HyperChem 程序及其应用
化学信息学——第八讲hyperchem 软件学习

HyperChem程序简介 7.5版新增功能 OpenGL绘图: 基础演示模式:已全部转换为全新的 OpenGL模式,使分子演示图形具有更高 质量支持自定义色彩,1600万种颜色代替 了传统的8种标准色。 混合演示:棒型,球型或组合型等分子 演示手段可以用于该分子的任意原子及链 段的演示,及全新的适用于原子的"管型" 演示。增强的蛋白质模型构建功能,支持 多种蛋白质二级结构演示,大分子的电子 密度近似方法。
13
工具 条 •使用图标工具可以简化操作 •从左至右分别是绘制、选择、旋转、平面 转动、平移
14
工作状态
•工作 区: •状态 行:
15
2、打开已存在的数据文件 •File-Open
16
浏览文件夹
17
C60分子
18
选择分子图形的显示方式 •Display- Labels可以选择原子、化学键等 标记方式:
39
计算几何构型 •COMPUTE---GEOMETRY
40
计算结果 •优化结束
41
测量键长、键角、二面角等结构参数 •数值显示在底部状态栏
42
单点能计算
•对给定体系优化构型后,可对优化后的构 型再进行单点计算。
43
单点能结果 •单点能结果
44
输出结果
45
计算结果的输出文件
• • • • • • • • • • • • • • • HyperChemlog start -- Sun May 28 21:38:15 2000. Geometry optimization, SemiEmpirical, molecule = (untitled). CNDO PolakRibiere optimizer Convergence limit = 0.0001000 Iteration limit = 50 Accelerate convergence = YES Optimization algorithm = Polak-Ribiere Criterion of RMS gradient = 0.0100 kcal/(A mol) Maximum cycles = 150 RHF Calculation: Singlet state calculation Number of electrons = 22 Number of Double Occupied Levels = 11 Charge on the System = 0 Total Orbitals = 22
HyperChem

分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
HyperChem
分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
谷晓明 物理化学HyperChem
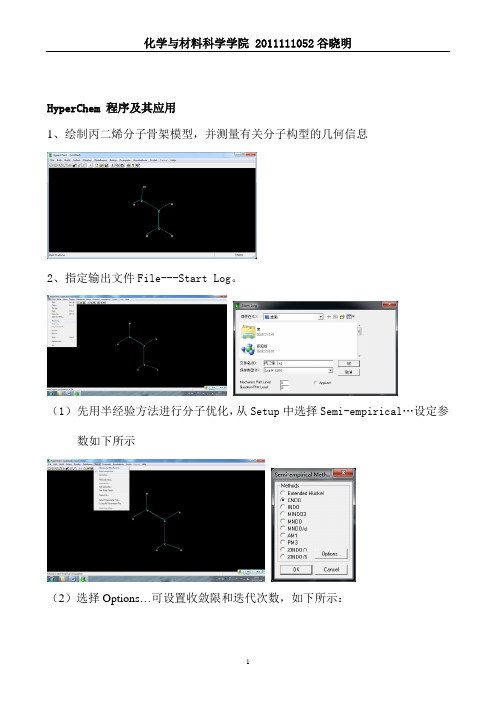
HyperChem 程序及其应用1、绘制丙二烯分子骨架模型,并测量有关分子构型的几何信息2、指定输出文件File---Start Log。
(1)先用半经验方法进行分子优化,从Setup中选择Semi-empirical…设定参数如下所示(2)选择Options…可设置收敛限和迭代次数,如下所示:(3)从Compute中选择Geometry Optimzation…进行集合构型优化:(4)优化完成之后,在Compute选择Single Point可进行单点计算。
3、采用从头算的方法:(1)Setup中选择Ab Initio…设定参数如下:(2)从Compute中选择Geometry Optimzation…进行集合构型优化:(3)完成集合构型优化后,从Compute选择Single Point可进行单点计算。
4、计算结束后,停止数据输出,从File---Stop Log。
5、分析有关分子的性质并简单分析讨论分子性质(1)采用从头算方法后,分析振动光谱:(该图显示谱线的位置、强度和振动模式)虚振动频率-185.84意味着,此结构不是一个稳定结构,而是一个过渡态。
(2)计算电子光谱最低能量跃迁π-π*在373.90,是禁阻跃迁允许的跃迁是116.84单态π-π*跃迁。
(3)分子偶极矩(4)轨道特征1、最高占据轨道2、最低空轨道(5)绘分子图,测电子光谱从Comput选择Plot Molecular Graphs1、2D图像2、3D图像6、结论与经验1、丙烯分子为一平面型分子,并且其振动频率存在虚频-185.84,意味着此平面结构不是一个稳定结构,而是一个过渡态。
2、半经验算法计算分子总能量为-16180.6852898 (kcal/mol),从头算方法计算分子总能量为-72576.4084722 (kcal/mol),所以计算方法的选择很重要。
3、计算分子的电子光谱能够得到该分子最低能量跃迁π-π*在373.90,是禁阻跃迁;允许的跃迁是116.84单态π-π*跃迁。
HyperChem程序及其应用
河北师范大学计算量子化学研究所蔡新华教授量子化学在线教学.100/qc/lzhx-0.htmHyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作区:状态行:2、打开已存在的数据文件File-Open选择分子图形的显示方式Display- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
HyperChem应用-水分子轨道计算

HyperChem应用水分子轨道计算构造水分子1. 在Display菜单,确保Show Hydrogens打开,Rendering对话框Sticks栏中的Perspe ctive关闭。
2. 关闭Default Element对话框中的Explicit Hydrogens,设置缺省元素为O。
3. 在工作区画一个氧原子。
4. 双击Selection激活Model Builder。
它自动为氧原子添加氢原子。
5. 设置Label添加元素符号。
结果是这样:∣H∣∣∣O╲╲╲H键角是109度。
结构校准1. 在Edit菜单选择Align Molecules。
2. 在Align对话框选择Secondary,With对话框选择Y Axis。
3. 关闭Minor Axis。
4. 选择OK。
得到这样的水分子:H H╲╱╲╱╲╱╲╱O5. 把这个结构保存为h2o.hin。
显示原子电荷1. 打开Labels对话框。
2. 把Charge作为Atom的选项,单击OK。
计算波函1. 在Setup菜单选择Semi-empirical。
2. 选择CNDO方法。
选择Options。
当然也可以选择其他方法。
3. 在Semi-empirical的Options的对话框中使用下面的值:Total charge: 0, Spin multiplicity: 1, Spin Pairing: RHF,State: Lowest, Convergence limit: 0.0001, Iteration limit: 50,Accelerate convergence: NO。
这意味着两次叠代计算的值的差小于0.0001 kcal/mol,叠代次数已经达到了最大的次数5 0次。
4. 选择OK回到工作区。
5. 选择Compute菜单中的Single Point。
结果:能量,梯度,和原子电荷如图所示:0.145 0.145╲╱╲╱╲╱╲╱-0.290窗口左下角:Energy=-320.414117, Gradient=124.385845, Symmetry=C2V(结果可能会有略微差别。
HyperChem程序及其应用
河北师范大学计算量子化学研究所蔡新华教授量子化学在线教学.100/qc/lzhx-0.htmHyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作区:状态行:2、打开已存在的数据文件File-Open选择分子图形的显示方式Display- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
HyperChem基本操作 单点计算

HyperChem基本操作单点计算单点计算,仅仅执行势能曲面上的一个单个点的计算。
例如对一个双原子分子来说,这可能是在点a原子间距离R=2.0埃的计算。
单点计算的结果给出系统在当前几何构型的势能,以及那个点的梯度。
在点b,c,d,或e的单点计算可能将给出更高的能量。
如果在关键的部分取足够多的点,利用Origin或者Matlab等数学工具软件,就能描出势能曲线,从而精确算出离解能De和核平衡距离re,利用公式就能得到震动参数ωe。
对多原子系统来说,状况更加复杂,但是本质是一样的。
Orbital中显示的轨道信息Alpha & Beta显示选择的轨道是alpha自旋还是beta自旋。
LUMO+显示选择轨道相对于LUMO的位置关系。
例如,选择能量在LUMO之上的轨道,文本框依次显示+1,+2,+3......;如果选择能量低于LUMO的轨道,文本框显示-1,-2,-3......。
HOMO-显示选择轨道相对于HOMO的位置关系。
例如,选择能量在HOMO之上的轨道,文本框依次显示-1,-2,-3......;如果选择能量低于HOMO的轨道,文本框显示+1,+2,+3......。
Number显示从能量最低轨道开始的选择轨道绝对数值。
对于UHF计算,轨道的alpha和beta列编号分开显示,对HOMO-和LUMO+选项也是这样。
Energy以eV为单位显示选择轨道的能量。
Symmetry显示选择轨道的不可约表示。
Labels在轨道显示窗口中,显示每个轨道的电子占据情况和每个轨道的能量。
按照约定俗成,向上的箭头代表alpha自旋,向下的箭头代表beta自旋。
对于RHF计算,最大轨道占据是2;对于UHF计算,最大轨道占据是1。
Zoom Out和轨道的放大如果轨道间距太密,在使用Labels时,文字符号会重合在一起,看起来很不方便。
通过鼠标左键圈出一个或相邻几个选择的轨道,这些轨道就会被放大。
放大之后,Zoom Out选项可以在窗口中重新回到显示全部轨道范围。
HyperChem软件的应用2011

5.模拟分子光谱 分子光谱(振动光谱、电子光谱)是结构 化学中非常重要的内容之一。 研究物质的分子光谱,可以使我们了解分 子中电子的运动,分子中各原子核的相对振动 以及整个分子的转动情况。分子光谱也是现代 研究分子结构和进行定性、定量分析的重要方 法之一。 HyperChem能够对分子进行振动分析,对振 动频率、振动模式、红外吸收强度、电子跃迁 等进行计算,
6. 数据库(Database) 数据库( ) 氨基酸、核酸、糖类。 氨基酸、核酸、糖类。
6. 其它模块:晶体构造器;糖类构造器,构 其它模块:晶体构造器;糖类构造器, 像搜寻, 特性, 像搜寻,QSAR特性,脚本编辑器。 特性 脚本编辑器。
ห้องสมุดไป่ตู้
2.建立与显示分子骨架模型: 结构输入和对分子操作。 除了能绘制分子的平面结构,还能够获得 分子的空间构型、键长、键角、扭转角、氢键 等参数。建立起3D或2D分子结构模型。 几何优化
3. 化学计算: 化学计算: 量子化学计算(从头计算,半经验方法, 量子化学计算(从头计算,半经验方法,密度 泛函计算):单点计算、 ):单点计算 泛函计算):单点计算、几何优化和过渡态寻 找计算; 找计算; 分子力学计算; 分子力学计算; 分子动力学模拟计算( 分子动力学模拟计算(Langevin,Metropolis , Monte Carlo模拟)。 模拟)。 模拟
4. 可以用来研究的分子特性: 可以用来研究的分子特性: 同位素的相对稳定性;生成热;活化能; 同位素的相对稳定性;生成热;活化能;原 子电荷; 能量间隔; 子电荷;HOMO-LUMO能量间隔;电离势; 能量间隔 电离势; 电子亲和力;偶极矩;电子能级; 电子亲和力;偶极矩;电子能级;过渡态结 构和能量;非键相互作用能; 构和能量;非键相互作用能;
HyperChem应用 乙烯最低电子激发态的从头计算

HyperChem应用乙烯最低电子激发态的从头计算优化乙烯的基态通过前面几个例子的学习,你已经对从头计算有了初步的了解,所以下面的一些指令将省略。
用STO-3G基组构造乙烯:1. 在File菜单中选择New,刷新工作区。
2. 确保Explicit Hydrogens没有选择。
3. 从Default Element中选择碳,并画一条C-C单键。
单击碳键中部使它变成双键。
4. 从Build菜单选择Add H和Model Build构造乙烯。
5. 从Setup菜单选择Ab Initio,并选基组为Minimal (STO-3G)。
同时令Total charge = 0,Spin multiplicity = 1,Spin pairing = RHF,Accelerate convergence =Yes,SCF Convergence limit = 0.0001. 按下CI按钮选择CI Method 为None。
优化乙烯基态:1. 选择Compute菜单的Geometry Optimization。
选择Polak-Ribiere方法,RMS gr adient 为0.01。
选择OK关闭对话框。
得到的结果为:C-C bond length:1.31埃,C-H bond length:1.08埃,H-C-H angle:115.7度。
计算相关能:1. 在Setup菜单选择Ab Initio,按下Options按钮并选择MP2 correlation energy。
单击OK回到工作区。
2. 在Compute菜单选择Single Point。
得到结果:SCF能量:-48364.64 kcal/mol ,MP2总能量:-48438.61 kcal/mol,包括-74.97kc al/mol相关能。
具体运算结果可能会与这个值有微小的差别。
乙烯基态轨道观察乙烯的轨道和轨道能量图:1. 从Compute菜单选择Orbitals。
HyperChem基本操作画原子[技巧]
![HyperChem基本操作画原子[技巧]](https://img.taocdn.com/s3/m/022c4b49302b3169a45177232f60ddccdb38e640.png)
HyperChem基本操作画原子1. 打开Element Table对话框。
这里有两种方法:在Build菜单中选择Default Element,或者双击Drawing工具。
Default Element对话框允许从周期表中选择缺省元素。
2. 如果单击Properties...按钮,将显示当前选择元素的物理属性。
也可以按下Shift键同时单击元素按钮,结果是一样的。
单击OK键,物理属性框消失。
3. 如果Allow Ions或者Explicit Hydrogens打开(用对勾选择),左键单击这些选项使其关闭。
4. 在缺省元素列表中选择Carbon,接着关闭元素对话框。
缺省元素将设置为碳。
当然也可以把打开的Default Element对话框移走,这样可以看到HyperChem工作区。
当画原子非常多的分子时,这是非常有效的。
5. 左键单击Drawing工具,把指针移到工作区。
6. 左键单击工作区左下角,将出现一个小圈,代表未成键的碳原子。
7. 在工作区不同位置画更多的原子。
画价键1. 把指针移到刚才画的第一个碳上。
2. 按下鼠标左键。
这是价键在第一个原子的位置。
3. 保持鼠标按钮按下的同时拖向工作区的顶端。
4. 放开鼠标按钮。
这是价键在第二个原子的位置。
一条线代表两个碳原子之间的价键。
5. 用仍旧停留在价键末端的指针, 用左键拖向工作区右下角。
6. 放开鼠标按钮。
这是第三个原子的位置。
7. 在空白工作区画六个价键,形成一个环。
现在你清楚了如何画原子和分子,并且学会了一些基本技巧。
选择原子在这个练习中,通过选择原子,你可以学到基本的选择技巧。
首先必须设置选择的级别[原子(atoms),基(residues),或分子(molecules)]。
这里设置为原子(atoms)。
1. 左键单击Select菜单。
2. 左键单击选择Atoms。
接下来,关闭Multiple Selections:1. 左键单击Select菜单。
分子模拟软件Hyperchem在固体物理教学中的应用

( )打开 stp 设置) 2 eu ( 菜单 , 中 Mo c a cai ( 选 l u r hnc 分子力学 ) , 图 2 示 el Me s 项 如 所
一
9 — 0
模 型 . 图 5所 示 如
图 1 C。 子 中碳 原 子 联 接 图 分
图2
图3
图 4 C 0 三 维 分 子 结构 图 6的
图 5 纳 米 碳 管 的 三 维 结 构
2 2 利 用 晶 体 构 造 器 功 能 显 示 晶 体 的 三维 结构 .
固体物理学 的学 习一般 是从 晶体结构开始 的 , 晶体 三维结构的掌握是后续学 习的重要 基础. 埘 这部分 内容涉及 到很多复杂 的空 间结 构 , 教师难 以用 语言描述清楚 , 为克服这 吲难 我们 可 以利用 H prhm 的 晶体 构造器 获得 各种 晶体 的三维结构 , yece 并
级 、 2电子相关能 、 I MP c 激发态能量 、 过渡态结构 和能 量、 非键相互作用能、 V吸收谱 、R吸收谱 、 u I 同位素对振动 的影 响、 团簇的
稳定性 、S E R谱 、 MR模拟、 N 磁场中分子计算 、 激发态几何优化 、 2相关结构优化等. MP
13 构 造 分 子模 型 .
第2 5卷 第 4期 (自然科 学版 )
Jun l f ins ntueo d ct n( a rl c n e) ora o aguIs t f uai N t a S i cs J it E o u e
Vo . 5 No 4 12 .
HyperChem实例应用

水中质子的从头计算用explicit hydrogens画H3O+:1. 在Build菜单选择Explicit Hydrogens。
2.选择Allow Ions。
3. 在Display菜单选择Labels打开Labels对话框。
4. 选择Symbols单击OK。
5. 在Build菜单选择Default Element打开周期表,设置氧元素为缺省元素。
6. 左键单击Drawing工具。
画H3O+1. 左键单击工作区创建氧原子。
2. 从氧原子画三个价键得到H3O。
在量子力学计算中添加正电荷:1. 在Setup菜单选择Ab Initio。
2. 单击Options按钮。
3. 设置总电荷(Total charge)值为1,单击OK关闭Options对话框。
4. 单击OK按钮关闭Ab Initio对话框。
这样就得到了H3O+。
选择基组1. 在Setup菜单选择Ab Initio。
2. 为基组选择Other。
3. 按下Assign Other Basis Set按钮。
4. 从列表中选择4-31G单击OK。
5. 为基组选择Minimal (STO-3G)。
6. 选择Apply Basis Set接着单击OK,或者直接选择OK关闭Ab Initio对话框。
观察每个原子应用的基组:1. 在Display菜单选择Labels。
2. 选择Basis Set,单击OK。
最小化能量结构计算H3O+的几何优化:1. 从Setup菜单选择Ab Initio。
2. 单击Options按钮,保证Total charge = 1,Spin multiplicity = 1,Spin pairing = RHF,Conver-gence limit = 0.01,Iteration limit = 50,Accelerate conver-gence= Yes,为几何优化选择合适的选项。
Single Point only 选项在几何优化中不起作用,因而可以是任何值。
乙烷-Hyperchem程序应用

Hyperchem程序应用-乙烷1、画乙烷分子模型:build-default element加氢:模型化:2、用半经验方法CNDO进行优化3、选用从头算计算方法进行单点计算:4、显示键长:5、显示原子电荷:6、显示键角、二面角:7、分子性质:8、乙烷分子2D、3D静电势图:9、等值面图:10、乙烷分子总电荷密度图(2D、3D):11、分子轨道图-最高占据轨道2D、3D图:12、分子轨道图-最低空轨道2D、3D图:13、分子结构模型表示:14、计算输出结果:HyperChem log start -- Sun Dec 12 13:20:41 2010.Geometry optimization, SemiEmpirical, molecule = (untitled).CNDOFletcherReeves optimizerConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESOptimization algorithm = Fletcher-ReevesCriterion of RMS gradient = 0.1000 kcal/(A mol) Maximum cycles = 120 RHF Calculation:Singlet state calculationNumber of electrons = 14Number of Double Occupied Levels = 7Charge on the System = 0Total Orbitals = 14Starting CNDO calculation with 14 orbitalsE=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=1 Diff=4413.54714]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=2 Diff=2.12522]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=3 Diff=0.13330]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=4 Diff=0.00917]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=5 Diff=0.00002]E=-1659.8192 Grad=64.946 Conv=NO(0 cycles 1 points) [Iter=1 Diff=27.89455] E=-1659.8192 Grad=64.946 Conv=NO(0 cycles 1 points) [Iter=2 Diff=2.23060] E=-1659.8192 Grad=64.946 Conv=NO(0 cycles 1 points) [Iter=3 Diff=0.18582] E=-1659.8192 Grad=64.946 Conv=NO(0 cycles 1 points) [Iter=4 Diff=0.01805] E=-1659.8192 Grad=64.946 Conv=NO(0 cycles 1 points) [Iter=5 Diff=0.00010] E=-1659.8192 Grad=64.946 Conv=NO(0 cycles 1 points) [Iter=6 Diff=0.00000] E=-1634.7422 Grad=146.122 Conv=NO(0 cycles 2 points) [Iter=1 Diff=11.78436] E=-1634.7422 Grad=146.122 Conv=NO(0 cycles 2 points) [Iter=2 Diff=0.88985] E=-1634.7422 Grad=146.122 Conv=NO(0 cycles 2 points) [Iter=3 Diff=0.06927] E=-1634.7422 Grad=146.122 Conv=NO(0 cycles 2 points) [Iter=4 Diff=0.00631] E=-1634.7422 Grad=146.122 Conv=NO(0 cycles 2 points) [Iter=5 Diff=0.00005] E=-1671.0494 Grad=3.886 Conv=NO(1 cycles 3 points) [Iter=1 Diff=0.00236]E=-1671.0494 Grad=3.886 Conv=NO(1 cycles 3 points) [Iter=2 Diff=0.00018]E=-1671.0494 Grad=3.886 Conv=NO(1 cycles 3 points) [Iter=3 Diff=0.00001]E=-1671.1287 Grad=2.282 Conv=NO(1 cycles 4 points) [Iter=1 Diff=0.00235]E=-1671.1287 Grad=2.282 Conv=NO(1 cycles 4 points) [Iter=2 Diff=0.00018]E=-1671.1287 Grad=2.282 Conv=NO(1 cycles 4 points) [Iter=3 Diff=0.00001]E=-1671.1643 Grad=1.471 Conv=NO(1 cycles 5 points) [Iter=1 Diff=0.00931]E=-1671.1643 Grad=1.471 Conv=NO(1 cycles 5 points) [Iter=2 Diff=0.00070]E=-1671.1643 Grad=1.471 Conv=NO(1 cycles 5 points) [Iter=3 Diff=0.00006]E=-1671.1063 Grad=3.987 Conv=NO(1 cycles 6 points) [Iter=1 Diff=0.00655]E=-1671.1063 Grad=3.987 Conv=NO(1 cycles 6 points) [Iter=2 Diff=0.00049]E=-1671.1063 Grad=3.987 Conv=NO(1 cycles 6 points) [Iter=3 Diff=0.00004]E=-1671.1665 Grad=1.618 Conv=NO(2 cycles 7 points) [Iter=1 Diff=0.03525]E=-1671.1665 Grad=1.618 Conv=NO(2 cycles 7 points) [Iter=2 Diff=0.00283]E=-1671.1665 Grad=1.618 Conv=NO(2 cycles 7 points) [Iter=3 Diff=0.00025]E=-1671.1665 Grad=1.618 Conv=NO(2 cycles 7 points) [Iter=4 Diff=0.00003]E=-1671.1250 Grad=5.375 Conv=NO(2 cycles 8 points) [Iter=1 Diff=0.01699]E=-1671.1250 Grad=5.375 Conv=NO(2 cycles 8 points) [Iter=2 Diff=0.00136]E=-1671.1250 Grad=5.375 Conv=NO(2 cycles 8 points) [Iter=3 Diff=0.00012]E=-1671.1250 Grad=5.375 Conv=NO(2 cycles 8 points) [Iter=4 Diff=0.00001]E=-1671.1765 Grad=1.170 Conv=NO(3 cycles 9 points) [Iter=1 Diff=0.00024]E=-1671.1765 Grad=1.170 Conv=NO(3 cycles 9 points) [Iter=2 Diff=0.00003]E=-1671.1899 Grad=0.985 Conv=NO(3 cycles 10 points) [Iter=1 Diff=0.00024]E=-1671.1899 Grad=0.985 Conv=NO(3 cycles 10 points) [Iter=2 Diff=0.00003]E=-1671.2013 Grad=0.804 Conv=NO(3 cycles 11 points) [Iter=1 Diff=0.00096]E=-1671.2013 Grad=0.804 Conv=NO(3 cycles 11 points) [Iter=2 Diff=0.00013]E=-1671.2013 Grad=0.804 Conv=NO(3 cycles 11 points) [Iter=3 Diff=0.00002]E=-1671.2177 Grad=0.462 Conv=NO(3 cycles 12 points) [Iter=1 Diff=0.00381]E=-1671.2177 Grad=0.462 Conv=NO(3 cycles 12 points) [Iter=2 Diff=0.00053]E=-1671.2177 Grad=0.462 Conv=NO(3 cycles 12 points) [Iter=3 Diff=0.00008]E=-1671.2252 Grad=0.440 Conv=NO(3 cycles 13 points) [Iter=1 Diff=0.00028]E=-1671.2252 Grad=0.440 Conv=NO(3 cycles 13 points) [Iter=2 Diff=0.00004]E=-1671.2266 Grad=0.298 Conv=NO(4 cycles 14 points) [Iter=1 Diff=0.03095]E=-1671.2266 Grad=0.298 Conv=NO(4 cycles 14 points) [Iter=2 Diff=0.00240]E=-1671.2266 Grad=0.298 Conv=NO(4 cycles 14 points) [Iter=3 Diff=0.00020]E=-1671.2266 Grad=0.298 Conv=NO(4 cycles 14 points) [Iter=4 Diff=0.00002]E=-1671.1543 Grad=5.510 Conv=NO(4 cycles 15 points) [Iter=1 Diff=0.02754]E=-1671.1543 Grad=5.510 Conv=NO(4 cycles 15 points) [Iter=2 Diff=0.00214]E=-1671.1543 Grad=5.510 Conv=NO(4 cycles 15 points) [Iter=3 Diff=0.00018]E=-1671.1543 Grad=5.510 Conv=NO(4 cycles 15 points) [Iter=4 Diff=0.00002]E=-1671.2268 Grad=0.117 Conv=NO(5 cycles 16 points) [Iter=1 Diff=0.00000]E=-1671.2268 Grad=0.058 Conv=NO(5 cycles 17 points) [Iter=1 Diff=0.00000]E=-1671.2268 Grad=0.005 Conv=NO(5 cycles 18 points) [Iter=1 Diff=0.00000]E=-1671.2268 Grad=0.120 Conv=NO(5 cycles 19 points) [Iter=1 Diff=0.00000]E=-1671.2268 Grad=0.005 Conv=YES(6 cycles 20 points) [Iter=1 Diff=0.00000]ENERGIES AND GRADIENTTotal Energy = -11813.3264338 (kcal/mol)Total Energy = -18.825358113 (a.u.)Binding Energy = -1671.2268529 (kcal/mol)Isolated Atomic Energy = -10142.0995808 (kcal/mol)Electronic Energy = -28506.1734829 (kcal/mol)Core-Core Interaction = 16692.8470491 (kcal/mol)Heat of Formation = -1016.8348529 (kcal/mol)Gradient = 0.0047345 (kcal/mol/Ang)MOLECULAR POINT GROUPD3DEIGENV ALUES(eV)Symmetry: 1 A1G 1 A2U 1 EU 1 EU 2 A1G Eigenvalue: -40.735771 -29.080317 -23.689325 -23.689322 -18.800663Symmetry: 1 EG 1 EG 2 EU 2 EU 2 A2U Eigenvalue: -15.713766 -15.713758 7.124663 7.124665 7.752848Symmetry: 3 A1G 2 EG 2 EG 3 A2UEigenvalue: 8.382067 10.037918 10.037923 11.649675ATOMIC ORBITAL ELECTRON POPULATIONSAO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C1.010632 0.971430 1.035716 0.971430 1.010632AO: 2 Px C 2 Py C 2 Pz C 3 S H 4 S H0.971430 1.035716 0.971430 1.003597 1.003597AO: 5 S H 6 S H 7 S H 8 S H1.003597 1.003597 1.003597 1.003597NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Massx y z1 6 0.010792 -0.01428 -0.64301 0.00000 12.011002 6 0.010792 -0.01429 0.81444 0.00000 12.011003 1 -0.003597 1.02363 -1.06429 -0.00000 1.008004 1 -0.003597 -0.53324 -1.06429 0.89886 1.008005 1 -0.003597 -0.53324 -1.06430 -0.89885 1.008006 1 -0.003597 -1.05220 1.23572 -0.00000 1.008007 1 -0.003597 0.50467 1.23572 0.89886 1.008008 1 -0.003597 0.50467 1.23572 -0.89885 1.00800ATOMIC GRADIENTSAtom Z Gradients(kcal/mol/Angstrom)x y z1 6 0.00014 -0.01636 -0.000652 6 -0.00019 0.01630 -0.000473 1 -0.00006 -0.00074 -0.000214 1 -0.00017 -0.00037 -0.000145 1 0.00046 -0.00043 0.000996 1 0.00005 0.00076 -0.000227 1 0.00016 0.00036 -0.000178 1 -0.00039 0.00048 0.00086Dipole (Debyes) x y z TotalPoint-Chg. -0.000 0.000 -0.000 0.000sp Hybrid 0.000 0.000 0.000 0.000pd Hybrid 0.000 0.000 0.000 0.000Sum 0.000 0.000 0.000 0.000Single Point, AbInitio, molecule = (untitled).Convergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESFull MP2 correlation energy is requested.The initial guess of the MO coefficients is from eigenvectors of the core Hamiltonian. Shell Types: S, S=P.RHF Calculation:Singlet state calculationNumber of electrons = 18Number of Doubly-Occupied Levels = 9Charge on the System = 0Total Orbitals (Basis Functions) = 30Primitive Gaussians = 48Starting HyperGauss calculation with 30 basis functions and 48 primitive Gaussians.2-electron Integral buffers will be 3200 words (double precision) long.Two electron integrals will use a cutoff of 1.00000e-010Regular integral format is used.Computing the one-electron integrals ...Computing 2e integrals (s and p orbitals only): done 0%.Computing 2e integrals (s and p orbitals only): done 10%.Computing 2e integrals (s and p orbitals only): done 20%.Computing 2e integrals (s and p orbitals only): done 30%.Computing 2e integrals (s and p orbitals only): done 40%.Computing 2e integrals (s and p orbitals only): done 50%.Computing 2e integrals (s and p orbitals only): done 60%.Computing 2e integrals (s and p orbitals only): done 70%.Computing 2e integrals (s and p orbitals only): done 80%.Computing 2e integrals (s and p orbitals only): done 90%.101923 integrals have been produced.Computing the initial guess of the MO coefficients ...Iteration = 1 Difference = 166.4667285296Iteration = 2 Difference = 177.9310845840Iteration = 3 Difference = 1.0658833387Iteration = 4 Difference = 0.0788379499Iteration = 5 Difference = 0.0064340028Iteration = 6 Difference = 0.0000355075Computing MP2 energy with 9 occupied and 21 virtual orbitals ...Transfering the 2e integrals from AO to MO: done 0%.Transfering the 2e integrals from AO to MO: done 10%.Transfering the 2e integrals from AO to MO: done 20%.Transfering the 2e integrals from AO to MO: done 30%.Transfering the 2e integrals from AO to MO: done 40%.Transfering the 2e integrals from AO to MO: done 60%.Transfering the 2e integrals from AO to MO: done 80%.Energy=-49438.788874 MP2 Correlation Energy=-121.200863 Symmetry=D3DENERGIES AND GRADIENT========== SCF RESULTS ==========Total Energy = -49438.7888742 (kcal/mol)Total Energy = -78.785715293 (a.u.)Electronic Kinetic Energy = 49269.5391973 (kcal/mol)Electronic Kinetic Energy = 78.515998798 (a.u.)The Virial (-V/T) = 2.0034eK, ee and eN Energy = -76101.9249468 (kcal/mol)Nuclear Repulsion Energy = 26663.1360726 (kcal/mol)======== POST SCF RESULTS ========MP2 Correlation Energy = -121.2008626 (kcal/mol)MP2 Correlation Energy = -0.193145845 (a.u.)Total Energy (with MP2 energy) = -49559.9897368 (kcal/mol)Total Energy (with MP2 energy) = -78.9788611 (a.u.)Occupied and Virtual Orbitals in MP2 = 9, 21========== SCF RESULTS ==========MOLECULAR POINT GROUPD3DEIGENV ALUES(eV)Symmetry: 1 A2U 1 A1G 2 A1G 2 A2U 1 EU Eigenvalue: -303.679016 -303.668945 -28.084827 -22.511816 -16.265091 Symmetry: 1 EU 3 A1G 1 EG 1 EG 4 A1G Eigenvalue: -16.265085 -14.393775 -12.705372 -12.705358 7.440326 Symmetry: 2 EU 2 EU 3 A2U 2 EG 2 EG Eigenvalue: 8.671023 8.671030 9.123261 10.004039 10.004100 Symmetry: 4 A2U 5 A1G 3 EU 3 EU 3 EG Eigenvalue: 12.489616 25.331339 25.391115 25.391127 30.074318 Symmetry: 3 EG 5 A2U 6 A1G 4 EU 4 EU Eigenvalue: 30.074396 30.699768 34.373802 36.270630 36.270672 Symmetry: 4 EG 4 EG 6 A2U 7 A1G 7 A2U Eigenvalue: 36.664597 36.664612 38.297749 52.164272 62.871342ATOMIC ORBITAL ELECTRON POPULATIONSC 1 S C 1 S C 1 Px C 1 Py C 1 Pz1.988137 0.353586 0.530223 0.558487 0.530223C 1 S C 1 Px C 1 Py C 1 Pz C 2 S1.119103 0.571374 0.356604 0.571373 1.988137C 2 S C 2 Px C 2 Py C 2 Pz C 2 S0.353586 0.530223 0.558487 0.530223 1.119107C 2 Px C 2 Py C 2 Pz H 3 S H 3 S0.571374 0.356604 0.571372 0.456051 0.350912H 4 S H 4 S H 5 S H 5 S H 6 S0.456051 0.350911 0.456051 0.350913 0.456050H 6 S H 7 S H 7 S H 8 S H 8 S0.350912 0.456051 0.350911 0.456051 0.350913NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Mass(Mulliken) x y z1 6 -0.579110 -0.01428389 -0.64301264 0.00000122 12.011002 6 -0.579113 -0.01428757 0.81444120 0.00000129 12.011003 1 0.193037 1.02363050 -1.06429148 -0.00000332 1.008004 1 0.193038 -0.53323740 -1.06429136 0.89886373 1.008005 1 0.193036 -0.53324407 -1.06429577 -0.89885461 1.008006 1 0.193038 -1.05220199 1.23572016 -0.00000344 1.008007 1 0.193038 0.50466585 1.23571992 0.89886379 1.008008 1 0.193036 0.50467283 1.23572433 -0.89885455 1.00800Net Charge (Electrons):0.0000Dipole Moment (Debye):X: 0.0000 Y: -0.0000 Z: 0.0000 Ttl: 0.0000Quadrupole Moment (Debye-Ang):XX: -15.1880 YY: -15.7165 ZZ: -15.1880XY: -0.0000 XZ: 0.0000 YZ: -0.0000Octapole Moment (Debye-Ang^2):XXX: 0.6509 YYY: -4.0414 ZZZ: -0.0000XYY: 0.2245 XXY: -1.3018 XXZ: -0.0000XZZ: 0.2170 YZZ: -1.3018 YYZ: -0.0000 XYZ: 0.0000Hexadecapole Moment (Debye-Ang^3):XXXX: -29.1484 YYYY: -90.3215 ZZZZ: -29.1299XXXY: -1.4595 XXXZ: 0.0000 YYYX: 0.0578YYYZ: -0.0000 ZZZX: 0.0000 ZZZY: -0.0000XXYY: -18.7277 XXZZ: -9.7131 YYZZ: -18.7245XXYZ: -0.0000 YYXZ: 0.0000 ZZXY: 1.5339HyperChem log stop -- Sun Dec 12 13:58:11 2010.。
化学软件——HyperChem(分子模拟)介绍

化学软件——HyperChem(分子模拟)介绍HyperChem是一款以高质量,灵活易操作而闻名的分子模拟软件。
通过利用3D对量子化学计算,分子力学及动力学进行模拟动画,HyperChem 为您提供比其它 Windows 软件更多的模拟工具。
图形界面:图形界面,有半经验方法( AM1 , PM3 等), UHF , RHF 和 CI 和7.0 版新增加的密度泛函。
可进行单点能,几何优化,分子轨道分析,预测可见- 紫外光谱,蒙特卡罗和分子力学计算。
主要功能:1. 结构输入和对分子操作。
2. 显示分子。
3. 化学计算。
用量子化学或经典势能曲面方法,进行单点、几何优化和过渡态寻找计算。
可以进行的计算类型有:单点能,几何优化,计算振动频率得到简正模式,过渡态寻找,分子动力学模拟 Langevin 动力学模拟, Metropolis Monte Carlo 模拟。
支持的计算方法有:从头计算,半经验方法,分子力学,混合计算。
4. 可以用来研究的分子特性有:同位素的相对稳定性;生成热;活化能;原子电荷; HOMO-LUMO能量间隔;电离势;电子亲和力;偶极矩;电子能级; MP2 电子相关能; CI 激发态能量;过渡态结构和能量;非键相互作用能; UV-VIS 吸收谱; IR 吸收谱;同位素对振动的影响;对结构特性的碰撞影响;团簇的稳定性。
5. 支持用户定制的外部程序。
6. 其它模块: RAYTRACE 模块, RMS Fit , SEQUENCE 编辑器,晶体构造器;糖类构造器,构像搜寻,QSAR 特性,脚本编辑器。
7. 新的力场方法: Amber 2 , Amber 3 ,用于糖类的 Amber , Amber 94 ,Amber 96 。
8. ESR 谱。
9. 电极化率。
10. 二维和三维势能图。
11. 蛋白质设计。
12. 电场。
13. 梯度的图形显示。
14. 新增功能:密度泛函理论 (DFT) 计算; NMR 模拟;数据库; Charmm 蛋白质模拟;半经验方法TNDO ;磁场中分子计算;激发态几何优化; MP2 相关结构优化;新的芳香环图;交互式参数控制;增强的聚合物构造功能;新增基组。
乙烷-高斯程序应用

乙烷分子高斯程序应用1、用HyperChem程序画乙烷分子图,保存为*.PDB格式2、设置工作环境3、格式转化:4、分子构型进行优化(opt)5、进行振动分析、单点计算6、计算结果输出:优化结果输出:Final structure in terms of initial Z-matrix: CC,1,R2H,1,R3,2,A3H,1,R4,2,A4,3,D4,0H,1,R4,2,A4,3,-D4,0H,2,R6,1,A6,3,180.,0H,2,R7,1,A7,3,D7,0H,2,R7,1,A7,3,-D7,0V ariables:R2=1.52714912R3=1.0851582R4=1.08516911R6=1.08517148R7=1.08518242A3=111.18808808A4=111.18735176A6=111.18983868A7=111.18910141D4=119.99975739D7=-60.000242381|1|GINC-UNK|FOpt|RHF|6-31G(d)|C2H6|PCUSER|11-Jan-1911|0||# HF/6-31G*OPT||乙烷||0,1|C,0.0000021919,0.,-0.7635805116|C,-0.0000021919,0.,0.7635686082|H,1.0118036956,-0.0000000002,-1.1557871264|H,-0.5059007695,-0.8762601514,-1.155782425|H,-0.5059007693,0.8762601516,-1.155782425|H,-1.0118041011,0.0000000002,1.1558109401|H,0.5059009868,-0.8762605204,1.1558062284|H,0.5059009871,0.8762605202,1.1558062284||V ersion=x86-Win32-G94RevE.1|State=1-A'|HF=-79.2287541|RMSD=5.028e-010|RMSF=1.707e-004|Dipole=0.,0.,-0.0000069|PG=CS [SG(C2H2),X(H4)]||@HE WHO LAUGHS LAST PROBA BLY DIDN'T GET THE JOKE.Job cpu time: 0 days 0 hours 0 minutes 8.0 seconds.File lengths (MBytes): R WF= 5 Int= 0 D2E= 0 Chk= 2 Scr= 1Normal termination of Gaussian 94振动分析结果:1 2 3A" A' A" Frequencies -- 320.9807 887.6569 887.6878Red. masses -- 1.0078 1.0562 1.0562Frc consts -- .0612 .4903 .4904IR Inten -- .0000 2.4997 2.4996Raman Activ -- .0000 .0000 .0000Depolar -- .0000 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .00 .00 -.05 .00 .00 .00 .00 .052 6 .00 .00 .00 -.05 .00 .00 .00 .00 .053 1 .00 .00 .41 .16 .51 .00 .00 .00 -.224 1 .35 .00 -.20 .20 -.26 -.03 .03 .44 -.175 1 -.35 .00 -.20 .20 -.26 .03 -.03 -.44 -.176 1 .00 .00 .41 .16 .51 .00 .00 .00 -.227 1 -.35 .00 -.20 .20 -.26 .03 -.03 -.44 -.178 1 .35 .00 -.20 .20 -.26 -.03 .03 .44 -.174 5 6A' A' A" Frequencies -- 1062.1629 1337.0508 1337.0597Red. masses -- 3.1308 1.4544 1.4544Frc consts -- 2.0811 1.5319 1.5320IR Inten -- .0000 .0000 .0000Raman Activ -- 13.9063 3.1051 3.1046Depolar -- .3025 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .31 .00 .14 .00 .00 .00 .00 .142 6 .00 -.31 .00 -.14 .00 .00 .00 .00 -.143 1 .02 .37 .00 -.06 -.51 .00 .00 .00 -.254 1 -.01 .37 -.01 -.20 .25 .08 .08 .44 -.115 1 -.01 .37 .01 -.20 .25 -.08 -.08 -.44 -.116 1 -.02 -.37 .00 .06 .51 .00 .00 .00 .257 1 .01 -.37 -.01 .20 -.25 .08 .08 .44 .118 1 .01 -.37 .01 .20 -.25 -.08 -.08 -.44 .117 8 9A' A' A' Frequencies -- 1546.7873 1578.7780 1643.8192Red. masses -- 1.1984 1.2740 1.0212Frc consts -- 1.6893 1.8709 1.6259IR Inten -- .1468 .0000 .0000Raman Activ -- .0000 4.1660 38.8356Depolar -- .6187 .7157 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .09 .00 .00 -.11 .00 -.02 .00 .002 6 .00 .09 .00 .00 .11 .00 .02 .00 .003 1 -.17 -.37 .00 .18 .36 .00 -.12 -.28 .004 1 .08 -.37 .14 -.09 .36 -.15 .34 .14 -.265 1 .08 -.37 -.14 -.09 .36 .15 .34 .14 .266 1 -.16 -.37 .00 -.18 -.36 .00 .12 .28 .007 1 .08 -.37 -.14 .09 -.36 -.15 -.34 -.14 -.268 1 .08 -.37 .14 .09 -.36 .15 -.34 -.14 .2610 11 12A" A" A' Frequencies -- 1643.8212 1649.7178 1649.7193Red. masses -- 1.0212 1.0628 1.0628Frc consts -- 1.6259 1.7041 1.7041IR Inten -- .0000 5.7101 5.7108Raman Activ -- 38.8364 .0001 .0001Depolar -- .7500 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .00 -.02 .00 .00 -.05 .05 .00 .002 6 .00 .00 .02 .00 .00 -.05 .05 .00 .003 1 .00 .00 .49 .00 .00 .52 .12 .22 .004 1 -.26 .24 .04 -.28 .19 .04 -.36 -.11 .285 1 .26 -.24 .04 .28 -.19 .04 -.36 -.11 -.286 1 .00 .00 -.49 .00 .00 .52 .12 .22 .007 1 -.26 .24 -.04 .28 -.19 .04 -.36 -.11 -.288 1 .26 -.24 -.04 -.28 .19 .04 -.36 -.11 .2813 14 15A' A' A" Frequencies -- 3203.2175 3209.3460 3253.3299Red. masses -- 1.0340 1.0379 1.1032Frc consts -- 6.2510 6.2986 6.8798IR Inten -- 72.4201 .0057 .0008Raman Activ -- .0175 221.0308 150.1856Depolar -- .0191 .0193 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .03 .00 .00 -.04 .00 .00 .00 -.072 6 .00 .03 .00 .00 .04 .00 .00 .00 .073 1 .38 -.14 .00 -.39 .14 .00 .00 .00 -.014 1 -.19 -.14 -.33 .19 .14 .33 .24 .18 .405 1 -.19 -.14 .33 .19 .14 -.33 -.24 -.18 .406 1 .39 -.14 .00 .38 -.14 .00 .00 .00 .017 1 -.19 -.14 .34 -.19 -.14 .33 .24 .18 -.408 1 -.19 -.14 -.34 -.19 -.14 -.33 -.24 -.18 -.4016 17 18A' A" A' Frequencies -- 3253.3981 3278.4462 3278.5130Red. masses -- 1.1032 1.1037 1.1037Frc consts -- 6.8801 6.9891 6.9894IR Inten -- .0008 101.4189 101.4133Raman Activ -- 150.1842 .0011 .0011Depolar -- .7500 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 -.07 .00 .00 .00 .00 .07 .07 .00 .002 6 .07 .00 .00 .00 .00 .07 .07 .00 .003 1 .54 -.20 .00 .00 .00 .01 -.54 .21 .004 1 .12 .10 .24 -.24 -.18 -.40 -.13 -.10 -.245 1 .12 .10 -.24 .24 .18 -.40 -.13 -.10 .246 1 -.54 .20 .00 .00 .00 .01 -.54 .21 .007 1 -.12 -.10 .24 .24 .18 -.40 -.12 -.10 .248 1 -.12 -.10 -.24 -.24 -.18 -.40 -.12 -.10 -.24Zero-point vibrational energy 209475.1 (Joules/Mol)50.06574 (Kcal/Mol)WARNING-- EXPLICIT CONSIDERATION OF 1 DEGREES OF FREEDOM AS VIBRATIONS MAY CAUSE SIGNIFICANT ERRORVIBRATIONAL TEMPERA TURES: 461.82 1277.13 1277.18 1528.21 1923.71 (KELVIN) 1923.72 2225.47 2271.50 2365.08 2365.082373.56 2373.56 4608.69 4617.51 4680.794680.89 4716.93 4717.02Zero-point correction= .079785 (Hartree/Particle)Thermal correction to Energy= .083192Thermal correction to Enthalpy= .084136Thermal correction to Gibbs Free Energy= .056714Sum of electronic and zero-point Energies= -79.148969Sum of electronic and thermal Energies= -79.145563Sum of electronic and thermal Enthalpies= -79.144618 Sum of electronic and thermal Free Energies= -79.172040。
Hyperchem程序安装

1、将文件解压,在系统盘外的其他盘创建一名为hyperchem的文件夹
2、点击HyperChem6Evaluation.exe
按提示进行。当出现
选择custum-----
通过browse选择指定的安装文件夹
当出现
不要按确定而是关闭窗口;
当当出现
不要按确定而是关闭窗口;
当出现
若你的计算机原已有acrobat reader软件,则选否,否则选是。
当出现
记住不要作任何勾选,按finish
3、进行程序crack
方法:进入hyperchem安装文件夹,打开programs文件夹,
进入hyperchem软件解压包中的crack文件夹,将其中的所有文件选定并复制,并粘贴到上述programs文件夹。当出现是否复盖时选“是”。将programs文件夹中的绿色chem.exe图标发送到桌面快捷方式。OK!
4、点击桌面快捷方式绿色chem.exe图标即可运行了。
利用Hyperchem软件进行分子结构构建及性质计算

利用Hyperchem软件进行分子结构构建及性质计算实验目的1.初步了解分子模型方法的原理和应用。
2.学习使用Hyperchem软件构建简单的分子并使用适当方法优化结构。
3.学习使用Hyperchem软件计算简单分子的几何和电子性质。
实验原理化学的学习使我们认识了许多分子的分子式及二维结构,如何得到分子的三维结构,以及分子在空间的几何特征和电子特征,则可以借助于理论计算的工具和方法去模拟计算。
HyperChem软件是HyperCube公司开发的Windows界面程序。
是常用的分子设计和模拟软件。
它可以应用于构建简单及复杂的分子模型并进行综合计算与分析。
分子构建过程可以通过熟练各个菜单及工具栏的操作来实现,计算和分析需要我们了解常用的计算方法。
在本实验室中我们需要了解一下计算方法,这些方法位于HyperChem的Setup菜单下。
(1)分子力学(Molecular Mechanics)方法:分子力学又叫力场方法,目前广泛地用于计算分子的构象和能量。
适用于超大规模体系,超低精度计算。
分子力学的基本假设:玻恩-奥本海默近似,原子核的运动与电子的运动可以看成是独立的;分子是一组靠各种作用力维系在一起的原子集合。
这些原子在空间上若过于靠近,便相互排斥;但又不能远离,否则连接它们的化学键以及由这些键构成的键角等会发生变化,即出现键的拉伸或压缩、键角的扭变等,会引起分子内部应力的增加。
每个真实的分子结构,都是在上述几种作用达到平衡状态的表现。
分子力学从几个主要的典型结构参数和作用力出发来讨论分子结构,即用位能函数来表示当键长、键角、二面角等结构参数以及非键作用等偏离“理想”值时分子能量的变化。
不同的分子力场方法采用不同的势能函数。
MM+:适用于有机分子的计算。
Amber:适用于有机分子、蛋白质和核酸等大分子的计算。
(2)半经验计算(Semi-empirical)方法:是求解HF(Hartree-Fock)方程时采用各种近似,或者直接使用拟合的经验参数来近似求解自洽场。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
HyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作 区:状态 行:2、打开已存在的数据文件 File-OpenDisplay- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
左键、右键增减键序把2D骨架转换为3D图像Select选定原子后,状态行提示原子标号、坐标等结构信息。
保存为数据文件如需要修改原子骨架,比如把某个氢原子改为氯原子: 选定缺省元素后,在指定原子位置上双击即可。
移动、旋转、放大、缩小分子图像此时,可以用Build—Set设定初始数据,如键长、键角、形式电荷等等。
也可以用来直接测定分子构型数据三、优化分子构型1、丁二烯分子体系能量极小值的计算记录输出结果: FILE---START LOG….绘出丁二烯分子骨架模型:DRAW测量键长、键角、二面角等结构信息:SELECT用半经验方法优化分子构型 SETUP--CNDOCOMPUTE---GEOMETRY测量键长、键角、二面角等结构参数。
对给定体系优化构型后,可对优化后的构型再进行单点计算。
计算结果的输出文件HyperChem log start -- Sun May 28 21:38:15 2000.Geometry optimization, SemiEmpirical, molecule = (untitled).CNDOPolakRibiere optimizerConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESOptimization algorithm = Polak-RibiereCriterion of RMS gradient = 0.0100 kcal/(A mol) Maximum cycles = 150RHF Calculation:Singlet state calculationNumber of electrons = 22Number of Double Occupied Levels = 11Charge on the System = 0Total Orbitals = 22Starting CNDO calculation with 22 orbitalsE=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=1 Diff=7144.22066]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=2 Diff=5.36263]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=3 Diff=0.48300]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=4 Diff=0.05233]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=5 Diff=0.00750]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=6 Diff=0.00009]E=-2659.4619 Grad=53.592 Conv=NO(0 cycles 1 points) [Iter=1 Diff=4.99709] E=-2659.4619 Grad=53.592 Conv=NO(0 cycles 1 points) [Iter=2 Diff=0.38262] E=-2671.4854 Grad=0.022 Conv=NO(26 cycles 68 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.042 Conv=NO(26 cycles 69 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.033 Conv=NO(27 cycles 70 points) [Iter=1 Diff=0.00001] E=-2671.4854 Grad=0.019 Conv=NO(27 cycles 71 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.015 Conv=NO(28 cycles 72 points) [Iter=1 Diff=0.00001] E=-2671.4854 Grad=0.069 Conv=NO(28 cycles 73 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.011 Conv=NO(29 cycles 74 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.012 Conv=NO(29 cycles 75 points) [Iter=1 Diff=0.00000]E=-2671.4854 Grad=0.012 Conv=NO(30 cycles 76 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.009 Conv=NO(30 cycles 77 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.010 Conv=NO(30 cycles 78 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.022 Conv=NO(30 cycles 79 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.011 Conv=NO(31 cycles 80 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.017 Conv=NO(31 cycles 81 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.006 Conv=YES(32 cycles 82 points) [Iter=1 Diff=0.00000]ENERGIES AND GRADIENTTotal Energy = -20550.8323392 (kcal/mol)Total Energy = -32.749182076 (a.u.)Binding Energy = -2671.4854530 (kcal/mol)Isolated Atomic Energy = -17879.3468863 (kcal/mol)Electronic Energy = -55625.1226392 (kcal/mol)Core-Core Interaction = 35074.2903000 (kcal/mol)Heat of Formation = -1675.3134530 (kcal/mol)Gradient = 0.0062527 (kcal/mol/Ang)MOLECULAR POINT GROUPC2HEIGENVALUES(eV)Symmetry: 1 AG 1 BU 2 AG 2 BU 3 AG Eigenvalue: -43.673668 -37.506996 -29.111160 -28.857952 -23.531034 Symmetry: 3 BU 1 AU 4 BU 4 AG 5 AG Eigenvalue: -22.587774 -20.271618 -16.989029 -16.868931 -14.247993 Symmetry: 1 BG 2 AU 5 BU 6 AG 6 BU Eigenvalue: -13.322699 3.356511 6.598820 7.283803 7.489040 Symmetry: 2 BG 7 AG 7 BU 8 AG 8 BU Eigenvalue: 7.776278 8.189266 8.610060 10.810771 11.913039 Symmetry: 9 AG 9 BUEigenvalue: 13.934214 14.264333ATOMIC ORBITAL ELECTRON POPULATIONSAO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C 1.059899 0.950186 1.006745 1.012745 1.003647AO: 2 Px C 2 Py C 2 Pz C 3 S C 3 Px C 0.973639 1.009929 0.987255 1.003647 0.973639AO: 3 Py C 3 Pz C 4 S C 4 Px C 4 Py C 1.009929 0.987255 1.059899 0.950186 1.006745AO: 4 Pz C 5 S H 6 S H 7 S H 8 S H 1.012745 0.997504 0.993683 1.004769 1.004769AO: 9 S H 10 S H0.997504 0.993683NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Massx y z1 6 -0.029575 0.13401 -1.42160 0.00000 12.011002 6 0.025530 0.08094 -0.09964 -0.00000 12.011003 6 0.025530 -1.09703 0.72952 0.00000 12.011004 6 -0.029575 -1.15010 2.05149 -0.00000 12.011005 1 0.002496 1.07933 -2.00856 -0.00000 1.008006 1 0.006317 -0.75684 -2.08833 -0.00000 1.008007 1 -0.004769 1.05152 0.45978 0.00000 1.008008 1 -0.004769 -2.06762 0.17011 -0.00000 1.008009 1 0.002496 -2.09542 2.63844 0.00000 1.0080010 1 0.006317 -0.25925 2.71822 0.00000 1.00800ATOMIC GRADIENTSAtom Z Gradients(kcal/mol/Angstrom)x y z1 6 0.00121 -0.01081 0.000122 6 0.00599 0.01255 -0.000213 6 -0.00607 -0.01263 0.000224 6 -0.00120 0.01073 -0.000125 1 0.00859 -0.00497 -0.000026 1 -0.00946 -0.00870 -0.000037 1 0.00169 0.00240 0.000048 1 -0.00161 -0.00235 -0.000059 1 -0.00867 0.00502 0.0000210 1 0.00953 0.00875 0.00003Dipole (Debyes) x y z TotalPoint-Chg. -0.000 0.000 0.000 0.000sp Hybrid -0.000 0.000 0.000 0.000pd Hybrid 0.000 0.000 0.000 0.000Sum -0.000 0.000 0.000 0.000Single Point, SemiEmpirical, molecule = (untitled).CNDOConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESSingle Point, SemiEmpirical, molecule = (untitled).CNDOConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESRHF Calculation:Singlet state calculationNumber of electrons = 22Number of Double Occupied Levels = 11Charge on the System = 0Total Orbitals = 22Starting CNDO calculation with 22 orbitalsIteration = 1 Difference = 7100.34970Iteration = 2 Difference = 5.91366Iteration = 3 Difference = 0.55648Iteration = 4 Difference = 0.06489Iteration = 5 Difference = 0.00841Iteration = 6 Difference = 0.00014Iteration = 7 Difference = 0.00001Energy=-2671.485453 Gradient=0.006301 Symmetry=C2HENERGIES AND GRADIENTTotal Energy = -20550.8323392 (kcal/mol)Total Energy = -32.749182076 (a.u.)Binding Energy = -2671.4854529 (kcal/mol)Isolated Atomic Energy = -17879.3468863 (kcal/mol) Electronic Energy = -55625.1216733 (kcal/mol)Core-Core Interaction = 35074.2893341 (kcal/mol)Heat of Formation = -1675.3134529 (kcal/mol) Gradient = 0.0063010 (kcal/mol/Ang)MOLECULAR POINT GROUPC2HEIGENVALUES(eV)Symmetry: 1 AG 1 BU 2 AG 2 BU 3 AG Eigenvalue: -43.673672 -37.507000 -29.111160 -28.857954 -23.531034Symmetry: 3 BU 1 AU 4 BU 4 AG 5 AG Eigenvalue: -22.587776 -20.271622 -16.989029 -16.868935 -14.247996Symmetry: 1 BG 2 AU 5 BU 6 AG 6 BU Eigenvalue: -13.322702 3.356504 6.598815 7.283800 7.489039Symmetry: 2 BG 7 AG 7 BU 8 AG 8 BU Eigenvalue: 7.776274 8.189266 8.610056 10.810770 11.913034Symmetry: 9 AG 9 BUEigenvalue: 13.934207 14.264326ATOMIC ORBITAL ELECTRON POPULATIONSAO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C 1.059899 0.950186 1.006744 1.012745 1.003647AO: 2 Px C 2 Py C 2 Pz C 3 S C 3 Px C 0.973639 1.009930 0.987255 1.003647 0.973639AO: 3 Py C 3 Pz C 4 S C 4 Px C 4 Py C 1.009930 0.987254 1.059899 0.950186 1.006744AO: 4 Pz C 5 S H 6 S H 7 S H 8 S H 1.012746 0.997504 0.993683 1.004769 1.004769AO: 9 S H 10 S H0.997503 0.993682NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Massx y z1 6 -0.029575 0.13401 -1.42160 0.00000 12.011002 6 0.025530 0.08094 -0.09964 -0.00000 12.011003 6 0.025530 -1.09703 0.72952 0.00000 12.011004 6 -0.029575 -1.15010 2.05149 -0.00000 12.011005 1 0.002496 1.07933 -2.00856 -0.00000 1.008006 1 0.006317 -0.75684 -2.08833 -0.00000 1.008007 1 -0.004769 1.05152 0.45978 0.00000 1.008008 1 -0.004769 -2.06762 0.17011 -0.00000 1.008009 1 0.002497 -2.09542 2.63844 0.00000 1.0080010 1 0.006318 -0.25925 2.71822 0.00000 1.00800ATOMIC GRADIENTSAtom Z Gradients(kcal/mol/Angstrom)x y z1 6 0.00089 -0.01031 0.000152 6 0.00652 0.01230 -0.000253 6 -0.00671 -0.01284 0.000254 6 -0.00129 0.01086 -0.000095 1 0.00889 -0.00516 -0.000066 1 -0.00948 -0.00880 -0.000037 1 0.00169 0.00244 0.000048 1 -0.00152 -0.00235 -0.000069 1 -0.00861 0.00493 -0.0000010 1 0.00963 0.00894 0.00005Dipole (Debyes) x y z Total Point-Chg. -0.000 0.000 0.000 0.000 sp Hybrid -0.000 -0.000 0.000 0.000 pd Hybrid 0.000 0.000 0.000 0.000 Sum -0.000 0.000 0.000 0.0002、二氧化碳分子例用分子力学方法优化分子结构用半经验方法CNDO 优化分子结构选择优化方法和优化收敛限。