA早晚会来查,一定会查,如果没有做,到时候自己解释,而且解 释也没有用,如果FDA审核员判刑,恐怕万一产品以后无法出口,谁 也负责不了,配合的供应商也是如此。
原料药FDA现场GMP符合性要求与检查实践-精品文档

• • • • • FDA的国外检查 FDA对原料药检查的依据 FDA检查方式 FDA的系统检查法 FDA检查重点及检查实践
1
FDA的国外检查
1955年开始进行第一次国外检查(抗生素)。 1961年国外检查达13 次,以后继续增加。 1971年成倍增加, 达到80 次。 此后,在整个70 年代里不断增加。到了80年代 及至90年代初,达到了每年检查160次之多。 1993年FDA计划要进行340次检查 2000年进行了48次检查,批准了28项。在不批准 的项目中有14个是问题很大的。发出了11封警告 信(2019年发出了9封)。 从1981年起, FDA先后派了一些检查官来我国检查一 些申报原料药的药厂。 截至2019年10月,我国原料药获得美国FDA的DMF II文件登记号为213项,涉及产品150余种,文件持 有者达100余家.
OOS调查 4% 设备确认 4% 工艺验证 4% 质量系统 5% 原料控制 5% 生产工艺 稳定性研究 7% 6% 实验室记录 7% 其它 19% 实验室控制 14% 各种SOPs不 足 9% 生产批记录 9% 设备清洗 7%
14
2019年API生产商CGMP检查存在的缺陷
工艺验证 4% 分析方法 4% 水系统 4% 环境控制 4% 设备确认 6% 其它 20% 实验室控制 13% OOS调查 10% 实验室记录 8% 各种SOPs不 足 生产工艺控制 8% 设备清洗 生产记录 7% 6% 6%
2
FDA对国外API检查
天然原料药 NEC 7% 发酵非无菌 化学合成 6% 无菌 2% 生物技术 原料药 1% 化学合成非无 菌 81% 天然原料药NEC 生物技术原料药 发酵非无菌 其它 其它 3%
化学合成 无菌 化学合成非无菌
FDA批准前现场检查的政策要求及实施概述(1)

摘要鉴于美国药品市场在全球市场中的特殊地位,和美国监管法规FDA的cGMP 在全球法规监管政策中的指导性作用,学习和掌握该法规的要求是中国制药企业在实施国际化战略的必由之路。
批准前现场检查是美国药品审批的重要步骤之一,其目的是检查企业的现场GMP状态和检查现场原始数据是否真实及和申报资料一致,现场检查的意见对药品申请是否获得批准至关重要。
本文通过对美国FDA的药品申请批准前检查的政策要求的阐述,并分析总结若干国内企业在接受批准前检查过程中的经验教训,并将中美两国药品批准前检查政策进行了对比分析,希望藉此帮助国内企业更好地理解美国批准前检查政策的要求,在贯彻实施的过程中注意一些关键因素的把握,从而为企业顺利通过美国药品审批提供一些借鉴。
论文简要介绍了美国FDA药品申请批准前检查的政策发展历史和法规依据,并阐述了药品申请批准前检查的目的、范围、方式、实际执行流程、检查政策的基于体系的检查方式的特点,以及六大体系在检查过程中的关注重点等。
并通过国内三个企业通过FDA的批准前检查的案例分析,总结批准前检查流程中的关键环节和如何进行检查后的整改措施及检查成果和教训,强调在完成批准前检查后的维持GMP状态的重要性。
通过分析国际药品市场、中国药品市场、中国制药企业的现状及自身优缺点,论证国际认证特别是美国FDA认证是中国企业发展壮大的必由之路,及获得国际认证后的重要意义,鼓励国内企业坚定国际化战略思维。
鉴于国际法规政策的多样性,论文专门将中美两国的批准前政策的异同点进行了对比分析,并讨论了不同规定的优缺点,方便国内企业更好地理解美国政策。
国家新版的GMP即将推行,其宗旨和美国的cGMP要求更加接近,此对比分析对于国内企业理解和遵循新的中国法规也有积极意义。
关键词:药品申请批准前检查现行药品生产质量管理规范批准前检查政策基于体系的检查The Requirement and E nforcement of FDA’s Pre-approval Inspection(PAI)PolicyAbstractGiven the special position of American drug market in the global market, and the guiding function of American supervise regulations(FDA ‘s cGMP)in global regular policy, it is inevitable for Chinese drug companies to learn and master the requirements of this regulations in order to execute their globalization strategies. Pre-approval inspection is an important step of American drug approval, whose purpose is to ensure that the on-spot GMP and on-spot statistics of companies are true and the same as written in the application material. Therefore, the opinion from the on-spot check is vital to the approval of the application. This thesis, with an illustration of the checking policy requirements prior to FDA drug application approval in U.S.A. , through an analysis of some lessons and experience of some domestic companies during the checking, and a comparison of the checking policies between the two countries, aims to help domestic companies gain a better understanding of the American pre-approval inspection policy, and pay attention to some crucial factors during the execution process, thus to provide some guidance for the companies to get a smooth approval from the American drug approval.The thesis gives a brief introduction of the policy history and the regulation resources of American FDA pre-approval inspection, and an illustration of the purpose, the range, the method, and the actual execution procedure of the check. It lays emphasis on the features of the checking method and the checking policy, and the different focuses of the six systems.By analyzing the current situation and their own advantages and disadvantages of the international drug market, Chinese drug market, and Chinese drug companies, the author draws a conclusion that the international qualification, especial American approval is an inevitable path for Chinese companies to take for their development. Thesignificance of obtaining international 认证encourages domestic companies to become international.With an brief introduction of some necessary steps before the application check, and the sample analysis of the actual check of three domestic companies, the thesis illustrates the critical steps during the process and how to improve after the check. It emphasizes the importance to sustain the GMP state after the check by focusing on the check result, the lesson, and the staff training.Given the variety of international regulations and policies, the author gives a special comparison and analysis of the similarities and differences of the approval polices between China and U.S.A.. and discusses their merits and demerits, therefore hopes to give the domestic companies a better understanding of American policies.The thesis does not give too much pages to the methods and skills about how to cope with the check, with the hope that the companies can regard the basic requirements of GMP as their real target, and perfect and maintain their GMP system through the check. We found that the new Chinese GMP which is to be practice recently is highly similar with American’s cGMP, therefore this analysis is also helpful for Chinese pharmaceutical companies to understand and follow Chinese policy.Key Words: drug, application Pre-approval inspection cGMPChinese PAI policy System-based inspection目录第1章前言 (1)第2章美国FDA药品申请批准前GMP检查(PAI)的要求 (2)2.1 FDA的cGMP检查的目的、分类,检查范围和方式 (4)2.2 美国FDA的药品申请批准前GMP检查的执行流程 (6)2.3 美国FDA的药品申请批准前GMP检查的 (9)第3章中国企业通过FDA的cGMP检查的意义 (15)3.1全球市场情况分析 (15)3.2中国市场分析 (18)cGMP认证的意义分析 (19)第4章制药企业如何准备FDA药品申请批准前检查以及实施检查后的整改22 4.1制药企业如何准备FDA的批准前检查 (22)4.2迎接和陪同FDA的批准前检查 (23)第5章中国企业在准备和应对cGMP批准前检查过程中案例分析 (24)第6章中国企业通过FDA的cGMP现场检查后维持cGMP状态及应对批准后检查(Post- AI)的必要措施 (34)6.1维持良好的GMP状态的常规性条件 (34)6.2在职员工的cGMP培训 (37)第7章中美两国GMP批准前检查政策的异同点对比和先进性分析 (38)7.1中国GMP批准前检查政策的简要介绍 (38)7.2中美两国批准前检查政策的异同点对比和先进性分析 (43)7.3针对中国SFDA的批准前检查政策的建议 (49)结束语 (50)参考文献 (51)致谢 (53)第一章前言制药行业是一个非常特殊的行业,其产业应用的科学基础涵盖物理、化学、生物学、微生物学、医学、材料学、矿物学、机械、电子、光学、流体力学、计算机等多种学科;由于其产品的使用和人类健康息息相关,所以这也是一个被高度关注,关乎国家政治稳定性的特殊行业。
体外诊断试剂的政策法规

体外诊断试剂的政策法规全文共四篇示例,供读者参考第一篇示例:体外诊断试剂是医疗设备领域中一类应用广泛的产品,其在医疗诊断中扮演着至关重要的角色。
体外诊断试剂可以帮助医生诊断疾病、监测疗效、筛查疾病等,对于提高诊断的准确性和效率起着重要作用。
为了确保体外诊断试剂的质量和安全性,各国都制定了一系列的政策法规来规范和监管。
本文将重点介绍体外诊断试剂的政策法规及其相关内容。
一、FDA(美国食品药品监督管理局)对体外诊断试剂的监管在美国,FDA对体外诊断试剂的监管非常严格。
根据美国《药品和化妆品法案》,所有在美国市场销售的体外诊断试剂都必须经过FDA的审批。
FDA要求厂商在向FDA提交上市申请时,必须提供充分的临床数据和试验报告,以证明产品的有效性和安全性。
FDA还会对生产工艺、质量管理体系和产品标签等方面进行严格监督,确保体外诊断试剂的质量和安全性。
FDA还颁布了一系列的法规和指南,规定了体外诊断试剂在研发、生产、销售和使用过程中的要求和规范。
《体外诊断试剂法规》(21 CFR Part 809)、《体外诊断试剂注册和上市报告指南》等文件对体外诊断试剂的注册、标识、报告、质量管理等方面做出了详细规定,确保体外诊断试剂符合美国法律法规的要求。
二、欧盟对体外诊断试剂的监管在欧盟,体外诊断试剂的监管主要由欧盟委员会和各成员国的监管机构共同负责。
欧盟委员会根据《体外诊断试剂法规》(98/79/EC)对体外诊断试剂的注册、标识、质量管理、审评等方面做出了详细规定。
根据该法规,体外诊断试剂必须通过欧盟指定的评估机构进行审评,并获得CE认证方可在欧盟市场销售和使用。
在中国,体外诊断试剂的监管由国家药监局和各级地方药监部门共同负责。
中国的《医疗器械监督管理条例》、《医疗器械注册管理办法》等文件对体外诊断试剂的注册、生产、销售等方面做出了详细规定。
根据中国的相关法规,体外诊断试剂必须取得《医疗器械注册证》方可在中国市场销售和使用。
FDA检查员手册

FDA检查员指导手册7356.002F原料药生产检查(药品质量保证)第一部分背景总则法案的501(a)(2)(B)条款要求所有药品的生产都必须遵守现行GMP的要求,而原料药也不例外。
对于原料药和制剂这两者的要求,法案并没有区别对待,而任何原料药或制剂方面的GMP缺陷都构成了对法案的偏离。
对于原料药或药物成分来说,FDA并没有为此而专门发布cGMP法规文件(就像我们现在有的制剂cGMP法规一样)。
因此,本文提到的“cGMP”指的是法案要求,而并非美国联邦法规(CFR)第21部分210和211条款中关于制剂的要求。
其实,FDA早就意识到cGMP对制剂的要求(美国联邦法规第21部分210、211条款)在理念上对于原料药生产来说同样适用且有效。
这些理念包括使用合适的设备;聘用经过培训且通过资质确认的人员;建立充分合理的书面程序和控制,确保生产工艺和控制的有效性,从而保证产品质量;建立一套中间体和最终药品检测方法的体系,确保药品在规定的使用期限内保持质量的稳定性。
2001年,FDA在人用药物注册技术要求国际协调会议(ICH)上与其他政府监管部门共同努力,采用了针对API行业cGMP的国际性指南,也就是ICH Q7A,活性药物成分的药品质量管理的指南。
ICH Q7A正体现了FDA对于原料药现行GMP体系的要求。
因此,遵循该指南要求的API及其相关生产和检验设施是符合法定cGMP要求的。
然而,只要是能符合法案501(a)(2)(B)的要求,并能确保API符合其纯度、均一性和质量特性的方法都可以采用。
在本程序中所使用的术语“活性药物成分”(原料药)的含义与ICH Q7A 中的定义一致。
在ICH Q7A中活性药物成分被定义为“旨在用于药品生产的任何物质或混合物,当用于药品生产时,这些物质即成为药品中的活性成分。
这种物质被用来提供药学活性或在诊断、治疗、止痛、缓解、处理或疾病预防中起着直接作用或用于影响机体结构和功能。
美国FDA对血管内导管的相关要求

美国FDA对血管内导管的相关性能要求简述一、美国FDA对血管内导管性能的一般技术要求(一)对导管性能的技术要求在物理性能要求方面,美国FDA指南文件包括九方面的要求,即管体拉伸强度、管体和导管座连接部分连接强度、导管刚性、导管尖端和管体的连接强度、导管伸长率、导管座泄漏、导管正压涨破力(导管内正压)、导管塌陷力(导管内负压)和导管弯曲疲劳耐受性。
与YY 0285相比,FDA对血管内导管增加了六个方面的附加性能要求,即管体刚性、导管伸长率、导管爆裂力、导管塌陷力、导管弯曲疲劳耐受性以及导管尖端和管体的连接强度。
FDA附加的上述性能要求与产品的原材料、生产加工过程以及临床使用息息相关。
譬如管体刚性,导管过硬可能会导致血管壁的机械损伤和破坏,从而促进血栓的形成并诱发相关的并发症,因此部分导管要求导管尖端刚性较小,而后端刚性较大(先进入血管端较软,以避免对血管及其他组织的损伤;后端刚性较大以产生较大的扭矩利于操作)。
导管伸长率是指在拉力作用下,导管的伸长量占原来长度的百分率,伸长率大且弹性恢复率大,表明导管材料的变形适应性较好。
若导管伸长率过大可能易导致管腔变小从而影响流量,同时还会影响导管插入深度的判断,而伸长率过小则表示材料分子柔顺性差,会影响导管的形态记忆,换言之导管容易打折。
特别是对于外周中心静脉导管而言,由于该类导管进入人体较深,长度大于其它血管内导管,伸长率指标尤为重要。
血管造影导管和其它一些给药导管,由于要输送造影剂和药物,有时需要在较短的时间内高压注入一定量的药物,会要求导管有一定的耐高压能力,这就要求导管有一定的正压涨破力。
导管正压涨破力指标直接关系到导管壁的强度,对于快速大量输注,尤其是用小型注射器在导管内注射液体具有重要的临床意义。
导管塌陷力是要求导管在一定负压条件下不塌陷,保证临床抽吸血液时导管保持畅通,这对于高流量血管内导管如血液透析导管特别重要。
为保证产品上述性能,部分生产企业在管身内加入加强丝,一方面增强正压耐受性,除使管壁更薄,管腔更小以外,又具有了较强的径向支撑强度,保持了良好的管腔通畅性。
FDA食品级材料测试要求限量标准 列表
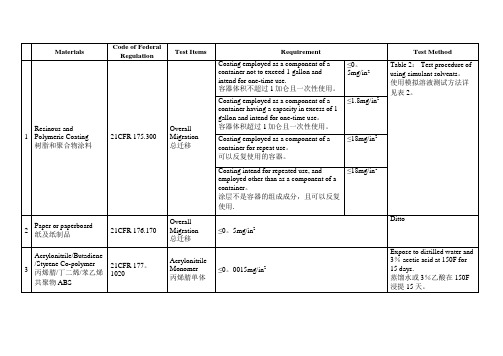
≤18mg/in2
≤0。5mg/in2
≤0。0015mg/in2
Test Method Table 2: Test procedure of using simulant solvents。 使用模拟溶液测试方法详 见表 2。
Ditto
Expose to distilled water and 3% acetic acid at 150F for 15 days. 蒸馏水或 3%乙酸在 150F 浸提 15 天。
≤0。2mg/in2 (distilled water )
≤0。2mg/in2 (n— heptane )
≤0.2mg/in2 (ethyl ethanol )
Used for packaging,
transporting or holding alcoholic foods that do not exceed 95 percent by volume。 接触含量不超过 95%的酒精 饮料。
Used in contact with fatty foods 接触油性食品
≤0。05mg/in2 (ethanol ) Container capacity: Less than or equal to 50ml ≤1%w/w
≤0.5%w/w
≤0。5%w/w
≤20mg/in2 (first 7h of extraction) ≤1mg/in2 (succeeding 2h of extraction) ≤175mg/in2 (first 7h of extraction) ≤4mg/in2 (succeeding 2h of extraction)
Materials
Code of Federal Regulation
美国食品级接触材质的FDA认证检测要求

美国食品级接触材质的FDA认证、FDA检测要求为:No. Material材质TestItem 测试项目1 175.300有机涂层制品要求去离子水萃取法8%酒精萃取法正庚烷萃取法2 176.170Paper and paper cardboard纸制品要求氯仿可溶萃取物(去离子水浸取法)氯仿可溶萃取物(8%酒精浸取法)氯仿可溶萃取物(50%酒精浸取法)氯仿可溶萃取物(正庚烷浸取法)3 177.1010 for Acrylic丙烯酸树脂要求总提取物,去离子水、8%酒精,正己烷高锰酸钾可氧化萃取紫外吸收(水,8%乙醇,50%乙醇中)紫外吸收(正庚烷中)4177.1210食品容器的密封圈,密封衬垫要求,如硅橡胶圈氯仿可溶萃取物(去离子水浸取法)氯仿可溶萃取物(8%酒精浸取法)氯仿可溶萃取物(正庚烷浸取法)5 177.1240对苯二甲酸1,4-亚环己基二亚甲基酯与间苯二甲酸1,4-亚环己基二亚甲基酯共聚物Water extractives at refluxing temperature for 2 hoursEthyl acetate extractives at refluxing temperature for2 hoursN-hextane extractives at refluxing temperature for 2hours6 177.1340 forEMA乙烯- 丙烯酸甲酯氯仿可溶萃取物(去离子水浸取法)氯仿可溶萃取物(8%酒精浸取法)氯仿可溶萃取物(50%酒精浸取法)氯仿可溶萃取物(正庚烷浸取法)7 177.1350 for EVAEVA要求氯仿萃取(在4种不同模拟液中:水,8%乙醇,50%乙醇,正庚烷)Vinylidene fluoride & hexafluropropene黄原胶(涂层)8 177.1460 for Melamine resins三聚氰胺树脂(密胺)要求氯仿萃取(在3种不同模拟液中:水,8%乙醇,正庚烷))9 177.1500 for Nylon尼龙塑料要求,如尼龙n6, 尼龙66, 尼龙610,尼龙6/12, 尼龙12T等(需要确定尼龙类型。
FDA质量体系规范-中文版 (QSR820)

FDA质量体系规范-中文版 (QSR820)1.1 概述§820.1 范围(a) 适用性(1) 在这个质量体系规范中描述了现行的生产管理规范的要求(CGMP). 本规范要求规定了所有医用器械成品在设计,制造,包装,标签,贮存,安装和服务中使用的方法,设施和控制.这些要求是为了确保医疗器械成品的安全和有效,并遵从美国食品,药品和化妆品法.本规范提出了适用于医疗器械成品制造商的基本要求.如果某制造商只进行本规范规定的一部分操作,而不进行其他操作,则该制造商仅需执行适用于他所进行操作的那些要求.有关Ⅰ类器械,设计控制仅按在§820.30(a)(2)中列出的要求进行.这个规范不适用于成品组件和零件的制造商,但鼓励这样的制造商使用规范的适当规定作为指导.人类血液制品和血液成分的制造商不属于本规范的管理范围,但属于606的管理范围.(2) 这一规范的规定适用于本规范定义的医疗器械成品,即使用的对象是人的,在美国各州或领地,哥伦比亚特别区或波多黎哥联邦制造,进口或出口的器械成品.(3) 在本规范中,几次使用了短语”适当的地方”. 当要求以”适当的地方”来限制时,如果制造商没有合理的理由来证明不适宜,就认为此要求是”适当的”. 如果不贯彻”适当的”要求,就会导致产品达不到要求或制造商不能采取某些必要的正确措施.(b) 范围这一的质量体系规范和增补在这一规范其他节的条文明确注明用于其他方面的条文除外. 在不可能执行全部适用条文(包括这一节的和这一规范其他节的条文)的情况下,特定的运用于有问题器械的条文将取代其他一般的适用条文.(c) 权威性根据联邦法501, 502, 510, 513, 514, 515, 518, 519, 520, 522, 701, 704,801, 803, (21U.S.C.315, 352, 360, 360c, 360d, 360e, 360h, 360i, 360j, 360l, 371, 374, 381, 383) 建立和提出了820规范的权威性. 如果没能执行适用规范,可能会导致产生伪劣器械, 根据法规501(h), 对这样的器械和未能执行规范的人都要进行处罚.(d) 外国制造商如果某制造商提供给美国的进口器械,拒绝接受FDA对外国设备进行是否执行本规范的检查,就会出现法规的801(a)节中的后果. 用这样的设备生产出的任何器械,在设计,制造,包装, 标签,贮存,安装或服务方面,使用方法,设备和控制都未遵从法规的520(f)节和本规范的要求. 根据法规501(h), 用这样的设备制造出来的器械都属于伪劣产品.(e) 豁免或更改(1) 任何希望豁免或更改执行某些器械质量体系要求的申请,都要遵从法规520(f)(2)的要求. 申请豁免或更改的过程将依据这一规范§10.30的程序进行,即FDA的管理程序.以下地址可提供指导: 器械和放射卫生中心, 小制造商处(HFZ-220), 1350 Picca rd Dr., Rockville, MD20850, U.S.A. 电话: 1-800-638-2041或1-301-443-6597, FAX301-443-8818.(2) 当FDA判定某种更改有益于公众健康时, 就会起草并认可这项更改. 这种更改仅能在一段时间内维持有效, 即当器械仍能满足公众健康需要, 并且如果没有更改,器械不可能制造得非常有效的一段时间内.§820.3 定义(a)法指的是美国食品,药品和化妆品法修正案(secs.201~903, 52Stat.1040 et seq., 修正版21U.S.C.321~394). 在法201中的全部定义都适用于本规定.(b)投诉指的是以某些书面的,电子的或口头的形式表达意见, 认为在配发后的器械在鉴定,质量,耐久性,可靠性,安全性,有效性或性能方面有缺陷.(c)组成指的是原材料,物质,小件,零件,软件,硬件,标签或有包装和标签的成品器械的零配件.(d)控制编号指的是有区别的符号,如字母或数字的不同组合,或以原制造,包装,标签和分发的单个或批量成品的区别符号来分辨.(e)设计历史文件(DHF)指的是描述某医疗器械成品设计过程的有关记录.(f)设计输入是指作为器械设计基础对器械的物理和特性要求.(g)设计输出是指各设计阶段的设计成果和最终的总设计成果. 完成设计输出包括器械,包装和标签,器械主记录.(h)设计评审是指依照依照文件进行广泛,系统的设计评审, 以评价设计要求的适当性,并评价设计达到这些要求的能力,查明问题所在.(i)器械历史记录(DHR)是指医疗器械成品制造过程的记录.(j)器械主记录(DMR)是指包括医疗器械成品的程序和规范的完满记录.(k)建立是指定义,文件(书面的或电子的)和执行情况.(l)器械成品是指适于使用或具有功能的器械或器械附件, 不论是否经过包装,贴标签或灭菌.(m)批是指一种或几种组成或成品器械具有单一类型,型号,类别,尺寸,成分或软件版本,必须在相同条件下制造,并在规定的限度内具有相同的特征和质量.(n)管理职责是指制造商的高级雇员有权建立或改变制造商的质量方针和质量体系.(o)制造商是指设计,制造,构造,装配或加工成品器械的人. 制造商包括但不局限于那些从事灭菌,安装,再贴标签,再制造,再包装或Specification开发商和从事这些工作的外国实体的最初代理人.(p)生产过程副产物是指促进生产过程所用的材料或物质,制造加工过程中的伴随组分或副产品,以残余物或混杂物的形式存在.(q)不合格是指未达到特定的要求.(r)产品是指组成,制造材料,加工过程中器械,成品器械及返回器械.(s)质量是指使器械安全适用的总性质和特征,包括安全性和性能.(t)质量审核是指在规定的时间间隔,以足够的次数,对制造商质量体系进行有组织的自主的检查,检验质量体系行为和结果是否执行质量体系程序,以保证有效地执行程序,达到质量体系目标.(u)质量方针是指有关质量的机构方向和目标,是由负责的管理人员建立的.(v)质量体系是指检查质量管理的组织机构,职责,程序,处理和资源.(w)再加工是指对成品器械进行加工,调节,革新,再包装,再贮存,大大改变了成品器械的性能,安全性规范或用途.(x)返工指的是对不合格产品采取某些措施,以使他在获准配发之前达到指定的DMR要求.(y)规范是指生产,加工,服务或其他行为必须遵守的一些要求.(z)有效性是指通过检查和提供客观证据来证明能始终满足特定的用途.(1)过程确认是指通过客观的证据证明加工生产出的产物或产品始终达到预定的规范.(2)设计确认是指通过客观的证据证明器械规范与使用者的需要和设计的用途相一致.(aa)验证是指通过检查和提供客观证据来证明已经满足指定的要求.§820.5 质量体系各制造商应建立并保持一个质量体系,适合于他们设计或制造的医疗器械,并且达到本规范的要求.1.2 质量体系要求§820.20 管理职责(a)质量方针管理职能机构应建立质量方针目标和质量承诺,并保证质量方针在企业各级人员中的理解,贯彻和持续执行.(b)管理机构各制造商都应建立并维持一个适当的组织机构,以保证器械依照本规范进行设计和生产.(1)职责和权限各制造商都应任命有相应职责,权限和能独立行使职权的人员负责管理,执行和评价质量体系.(2)人员各制造商都应具备足够的合格人员,包括分派培训有素的人员从事管理,执行,评价和内部质量审核等工作,以达到本规范要求.(3)管理者代表管理职能机构应任命其中一员为管理者代表,并在文件中注明.管理者代表不论其他职责如何,必须履行下列职责和权力:i. 确保按本规范要求有效地建立和保持.ii. 向管理机构汇报质量体系进行情况,供其讨论.(c)管理评审管理职能机构应按照建立的程序,以足够的次数定期评审质量体系的适用性和有效性. 以保证质量体系达到本规范要求和制造商建立的质量方针和目标,评审日期和结果应形成文件.(d)质量策划各制造商应编制质量计划,确定与设计和制造的器械相关的质量实践,人员和措施,并建立达到质量要求的规划.(e)质量体系程序各制造商应建立质量体系的各种程序和实施指南,并形成文件.§820.22 质量审核各制造商应建立质量审核的程序,并进行管理,以保证质量体系符合建立的质量体系要求,确定该质量体系的有效性.质量审核应由与审核事物无直接责任的人执行.若有必要时,应采取措施纠正错误措施,包括对有缺陷的事物进行再审核.管理机构对各质量审核的结果及再审核的情况进行复核.提供质量审核日期和结果及再审核的有关文件.§820.25 全体工作人员(a)一般要求各制造商都应具有足够的工作人员,具备必需教育,背景,接受过培训并富有经验,以保证正确履行本节所要求的全部工作.(b)培训各制造商应建立必需培训的程序,保证全部工作人员在经过培训后能胜任他们各自的职责,并提供与培训有关的文件.(1)培训内容还包括使全体工作人员懂得由于错误执行指定工作可能会导致器械产生缺陷.(2)使从事验证和确认工作的全体工作人员能预见可能会发生的缺陷和错误.1.3 设计控制§820.30 设计控制(a) 总则(1) Ⅱ,III类器械的制造商,以及在本规范(a)(2)段列出的Ⅰ类器械制造商,应建立并保持控制器械设计的方法,以保证达到特定的要求.(2) 下列Ⅰ类器械也需要设计控制.i.计算机软件的自动化机械.ii.下面列出的器械:(b)设计和开发计划各制造商应建立并保持有关设计和开发行为的计划,并规定执行职责.计划应规定提供或输入设计和开发程序的不同组或行为的互换信息.对计划应进行检查,用现代化手段处理并证实设计和开发的进展.(c)设计输入各制造商应建立并保持关于保证器械的设计要求适当的程序,以器械用途为主,包括使用者和病人的需要.该程序应包括关于不完善,不清楚或抵触要求的处理办法.设计输入要求应记录在文件中,并由指定的人进行检查和认可,并提供认可这些要求的日期和个人签名的文件.(d)设计输出各制造商应建立并保持关于确定和提供设计输出文件的程序,并进行执行设计输入要求的适当评价.设计输出程序应包含或制定参照的认可标准,并保证那些设计输出是鉴定器械良好性能所必需的.设计输出应记录在文件中,在获准之前进行评审,并提供有关评审认可日期和签名的文件.(e)设计评审各制造商应建立并保持一套程序, 保证在器械设计开发的适当阶段,按计划评审设计结果,并提供正式文件.评审参加者应包括设计的专业人员对设计阶段负有责任的代表和与设计阶段五直接责任的人和必要的专家.设计评审的结果包括设计鉴定,评审人员和日期,都应记录在设计历史文件(DHF)中.(f)设计验证各制造商应建立并保持验证器械设计的程序.设计验证应证明设计输出达到设计输入要求.设计验证的结果,包括设计方法的鉴定,验证人员和日期,都应当记录在DHF文件中.(g)设计确认各制造商应建立并保持设计确认的程序.应在规定的操作条件下,对试制的单个,批量产品或等同物进行设计确认的确认.设计确认应保证器械满足使用者的需要,并具有预期用途,还应包括产品在实际或设想使用条件下的试验.设计确认还应包括软件确认及适当的时候的风险分析.有关设计确认的结果,包括对设计和设计方法的鉴定,执行人员和日期都应记录在DHF文件中.(h)设计转换各制造商应建立并保持一套程序以确保器械设计正确性体现在一定的生产规范中.(i)设计更改各制造商应建立并保持一套程序,对更改的设计在执行之前进行鉴定,提供有效性文件或适当的地方进行验证,评审和认可.(j)设计历史文件各制造商应建立并保持各种类型器械的DHF. DHF应包含或参照必要的原始记录,来证明设计开发过程与认可的设计计划一致,并遵守本规范要求.1.4 文件控制§820.40 文件控制各制造商应建立并保持本规范所要求的全部文件控制的程序.程序应提供下列内容:(a)文件认可和发布各制造商应在分发达到本规范要求的全部文件之前,委派专人检查适用性和认可情况.应提供有关认可文件的日期和个人签名的文件.达到本规范要求的文件适用于指定的,使用的或其他需要的地方,所有失效的文件应从使用条款中删除.(b)文件更改更改文件应由执行原文件检查和认可的同一职能部门内的人进行检查和认可,除非有另外明确指定人选. 认可的改动应及时地转达给有关人员.各制造商应保留更改文件的记录. 更改记录应包括修改内容,相关文件的鉴定,认可人的签名,认可日期及更改生效的日期.1.5 采购控制§820.50 采购控制各制造商应建立并保持确保所有购买的或收到的产品和服务符合指定要求的程序.(a)对供应商,承包商和咨询机构的评审各制造商应建立一套供应商,承包商和咨询机构必须达到的指定要求.各制造商应:(1)根据指定要求(包括质量要求),评价和选择潜在的供应商,承包商和咨询机构.评价应记录在文件中.(2)根据评价结果,确定对产品,服务,供应商,承包商和咨询机构实施控制的方式和程度.(3)建立和保持可接受的供应商,承包商和咨询机构的记录.(b)采购资料各制造商应对采购的或收到的产品和服务建立并保留关于是否达到质量要求的资料.可能的话,应包括一份协议,关于供应商,承包商和咨询机构同意告知制造商,他们的产品或服务的改变,是制造商可以判断这些改变是否会影响成品器械的质量.采购资料应依照§820.40得到认可.1.6 标识和可追溯性§820.60 标识各制造商为防止混乱应建立并保持在接收,制造,交付和安装各阶段的产品标识程序. §820.65 可追溯性对于生产外科植入人体,支持或维持生命的器械制造商和依照制造商提供的使用说明正确使用时,如果器械运行失败可对使用者造成严重伤害,则应建立并保持对每个或每批产品都有唯一性标识的程序.程序应促进纠正错误措施.这种标识应包括在设计历史文件中.1.7 生产和过程控制§820.70 生产和过程控制(a)总则各制造商应制定,实施,控制并监测生产过程,以保证器械遵守本规范. 在制造加工过程中可能会发生违反规范的地方,制造商应建立并保持必须的生产过程控制的程序,生产过程控制应包括:(1)提供指导文件,标准操作程序(SOP’s),限定方法和生产控制方式;(2)在生产过程中监测和控制加工参数和产品特征;(3)应遵守的指定参考标准或编号;(4)加工和加工设备的认可;(5)工艺要求应阐述在工艺文件中或用通过鉴定和认可的代表性样品来表现.(b)生产和过程的改变各制造商应建立并保持改变规则,方法,加工或步骤的程序.这些改变在执行之前应被验证或在适当时依照§820.75使改变有效,这些行为均应记录在文件中.改变应依照§820.40得到认可.(c)环境控制在有理由认为周围环境条件对产品质量有不利影响时,制造商应建立并保持适当控制环境条件的程序.应定期检查环境控制系统,以核实该系统,包括必需设备的适当性,并正发挥着良好作用.检查应记录在文件中.(d)工作人员如果有理由认为工作人员和产品或环境的接触对产品质量有不利影响时,各制造商应建立并保持对工作人员的健康,卫生习惯,行为和衣着的要求. 各制造商应保证在指定的环境下临时工作的其他人员接受适当的训练或由接受过训练的人进行监督.(e)污染控制各制造商应建立并保持防止对产品质量有不良影响的物质污染设备或产品的程序.(f)厂房应该设计适当厂房,具有足够的空间进行必须的操作,以防止混乱,并保证有序的操作.(g)设备各制造商应保证在制造加工过程中使用的全部设备都达到指定要求,并经过适当设计,建造,放置和安装以利于保养,调试,清洁和使用.(1)保养计划表各制造商应建立并保持调试,清洁和其他设备保养的计划表,以保证达到生产规范.保养行为,包括执行保养行为的日期和人员应记录在文件中.(2)检查各制造商应依照建立的程序进行定期检查,以保证完成设备保养计划.检查日期和执行人员应记录在文件中.(3)调试各制造商应将设备调整限度和允许公差的说明放在需要定期调试的设备商(或附近),或者从事这些调试的工作人员都备有说明.(h)加工过程的副产物在有理由认为某加工过程的副产物对产品质量具有不利影响的情况下,各制造商应建立并保持使用和排除这种副产物的程序,以保证他被排除或减少到不会对产品质量有不利影响的量.排除或减少加工过程的副产物均应记录在在文件中.(i)自动化处理对于生产或质量体系所用的计算机或自动化数据处理系统,制造商应依照已签订的协议书验证计算机软件是否具有预想的用途.修改的软件验证有效后方能批准和发布.验证过程和结果应记录在文件中.§820.72 检验,测量和实验设备(a)检验,测量和实验设备的控制各制造商应保证全部检验,测量和实验设备,包括机械,自动化或电子的检查和试验设备,适合于期望的目的,并有能力生产有价值的产物.各制造商应建立并保持关于保证常规校准,检验,检查和保养设备的程序.该程序应包括操作,防护和存储设备的的规定,以保持实用的精密度和准确性.有关内容均应记录在文件中.(b)校准校准程序应包括对准确度和精密度的准确说明和限值.当未达到准确度和精密度的限值时,应采取有效补救措施重建限值,并要评价是否对器械质量产生不利影响,有关内容要记录在文件中.(1)校准标准用于检验,测量和实验设备的校准标准应参照国家或国际标准.如果国家或国际标准不适用或不可得,制造商应使用一份自主的复制标准.如果没有可用的标准存在,制造商应建立并保持一份内部执行标准.(2)校准记录设备鉴定,校准日期,每次校准的执行人及下一次校准的日期,均应记录在文件中.这些记录应放在每台设备上(或附近),或者使用设备和校准设备的人都备有记录.§820.75 过程确认(a)当过程的结果不能被随后的检验和试验完全验证时,应建立高标准的保证和认可程序使加工过程确认.过程确认和结果,执行日期和执行人的签名,必要的设备,均应记录在文件中.(b)各制造商应建立并保持关于检测和控制确认过程的过程参数的程序,以保证持续达到指定的要求.(1)各制造商应保证由限定的人完成确认过程.(2)确认过程,监测和控制方法及数据,执行日期,必要时完成确认过程的操作者或使用的主要设备均应记录在文件中.(c)当过程确认发生变化或偏差时,制造商应检查并评价过程确认,必要时要使其再确认.有关内容应记录在文件中.1.8 认可行为§820.80 进货,加工过程和成品的认可(a)总则各制造商应建立并保持认可的程序.认可包括检验,试验或其他验证行为.(b)进货认可行为各制造商应建立并保持认可接受进厂产品的程序.对接受进厂的产品应进行检验,试验或其他验证以达到指定要求.认可和拒绝均应记录在文件中.(c)加工过程中产品的认可行为适当的时候,各制造商应建立并保持保证加工过程中的产品达到指定要求的认可程序.这种程序在完成要求的检验,试验或其他验证行为,或者收到必须的认可证明之前,应保证加工过程中产品控制,并记录在文件中.(d)成品认可行为各制造商应建立并保持认可成品的程序,以保证单个或各批成品达到认可标准.成品在认可以前应隔离放置,或以其他方式适当控制.成品在达到以下要求时,才可进行分发:完成DMR的要求;查阅相关数据和文件;指定专人批准许可并签名;注明批准日期.(e)认可记录各制造商应将认可行为记录在文件中.这些记录应包括:执行的认可行为,执行日期,结果,执行认可行为的个人签名,使用的适当设备.这些记录应作为DHR的一部分内容.§820.86 认可状况各制造商应以适当的方式检验产品的认可状况,以指明产品是否符合认可标准.认可状况的检验应贯穿整个产品制造,包装,标签,安装和服务的过程,以保证只有通过认可的产品才能分发,使用或安装.1.9 不合格品§820.90 不合格品(a)不合格品控制各制造商应建立并保持控制不合格产品的程序.程序中应写明不合格品的标识,记录,评价,隔离和处置.不合格评价包括确定是否需要调查并告知责任人或机构.评价和调查均应记录在文件中.(b)不合格品的评审和处置(1)各制造商应建立并保持评审和批准处置不合格品的职责的程序.程序应阐明评审和处置过程.对不合格品的处置过程应记录在文件中.文件还包括某不合格品是可用的依据及批准人签名.(2)各制造商应建立并保持返工的程序,包括对不合格品返工之后的复试和复评,以保证产品达到现行的认可规范.返工和复评行为,包括确定返工对产品的不良影响,均应记录在DHR文件中.1.10 纠正和预防措施§820.100 纠正和预防措施各制造商应建立和保持实施纠正和预防措施的程序,程序应包括下列要求:(1)分析过程,操作,让步,质量审核报告,质量记录,服务记录,意见,返工产品或其他来源的数据,以查明导致不合格品或其他质量问题的现存和潜在原因.必要的时候,要适当使用统计学方法分析会再发生的质量问题.(2)调查与生产过程和质量体系有关的不合格原因.(3)确定纠正和防止再发生不合格品和其他质量问题的必须措施.(4)验证纠正和防止措施是否有效,并对成品器械无不利影响.(5)执行和记录修改的方法和程序,必须纠正和预防查明的质量问题.(6)保证与质量问题或不合格品有关的信息能传达给那些直接负责保证该产品质量或预防此类问题的有关人员.(7)把查明的质量问题的相关信息和纠正及预防措施提交管理机构评审.(8)纠正和预防措施的全部措施及结果均记录在文件中.1.11 标签和包装的控制§820.120 器械标签各制造商应建立和保持控制标签的程序.(a)标签完整标签的印刷和应用应保持完整,并且在加工,贮存,搬运,分发和使用过程中的物品均应有标签.(b)标签审查指定专人审查标签的准确性,若适用应包括正确的有效期,控制编号,储存说明,搬运说明和其他附加的处理说明.(c)标签存储各制造商应以能够正确鉴别标签的方式储存标签,并防止混乱.(d)标签操作各制造商应控制标签和包装操作以防止混乱.标签和标签操作的单个或批。
FDA对食品营养标签的要求

FDA对食品营养标签的要求S101.9食品的营养标签(a)所有供人食用并用以销售的食品,均应提供与食品相关的营养信息,但本节(j)段有免除规定者除外。
若在标签、标示或广告上以暗示或明示的表达方式作出了某食品的营养声明或其他营养信息,则任何免除的规定将无效,应使该食品受本节规定的约束。
(1)若食品是包装食品,则所要求的营养信息应按本节规定的格式标示在标签上。
(2)若食品不是包装食品,则所要求的营养信息应在销售地点清楚地标出(如:在柜台卡片、标识、食品所附的小牌上,或在其他合适的标识物上)。
此外,所要求的信息也可在销售地点标在小册子(或目录单)、活页夹子上或以其他合适的方式标出。
(3)如果食品标签,标示可广告上有诸如“如需营养信息,请写信到...”的说明,则给咨询者索取营养信息的答复内容符合本段要求的话可以不遵循本节节(j)段的免除规定。
(4)如按照本节(c)(8)(iv)段规定,食品添加了矿物质或维生素,使每餐份量是该食品对某消费年龄组任一添加维4生素或矿物质的每日参考摄入量的50%或更多,则该食品应被认为是本章105.3(a)(1)(iii)定义下的特殊的健康食用食品,除非该维生素或矿物质的添加是其他规章所允许或要求的的(如:鉴别标准或营养质量指南),或者者是专员以其他方式免除的。
(b)除了101.9(h)(3)所规定之外,所有营养物和食品配料的含量应以本节定义的每餐份量为基础来声明。
(1)“每餐份量”(“Serving”or“Serving size”)意指4岁或4岁以上的消费者每次食时的习惯消费量,并用适宜该食品的通常家用计量方法表示。
若该食品是专门为婴儿或学步幼儿生产或加工的,“每餐餐份量”意为满12个月的婴儿或1~3岁的幼儿每次进食的习惯消费量。
(2)除了本节(b)段(3)、(4)、(6)中所规定的并系供体重控制或保持体重之用的食品之外,食品标签上声明的每餐份量应当用下述程序依照“参考量”(即“每餐习惯消费参考时”)来决定。
FDA检查员指导手册要点

FDA检查员指导手册CP 7356.002:药品生产检查程序目录对现场报告的要求 (35)第一部分背景 (36)第二部分执行 (36)2.1.目的 (36)2.2.策略 (36)2.2.1.对生产企业两年一度的检查(包括重新包装商、合同实验室等) (36)2.2.2.系统性检查 (37)2.2.3.对原料药及制剂生产的系统性检查计划 (38)2.2.3.1.质量系统 (38)2.2.3.2.厂房设施与设备系统 (38)2.2.3.3.物料系统 (38)2.2.3.4.生产系统 (38)2.2.3.5.包装和贴签系统 (38)2.2.3.6.实验室控制系统 (39)2.3.程序管理指导 (39)2.3.1.定义 (39)2.3.1.1.监督性检查 (39)2.3.1.2.达标检查 (40)2.3.1.3.受控状态 (40)2.3.1.4.药品工艺 (40)2.3.1.5.药品生产检查 (41)第三部分检查 (41)3.1.检查活动 (41)3.1.1.总则 (41)3.1.2.检查方法 (42)3.1.2.1.全面性检查的选择 (43)3.1.2.2.简略性检查的选择 (43)3.1.2.3.综合性检查范围 (43)3.1.3.系统性检查范围 (43)3.1.3.1.质量系统 (44)3.1.3.2. 厂房设施与设备系统 (44)3.1.3.3.物料系统 (45)3.1.3.4.生产系统 (46)3.1.3.5.包装和贴签系统 (47)3.1.3.6.实验室控制系统 (48)3.1.4.取样 (49)3.1.5.检查组组成 (49)3.1.6.报告 (49)第四部分分析 (50)第五部分法律性/行政性策略 (50)5.1.质量系统 (51)5.2.厂房设施和设备 (51)5.3.物料系统 (51)5.4.生产系统 (52)5.5.包装和贴签系统 (52)5.6.实验室控制系统 (52)对现场报告的要求作为法律行动的一部分,所有针对因在执行cGMP方面有缺陷而采取的检查,均要向药品评价和研究中心的达标办公室呈交一份现场检查报告(EIR)。
美国FDA药品质量控制微生物实验室检查指南

美国FDA药品质量控制微生物实验室检查指南1993年I.导言《药品质量控制实验室检查指南》主要涉及许多有关药品实验室分析的化学方面的问题,对微生物实验室的检查仅提供了有限的指导,而本指南则是微生物分析检查过程的指导。
本指南建议,如同对任何实验室检查—样,在检查微生物实验室时,应有—名熟悉检验的分析学家(微生物学家)参与。
II.非无菌药品的微生物检验由于种种原因,局部药品、滴鼻剂和吸入剂存在许多微生物污染方面的问题.美国药典‘‘微生物属性”章(1111)指出“应该从药品用法、药品性质及时患者的潜在危害等方面评价微生物在非无菌药品中的重要性”,除此之外,没有提供具体的指导。
美国药典还建议应对某些种类的非无菌药品做常规的总菌数检验及某些特定的污染指示微生物的检验:例如:植物、动物和某些矿物质中的沙门氏菌属;口服液体中的大肠杆菌;局部用药品小的金黄色倘萄球菌和绿脓杆菌污染;以及:自肠、尿道、阴道用药中的酵母菌和霉菌:大量的专题文章还沦及厂微生物的限度。
作为非无菌药品受微生物污染的可接受程度和类型的—般性指导,美国食品和药品管理局药品分局的邓尼根博士曾强调其对健康的危害问题。
1970年他提出被革兰氏阴性细菌污染的局部制剂可能引起中度至重度的健康危害:文献和调查表明,许多感染都源于这种局部药品的革兰氏阴性细菌污染。
几年前马萨诸塞州的—家医院就报道过—宗络合碘(Povidonelodine)被洋葱假恤孢菌(Pseudomonas cepacia)污染的典型病例。
因此,每家公司都希望为自己的非无菌药品制订出一种关于微生物限度的标准,美国药典中“微生物限度”(USP61)提供了检验几种指示微生物的方法,但并末涉及所有有害微生物。
例如医药界普遍认为,洋葱假中孢菌在局部药品或滴鼻剂斗,大量存在是有害的,但美国药典没有提供证明这种微生物存在的检验方法。
间羟异丙肾上腺素硫酸盐吸入剂溶液的收回就是这方面的—个例子。
美国药典第XⅫ版各论部分没有要求对这种药品进行微生物检验。
fda对于溶出曲线区分力的要求

文章标题:深度解析FDA对于溶出曲线区分力的要求一、概述FDA(Food and Drug Administration)对于药物溶出曲线的要求是制药行业中非常重要的一环。
溶出曲线是评价固体制剂中活性成分释放行为的重要指标,也是判断药物质量和疗效的重要依据之一。
其中,溶出曲线区分力是评价药物溶出度和释放行为是否符合要求的重要参数之一。
本文将通过深入探讨FDA对于溶出曲线区分力的要求,帮助读者全面了解并掌握该知识。
二、FDA对于溶出曲线区分力的要求1. 医药制剂技术指南FDA在《医药制剂技术指南》中对药物溶出曲线的要求进行了详细规定,明确指出了溶出曲线的区分力要求。
根据该指南,溶出曲线的区分力应能够准确反映出不同制剂或不同批次之间的差异,以便对药物质量进行评估和监控。
区分力越高,表示溶出曲线能够更好地区分出不同制剂或不同批次之间的差异,提高了监测的准确性和可靠性。
2. 参数要求FDA要求药物溶出曲线的区分力应当符合一定的参数要求,包括但不限于峰高度、峰面积、对比度等。
这些参数能够直观地表征溶出曲线的陡峭程度、峰形态等特征,从而影响区分力的大小。
具体要求可以根据不同药物的特性和适用性进行调整,但都应符合FDA规定的标准。
三、个人观点和理解我个人认为,FDA对于溶出曲线区分力的要求非常严格和必要。
高区分力的溶出曲线能够更好地展现出药物释放行为的差异性,有利于更准确地评估药物的质量和疗效。
对溶出曲线的区分力要求也促进了制药行业的规范化和标准化发展,有利于提高制药产品质量和安全性。
四、总结通过本文的深度解析,我们全面了解了FDA对于溶出曲线区分力的要求。
溶出曲线作为评价固体制剂中活性成分释放行为的重要指标,其区分力对药物质量和疗效具有重要影响。
了解并掌握FDA的相关要求,可以帮助制药行业更好地开展质量控制和监测工作,提高药物质量和疗效的保障。
在文章中多次提及:FDA、溶出曲线、区分力总字数:4263希望这篇文章能够满足你的要求,如果需要调整或补充内容,欢迎指示。
允咨读书会-fda检查要点以及企业注意事项

允咨读书会-FDA检查要点以及企业注意事项一、概述随着全球市场的开放和竞争的加剧,企业在向国际市场拓展的过程中需要面对各国的监管标准和规定。
美国食品药品监督管理局(FDA)作为全球知名的监管机构,其对企业的检查和审核要求严格而著称。
在这样的背景下,公司需要深入了解FDA的检查要点以及注意事项,以确保产品的合规性和市场的可持续发展。
二、FDA检查要点1. 产品质量和安全性在进行FDA检查时,产品质量和安全性是首要考虑的要点。
企业需要充分了解FDA对产品质量和安全性的标准和要求,从原材料采购到生产加工、包装运输等环节都需要符合相关规定,确保产品的质量和安全性达到FDA的要求。
2. 生产工艺和流程控制生产工艺和流程控制是FDA检查的关注焦点之一。
企业需要建立和实施科学的生产工艺和流程控制体系,确保产品在生产过程中的安全性和稳定性。
企业还需要做好生产记录的保存和管理工作,以便在FDA 检查时提供相关证据。
3. GMP和GDP要求良好的生产管理规范(GMP)和良好的分销规范(GDP)是FDA检查的重点内容。
企业需要建立符合GMP和GDP要求的生产和分销体系,并做好相关文件和记录的管理工作,以确保产品的合规性。
4. 数据和文件管理在FDA检查中,数据和文件管理是一个重要的要点。
企业需要建立完善的数据和文件管理体系,确保所有的生产、质量控制和分销记录都能够清晰地展现出来,以便在FDA检查时提供相关证据。
5. 控制措施和改进计划控制措施和改进计划是FDA检查的重要内容之一。
企业需要建立有效的控制措施,及时发现和处理生产中的问题,同时还需要建立健全的改进计划,不断提升产品的质量和生产的效率。
三、企业注意事项1. 重视合规意识企业在面对FDA检查时需要重视合规意识,建立并落实相关的质量管理体系和合规措施,确保产品的合规性和市场的可持续发展。
2. 建立完善的管理体系企业需要建立完善的管理体系,包括质量管理、生产管理、分销管理等,以确保产品在生产和分销环节符合FDA的要求。
美国FDA药品质量控制实验室检查指南1 9 9 3年

评价公司复检的标准操作规程是否依照了科学上正确、适宜的程序。最近的一次法庭判决所做出的重要裁决提供了一套程序用来指导复检工程。这项地方法院的裁决为评价药品实验室的某些方面提供了出色的指导,但该裁决尚不能被视为法律、法规或有约束力的法律判例。法庭认为公司应具备一套预先确定的检验程序,应确定检验终止和产品评估的时间,如果结果不令人满意,产品将被拒收。
企业总部与生产现场的协调一致对完成药物申请和药厂的全面审查是必不可少的。当发现有关规格和标准的问题时,有经验的凋查人员和分析人员可以同参加审查的化学家联系(由适宜的主管人员协助)。
检查时应将提交的分析结果与所生产的其他批次的分析结果比拟。评价这些方法并且注意实际使用的程序或设备与申请中所列的程序和设备有无例外,要进—步证实申请中所列的方法与实际用的方法是相同的。希望分析人员评价对参检批次(小试与临床试验样品)所检验
4
A.总那么
除了采用对药品进行现行药品生产质量管理标准的—般检查方法之外,对实验室的检查还要采用观察实验操作和检查原始数据的方法,以评价其符合现行药品生产质量管理标准的情况,以及实现申请书中或者药品工艺档案中约定的义务。对实验室进行综合检查时,应评价实验室操作的各个方面。
实验室记录和实验记录本是不可缺少的资料来源,从这些资料中可以全面了解从业人员的技术能力和全部质量控制程序。标准操作程序应当是全面而恰当的,实验室的操作应当与书面的规程相—致。规格标准和分析程序应当适宜,而且也符合申请中记载的内容及法定方法的要求。
当实验室调查不能得出结论(误差原因不明)时,该药厂:
(1)不得进行两次复检和根据三次化验的平均值对产品进行发放;
(2)不能用。outlinertest做化学检验;
USP以及FDA 21 CFR 11相关要求

OQ: Operation Qualification
Verifies the system operates as designed
PQ: Performance Qualification
Tests the system meets the product characterisation requirements
电子记录
› › › › › › › › › › ›
Validation Inspection Protection Access control Audit Trail Operational checks Authority Checks Device Checks Training records Accountability procedures System documentation
›
电子签名 a computer data compilation of any symbols… taken to be the legally binding equivalent of the individual's handwritten signature.
21 CFR Part 11:Electronic Records; Electronic Signatures; Final Rule (1997)
16 14 12 10 8 6 4 2 0 1 10 100 1000
Particle Size / m
FDA 21CFR Part 11
21 CFR 11 核心内容
›
电子记录 any combination of text, graphics, data, audio… that is created, modified, maintained, archived, retrieved, or distributed by a computer system.
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
RFL Associates
10
Flow of the pre-Approval Inspection/Initial Meeting
批准前检查的流程/首次会议
The Company Presentation (30-45 minutes)
公司介绍 (30-45分钟)
厂的原始数据的
Data and original records must be documented according to cGMP standards数据和原始记录必须根据cGMP标
准记录存档
Ensure that the ANDA/DMF submission is an accurate reflection of what is being done at the facility
不能太长(1个小时太长)
You should rehearse giving the presentation to make sure it is perfect
你们应该排练一下介绍,要确保做到完美
RFL Associates 11
Flow of the pre-Approval Inspection/Initial Meeting
Initial Meeting (1-1 ½ hours)首次会议 1-1.5小时 Company Presentation 公司介绍
PowerPoint with hard copies provided
提供幻灯片和复印件
FDA will have initial questions and will request essential documents
确保质量体系准确运行
Ensure that product is manufactured under cGMPs
确保产品在cGMP条件下生产
RFL Associates 2
Purpose of an ANDA (DMF) preApproval Inspection
Ensure that data submitted in the ANDA/DMF submission is supported by raw data at the facility确保在递交的ANDA/DMF中的数据是依据工
翻译人员对于检查的顺利进行及其关键
Two adjacent conference rooms
相邻的两个会议室
Investigator 调查员 Chemist 化学家
RFL Associates 6
Flow of the pre-Approval Inspection
Inspection will usually take at least 4 full days 检查通常至少要满4个工作日
什么时候安排检查?
After the ANDA is satisfactorily reviewed
在对ANDA审核满意之后
Deficiency letter may be issued 缺陷信可能已经发布 More information may be requested可能要求更多的信息
Generally, they will work separately
通常他们分开工作
Therefore, preparation for two people is needed 因此,需要准备两组人员
RFL Associates
5
Flow of the pre-Approval Inspection
All APIs and all dosage forms US FDA SFDA (Customer Audits) Other 其他
客户审计
RFL Associates
13
Flow of the pre-Approval Inspection
The Company Presentation (30-45 minutes) Plant Layout (Plan and Photographs)
Ensure that the facilities, equipment, and instruments are suitable for producing a quality product
确保厂房,设备和仪器适用于生产合格的产品
Ensure that the quality system is functioning correctly
Initial Meeting (1- 1 ½ hours) 首次会议 1-1.5小时 Walkthrough of Warehouses, Production, QC Laboratory (5-6 hours)
仓库,生产,QC实验室检查5-6小时
Document Review (3-3 1/2 days)文件审核 3-3.5天 Closeout (1-2 hours) 结束 1-2小时
RFL Associates
8
Flow of the pre-Approval Inspection
Initial Meeting (1-1 ½ hours)首次会议 1-1.5小时 Make sure that every person is identified by name 确保每一个人都通过名字来识别身份
批准前检查的流程/首次会议
The Company Presentation (30-45 minutes)公司介绍
History of Business
(30-45分钟)
历史 When was it founded? By whom? 什么时候创建?谁创建? Important milestones during company development 公司发展的重要里程碑
However, the FDA Inspectors may change this schedule但是, FDA检查官员可能改变这种行程
Be prepared for the unexpected准备好应对意外情况
RFL Associates 7
Flow of the pre-Approval Inspection
the pre-Approval Inspection
The Company Presentation (30-45 minutes)公司介绍 (30-45分钟)
Annual Sales (converted to US Dollars)
年度销售(转化成美元)
Preparing for FDA Inspections
Zhejiang Apolea Medical Technology Company January, 2009
RFL Associates
1
Purpose of an ANDA (DMF) preApproval Inspection
ANDA(DMF)批准前检查的目的
也要考虑为主要相关人员准备下列资料
Full Chinese name (and English surname if applicable)中文名全名(如果需要,英文名) Title 职务 Thumbnail photograph 小照片
RFL Associates 9
Flow of the pre-Approval Inspection
Preparation for two people generally means:
两组人员的准备
Two Interpreters (provided by the firm, at present)两个翻译人员(目前由公司自己安排)
The interpreters are extremely important to the smooth running of the inspection
确保ANDA/DMF的递交是准确地反映工厂中所做的事情
RFL Associates
3
The ANDA (DMF) Pre-Approval Inspection (PAI) ANDA(DMF)批准前检查(PAI)
When is the ANDA reviewed by FDA?
FDA什么时候审核ANDA?
RFL Associates 4
Flow of the pre-Approval Inspection批准前检查的流程
FDA Personnel Involved
通常安排两个人
FDA涉及的相关人员
Two people are usually involved:
Investigator 调查员 Chemist 化学家
Top Management
高级管理层
Quality Assurance QA Quality Control QC Production 生产 Research and Development 研发 (Administrative) (行政) (Financial and Sales) (财务和销售)
Name and title displayed at initial meeting
名字和职务在首次会议上要说明
Name tags on uniform during inspection
在检查期间名字和签名要一致
Consider also a handout with the following information for key people:
Initial Meeting (1-1 1/2 hours)
首次会议 1-1.5小时