阿奇霉素及其关键中间体的合成研究进展
阿奇霉素研究进展

ห้องสมุดไป่ตู้
感染 、 皮肤软组织感染 和生殖泌尿系感染等临床疾 病, 并且取得 了不错的临床疗效。已有研究发现, 阿 奇霉素罗红霉素相比, 其抗大肠杆菌、 肺炎链球菌的 抗菌能力强 5 倍 以上 , 而与红霉素相 比, 其抗流感嗜 血杆 菌 的能力 强 1 0倍 以上 。 1 . 2 药动 学研 究 阿奇 霉 素经 口服 给药后 , 可 以被 人体快 速 吸收 , 可 较 快 的 自血 液进 人 至 细胞 间质 进 而发挥 治疗 效果 。 目前 已有 药 理 学研 究 发 现 , 阿 奇 霉素生物利用率达 3 7 %I s ] 。阿奇霉素可在 口服 2 . 5 2 . 6 h 后达到血药峰浓度 , 且血药峰浓度达 0 . 4— 0 . 4 5 m g / L 。阿奇霉素在人体内分布广泛 , 在多种组 织 内血药浓度均 比同期血药浓度高 1 0— 1 0 0倍 , 由 此显示该药可与组织量有效结合。此外 , 阿奇霉素 在给药后可在巨噬细胞或多彩核 白细胞 内聚集 , 致 使阿奇霉素相较于 同类抗生素而言分布容积更广 , 细胞渗透性更广 , 更加便于 口服给药。另外, 由于阿 奇霉素在人体组织内的滞 留时间较长 , 释放较缓慢 , 所以患者单 剂服用两周之后仍 可检测到该药物原 形 J 。有研究发现, 阿奇霉素在用药 7天 内经尿排 泄率不足 6 %, 肾清 除率 为 1 . 6 7— 3 . 1 5 6 m l / s , 由此 可知只要阿奇霉素给药量足够多 , 通常只需每 日给 药一次便可发挥疗效 , 且药物能够较长时间的维持 其有 效浓 度 。 1 . 3 药效学研究 阿奇霉 素的作用机制是通过阻 碍细菌转肽过程来抑制细菌蛋 白质的合成。经体外 实验和临床研究表明阿奇霉素对 以下多种致病菌有 效: 金黄色葡萄球菌、 肺炎链球菌、 酿脓链球菌、 溶血 性链球菌等革兰阳性需氧菌。阿奇霉素对于耐红霉 素 的革兰 阳性 细菌 菌 株 呈 现 交 叉 耐药 性 , 其 中包 括 粪链球菌以及耐 甲氧西林 的多种葡萄球菌。此外 , 阿奇霉素对流感嗜血杆菌 、 莫拉菌、 脑膜炎奈瑟菌等 革兰 阴性 菌 的作用 最强 。
阿奇霉素的合成工艺研究

阿奇霉素的合成工艺研究张建州(郑州师范高等专科学校化学系,河南郑州450044) 摘 要:以红霉素6,9-亚胺醚为原料,采用硼氢化钾还原,以简单方法制备结晶9-脱氧-9a-氮杂-9a-同型红霉素A水合物,然后再将其甲基化得到阿奇霉素,收率为92%,并确定了还原反应的温度、时间、以及甲基化水解反应的试剂和催化剂。
关键词:阿奇霉素;红霉素6,9-亚胺醚;结晶9-脱氧-9a-氮杂-9a-同型红霉素A水合物Study on the Synthesis Route of AzithromycinZHA N G Jian2z hou(Department of Chemistry,Zhengzhou Teachers College,Zhengzhou 450044,China) Abstract:By using erythromycin6and92imino ether,a straightforward procedure was taken to obtain92 deoxo29a2aza29a2homoerythromycin A that was methlated to obtain azithromycin with92%high field.Tempera2 ture of reducing reaction,time of reducing reaction,field of NaBH4different mole equivalent,dissolvent of metla2 te hydrolyse reaction and catalyst of metlate hydrolyses reaction were ascertained.K ey w ords:azithromycin;erythromycin6;92imino ether;92deoxo29a2aza29a2homoerythromycin A hydrate 阿奇霉素(Azithromycin AZI)(3)是对红霉素A结构进行修饰后产生的衍生物;是新型红霉素的两个最具代表的药物之一,是将9位酮基肟化后进行Beckman重排和N上甲基化反应,内脂环被插入了一个氮原子而扩大的15圆氮杂环内脂类抗生素。
阿奇霉素及其关键中间体的合成研究进展
第 38卷 第 4 期 2009 年 4 月
化工技术与开发 T echnology & Development of Chemical Industry
Vol 38 No 4 Apr 2009
阿奇霉素及其关键中间体的合成研究进展
赵佳苗1, 施介华1, 2 , 许 超1
( 1. 浙江工业大学药学院, 浙江 杭州 310032; 2. 浙江工业大学绿色合成技术国家重点实验室培育基地, 浙江 杭州 310032)
及皮肤软组织感染等, 美国 FDA 批准替代青霉素类 药物作为抗感染药物的一线药物, 还可以治疗艾滋 病患者分支杆菌感染。阿奇霉素的另一个突出优点 是具有独特的药代动力学性质, 吸收后可被转移到 感染部位, 达到很高的组织浓度, 一般可以比细菌外 浓度高 300 倍。阿奇霉素的化学稳定性增强, 降低 了红霉素因为酸性降解而失去活性的问题, 提高了 血药浓度, 大大延长了半衰期。阿奇霉素制剂在目 前国内的抗感染类药物的应用中前景非常广阔。
关键词: 红霉素 A ; 红霉素 A 肟; 红霉素 A6; 9 亚胺醚; 9 脱氧 a 氮杂 9a 类红霉素 A ; 阿奇霉素; 合成
中图分类号: T Q 465 5
文献标识码: A
文章编号: 1671 9905( 2009) 04 0028 07
阿奇霉素( azit hromycin) 是一个 15 环含氮大环 内酯类 药物, 是新型 红霉 素的 最具代 表的 药物 之 一[ 1] , 由克罗地亚的普利瓦( Pliva) 制药公司在 20 世 纪 70 年代末开发, 1981 年美国辉瑞( P fizer) 公司获 得其专利使用权, 并开 始在全世界销 售, 商品 名为 Z it hromax( 希舒美) 。
阿奇霉素有关物质检测方法研究进展

阿奇霉素有关物质检测方法研究进展张静霞;唐克慧;李喆宇;王宇驰;张春然;徐明琴;邓思思【摘要】本文对阿奇霉素有关物质的各检测方法的研究进展进行了综述.常用的检测法有薄层色谱法(TLC)、高效液相色谱法(HPLC)、高效液相-质谱联用技术(LC-MS)及气相色谱法(GC).GC法和固相萃取—微柱高效液相色谱法用于挥发性残留溶剂的检测.【期刊名称】《中国抗生素杂志》【年(卷),期】2015(040)011【总页数】5页(P876-880)【关键词】阿奇霉素;有关物质;检测方法【作者】张静霞;唐克慧;李喆宇;王宇驰;张春然;徐明琴;邓思思【作者单位】成都大学四川抗菌素工业研究所,成都610052;成都大学四川抗菌素工业研究所,成都610052;成都大学四川抗菌素工业研究所,成都610052;成都大学四川抗菌素工业研究所,成都610052;成都大学四川抗菌素工业研究所,成都610052;成都大学四川抗菌素工业研究所,成都610052;成都大学四川抗菌素工业研究所,成都610052【正文语种】中文【中图分类】R978.1阿奇霉素化学名为9A-甲基-9-脱氧-9A-氮杂-9A-红霉素A,是由红霉素A经过化学修饰衍生而得到的,结构如图1,属第三代大环内酯类抗生素。
该抗生素主要用于呼吸道感染、软组织感染、泌尿系统感染和皮肤感染等,与其他抗生素相比,阿奇霉素具有给药次数少、疗程短、副作用少的优点,因此更适合于混合感染的治疗,临床应用更广泛。
在我国,阿奇霉素目前已经是大环内酯类抗生素中剂型最全面、临床应用最量大面广的品种。
随着阿奇霉素临床用药的不断增加,建立合适的方法对其有关物质进行有效控制,提高本品用药的安全性保障显得尤为重要。
而目前,国内外药典及文献报道方法又都存在缺陷,如检测灵敏度低,方法重复性不好,峰形差,同品牌不同批次的色谱柱重现性差;国外药典方法对色谱条件要求苛刻,如需要特定的色谱柱,并且在测定含盐的阿奇霉素品种时,主峰出峰时间非常迟后,且流动相中的盐容易析出造成色谱柱堵塞等。
阿奇霉素及其关键中间体的合成研究进展

及 皮肤 软组织 感染 等 , 国 F A批准 替代 青霉 素类 美 D 药 物作 为抗感染 药 物 的一 线药 物 , 可 以治疗 艾 滋 还 病 患者 分支杆 菌感 染 。阿奇霉 素 的另一个 突 出优点
l 1 J
,
由克罗 地亚 的普利 瓦 (l a制 药公 司在 2 Pi ) v 0世
醚 ( rtrmyi 6 9eoehr , 后 1 E yho c A ,-n l e) 然 n t 2位 的 羟 基进 攻 C一 C 8 9双 键 , 生成 6 9 9 1一 缩 酮 ( r— , : ,2螺 E y trmyi 6 9 9 1一 i kt1。在 更 强 的 酸 性 ho c nA , : ,2s r ea) po
赵 佳 苗 施 介 华 一许 , , ,
(. 1浙江工业大学药学院 , 江 杭州 浙 摘
超
303 ) 10 2
3 0 3 ; . 江工业大学绿 色合成技术 国家重点实验室培育基地 , 江 杭州 1 02 2 浙 浙
要: 以红霉素 A为原料 经肟 化反应 、 贝克曼 ( e 咖 ) Bc 重排反应 、 还原反应 和 N一甲基化还 原反应 制备大
酸, 中等极性 的异丙醇代替 甲醇制备 红霉素 A肟 , 使红霉素 A肟收率大幅度提高 , 达到 9 . %。并首 89 次提及 了红霉素 A肟合成过程 中 E式和 z式 的比 例 问题 , 讨论了不同的重结 晶条件对于产品收率 纯
度、 晶型特别 是 E Z比例 的影 响 , 为用异丙醇 一 / 认 水体系重结 晶可提高产品 E Z比例 。 / 20 0 1年 , 岩 等 人 【J 用 盐 酸 羟 胺 一三 乙 胺 石 l采 0 乙酸体系将红霉素 A转化为红霉 素 A肟 , 主要考 察 了酸度 对 反应 的影 响 。在 最 佳 反 应 条 件 下 , 霉 红
阿奇霉素的合成研究

ratio of acetone
and water is 1.45:1,the molar ration
hydrochloric aid and the salt of Erythromycin
A(E)oxime
is 1:1 5;The reaction time
is 3 hours and the reaction temperature is 0~5。C.The yield of Erythromycin A 6,9一iminoether is 82.86%.and the content of HPLC is 90.1 3%when reacting under these conditions. Three different kinds of method have been studied in the reduction
学位论文作者签名:互飞
签字日期:
枷7年 f月吗日
学位论文版权使用授权书
本学位论文作者完全了解苤鲞态堂有关保留、使用学位论文的规定。 特授权墨盗盘堂可以将学位论文的全部或部分内容编入有关数据库进行检
索,并采用影印、缩印或扫描等复制手段保存、汇编以供查阅和借阅。同意学校
向国家有关部门或机构送交论文的复印件和磁盘。 (保密的学位论文在解密后适用本授权说明)
tO
of the
Erythromycin A 6,9-iminoether with potassium hydride.The effect of the acid
reaction has been studied.And the most optimization condition is:the molar ratio of Erythromycin A 6,9一iminoether and potassium hydrid is 1:3,acidized with
阿奇霉素的最新研究进展

心等。对于其 机制的研究 , 有推测跟
红霉素相似 ,可能依赖 C a 2 + 泵 系统 直接作用于 胃肠道 平滑肌 口;有认
由于阿奇霉 素味道苦涩 、 水溶性差 , 为 了制剂方便 ( 如增 加溶解度) 或改善副作用( 减少刺激性) , 有 时候需要将 药物转
阿奇霉 素化学结构 中有可被质子化 的叔胺基 , 对组 织的
亲和力强 , 在 白细胞 、 巨噬细胞及成纤维细胞 内浓度较高 。 通
用。
效果非常理想 。 国外研究发现阿奇霉素对家畜有促生长 的作 用, 提示存在应用 于家禽临床药物 的潜能 。但 国内 目前还没 有全面开展阿奇霉索在养殖动 物上的应用研究 , 缺乏兽 医l 临 床上 的全 面了解 ,为此 中国农业部在 2 0 0 5年 8月发布 5 6 0 号令取 消阿奇霉素地方标 准 ,宣布 自 1 O月 2 8日起 至 2 0 1 0
化成盐 、 酯, 它们的活性成分没有根本改变。 比较常见 的有普 通片 、 缓释片 、 分散片 , 还可 以将将药物制成栓 剂和凝胶剂等
半固体制剂 。脂质体作为 一种新 型药物载体 , 能使药物靶 向 定位于特 定的组织 , 激 活机体 自身的免疫 功能 , 减少 或降低
药物的毒性 。
为红霉素和 阿奇霉 素同为 胃动素受 体激动剂 ,可作用 于 胃窦平滑肌 和 肠神经系统 的 胃动素受体 。研究发
子, 彻底 解决红霉素 因酸缩 酮化形成 8 . 9 一脱水红霉 素 一 6 . 9 一 半缩酮 的致 命缺点 ,有效地 阻止 了红霉素 的酸催化失 活反
临床 上也 发生过 过敏 性休 克现像 。
阿奇霉素的合成工艺研究
阿奇霉素的合成工艺研究张建州(郑州师范高等专科学校化学系,河南郑州450044) 摘 要:以红霉素6,9-亚胺醚为原料,采用硼氢化钾还原,以简单方法制备结晶9-脱氧-9a-氮杂-9a-同型红霉素A水合物,然后再将其甲基化得到阿奇霉素,收率为92%,并确定了还原反应的温度、时间、以及甲基化水解反应的试剂和催化剂。
关键词:阿奇霉素;红霉素6,9-亚胺醚;结晶9-脱氧-9a-氮杂-9a-同型红霉素A水合物Study on the Synthesis Route of AzithromycinZHA N G Jian2z hou(Department of Chemistry,Zhengzhou Teachers College,Zhengzhou 450044,China) Abstract:By using erythromycin6and92imino ether,a straightforward procedure was taken to obtain92 deoxo29a2aza29a2homoerythromycin A that was methlated to obtain azithromycin with92%high field.Tempera2 ture of reducing reaction,time of reducing reaction,field of NaBH4different mole equivalent,dissolvent of metla2 te hydrolyse reaction and catalyst of metlate hydrolyses reaction were ascertained.K ey w ords:azithromycin;erythromycin6;92imino ether;92deoxo29a2aza29a2homoerythromycin A hydrate 阿奇霉素(Azithromycin AZI)(3)是对红霉素A结构进行修饰后产生的衍生物;是新型红霉素的两个最具代表的药物之一,是将9位酮基肟化后进行Beckman重排和N上甲基化反应,内脂环被插入了一个氮原子而扩大的15圆氮杂环内脂类抗生素。
阿奇霉素临床应用研究进展

阿奇霉素临床应用研究进展摘要大环内酯类抗生素是目前最常用的杀菌消炎一线治疗药物之一,它的作用机理是通过与细菌核糖体50s亚基特殊靶位及某种核糖体蛋白质结合,阻断转肽酶作用,干扰mRNA翻译过程,从而抑制细菌蛋白质的合成。
第一个大环内酯类抗生素红霉素已应用40余年,由于对酸不稳定等性质限制的它的临床应用。
阿奇霉素是将红霉素A9—酮基酯化后经becklman重排,N-甲基化等一系列反应得到的15元氮杂化合物,也是氮环内酯类的第一个品种,这种结构上的差异阻碍了内部形成半酮缩醛反应,结果证明阿奇霉素和红霉素相比,其对酸的稳定性大大加强了。
阿奇霉素和其它大环内酯类抗生素相比,具有独特的药物动力学特性:口服吸收快,对酸稳定,半衰期长,有较高的生物利用度,而且感染部位组织及细胞内浓度远高于血液浓度;抗菌谱广;不良反应轻微,发生率低。
可以提高临床的治疗效果,改善病人的生活质量。
关键词:大环内酯类抗生素、阿奇霉素、临床应用阿奇霉素的药动学阿奇霉素对胃酸稳定,虽然口服生物利用度仅37%,单次服用0.5g后2~3h 血药峰浓度约0.4μg/ml,但组织分布好,蛋白结合率低(7%~23%),消除半减期12~14h,服药后12~30h在前列腺、扁桃体、肺组织、胃组织、女性生殖器组织中的浓度分别达2.6μg/g、4.5μg/g、3.6μg/g、6.1μg/g和2.7~3.5μg/g.50%以上的药物以原形由胆汁排泄,部分为去甲基的代谢产物。
药物在组织中滞留时间较长,释放缓慢,单剂服药后14日,医|学教育网搜集整理仍可在尿中测得原形药物。
1周内经尿排泄率低于6%,肾清除率为1.67~3.156ml/s。
阿奇霉素在组织中的浓度明显高于血药浓度的原因主要由于药物通过中性粒细胞与吞噬细胞主动由局部组织移至上述细胞内。
在感染部位储存药物的中性粒细胞受细菌刺激释放药物而起抗菌作用。
阿奇霉素的药理学阿奇霉素是将红霉素A9-酮基脂化后经Becklman重排,N-甲基化等一系列反应得到的15元氮杂化合物,也是氮环内酯类(Azalids)中的第一个品种,这种结构上的差异阻碍了内部形成半酮缩醛的反应,其中酯键水解成中性的红霉支糖是其重要的分解途径,阿奇霉素中酯键所连接的红霉支糖水解活化能为15.6kal.mol-1在37·C、PH2的溶液中(离子强度=0.02),阿奇霉素降解10%需21min,而同样情况下,红霉素仅需3.7s这些结果证明阿奇霉素与红霉素相比,其对酸的稳定性大大增强。
一种阿奇霉素的制备方法及其中间体的制备方法[发明专利]
![一种阿奇霉素的制备方法及其中间体的制备方法[发明专利]](https://img.taocdn.com/s3/m/69a235ade45c3b3566ec8bb0.png)
专利名称:一种阿奇霉素的制备方法及其中间体的制备方法专利类型:发明专利
发明人:吴范宏,杨波,杨雪艳,俞晓东
申请号:CN200910198992.2
申请日:20091118
公开号:CN101712703A
公开日:
20100526
专利内容由知识产权出版社提供
摘要:本发明公开了一种阿奇霉素的制备方法,其包括如下步骤:在酸性水溶液中,硼氢化钾的作用下,将如式II所示的红霉素A6,9-亚胺醚进行还原反应,之后进行甲基化反应,再在水与卤代烷烃的混和溶剂中进行水解反应,即制得如式I所示的阿奇霉素;该方法的还原剂用量适中且收率高,显著降低制备阿奇霉素的成本。
本发明还涉及一种红霉素A6,9-亚胺醚以及红霉素A9-肟的制备方法。
申请人:上海华理生物医药有限公司
地址:200231 上海市徐汇区华泾路1305弄18号B座四楼
国籍:CN
代理机构:上海智信专利代理有限公司
代理人:吴林松
更多信息请下载全文后查看。
阿奇霉素合成与检测的研究进展

阿奇霉素合成与检测的研究进展
淡保松;张大伟;冯良荣;邱发礼
【期刊名称】《化工进展》
【年(卷),期】2008(27)11
【摘要】合成阿奇霉素的关键步骤是红霉素亚胺醚的还原.本文对催化还原法和化学还原法存在的主要问题进行了分析,提出了进-步研究的设想.综述了产物的定性和定量分析.
【总页数】7页(P1793-1799)
【作者】淡保松;张大伟;冯良荣;邱发礼
【作者单位】中国科学院成都有机化学研究所,四川成都610041;中国科学院研究生院,北京100039;中国科学院成都有机化学研究所,四川成都610041;中国科学院研究生院,北京100039;中国科学院成都有机化学研究所,四川成都610041;中国科学院成都有机化学研究所,四川成都610041
【正文语种】中文
【中图分类】TQ110.2
【相关文献】
1.阿奇霉素及其关键中间体的合成研究进展 [J], 赵佳苗;施介华;许超
2.阿奇霉素有关物质检测方法研究进展 [J], 张静霞;唐克慧;李喆宇;王宇驰;张春然;徐明琴;邓思思
3.禁(限)用合成麝香的安全性评估与检测方法研究进展 [J], 李延川
4.食品中人工合成甜味剂的检测研究进展 [J], 李鋆婧;苏爽月;赵宇
5.OPO酶法合成和其在婴幼儿配方乳粉中的检测研究进展 [J], 安雪征;吕林燚;祁娜;杜旷;张双;杜鉴;齐原;田云翼
因版权原因,仅展示原文概要,查看原文内容请购买。
阿奇霉素及其关键中间体的工艺改进

阿奇霉素及其关键中间体的工艺改进
刘云霞;朱文祥;李凡华
【期刊名称】《山东化工》
【年(卷),期】2013(42)1
【摘要】以硫氰酸红霉素为起始原料,合成红霉素A肟,通过对红霉素A肟的水相乳化分散处理,可以显著提高贝克曼重排的转化率和选择性,转化率可达99.5%,选择性≥96%.本文着重介绍改进工艺后贝克曼重排对阿奇霉素收率以及质量的影响,阿奇霉素的总收率达到78%,质量符合USP要求,已实现工业化并取得良好经济效果.【总页数】3页(P60-62)
【作者】刘云霞;朱文祥;李凡华
【作者单位】菏泽市方明制药有限公司,山东菏泽274000;菏泽市方明制药有限公司,山东菏泽274000;菏泽市方明制药有限公司,山东菏泽274000
【正文语种】中文
【中图分类】TQ465
【相关文献】
1.艾氟康唑及其关键中间体合成工艺改进 [J], 张之建
2.Trk激酶抑制剂关键中间体的合成工艺改进 [J], 张丹君; 许亚文; 谢蓉蓉; 强浩; 李政
3.Trk激酶抑制剂关键中间体的合成工艺改进 [J], 张丹君; 许亚文; 谢蓉蓉; 强浩; 李政
4.替格瑞洛关键中间体的工艺改进 [J], 孙光祥;吴路新;张云然
5.巴洛沙韦关键中间体的合成工艺改进 [J], 于立国;孙光祥;张云然;陶维洁
因版权原因,仅展示原文概要,查看原文内容请购买。
去克拉定糖阿奇霉素的合成研究

去克拉定糖阿奇霉素的合成研究李亚林;苏永青;张志霞【摘要】研究了去克拉定糖阿奇霉素的制备工艺,探讨了重结晶溶剂种类、催化剂种类、温度、pH值等对去克拉定糖阿奇霉素反应效果的影响.结果表明,以水为反应体系,在室温下用盐酸调节pH值至1.0,反应结束后用氨水调节pH值至10.0,粗品用丙酮和水混合体系精制,HPLC检测产品纯度可达99.9%.【期刊名称】《石家庄职业技术学院学报》【年(卷),期】2015(027)004【总页数】6页(P9-14)【关键词】阿奇霉素;去克拉定糖阿奇霉素;HPLC;pH值;精制【作者】李亚林;苏永青;张志霞【作者单位】石药集团欧意药业有限公司十车间,河北石家庄 052165;石药集团欧意药业有限公司十车间,河北石家庄 052165;河北师范大学附属民族学院,河北石家庄 050024【正文语种】中文【中图分类】R914阿奇霉素作为第二代红霉素类产品,是第一个半合成氮杂十五元环大环内酯抗生素,被誉为“21世纪抗感染药重磅炸弹”.其化学名为9-去氧-9a-氮杂-9a-甲基-同型红霉素A,是由红霉素A衍生的广谱抗菌素化合物.它由南斯拉夫Pliva制药公司首先创制,美国Pfizer制药公司开发上市.阿奇霉素与红霉素相比,具有半衰期长、给药次数少、疗程较短、不良反应发生率低等优势[1].红霉素作为第一代大环内酯类抗生素,比青霉素抗菌效果更加广泛,同时药物副作用较小,产品上市后,成为对青霉素过敏者的首选替代药物.红霉素虽然没有过敏反应,但其在胃酸作用下迅速降解而失去药效,因此吸收不稳定,生物利用率低,降解产物还会引起胃肠道的不良反应,极大地限制了它的使用.鉴于此,科学家们对红霉素的结构进行了修饰,衍生了第二代大环内酯类抗生素,克拉霉素、阿奇霉素、罗红霉素等克服了红霉素酸性降解的问题,提高了抗菌活性,它们的出现是20世纪末医药界最重大的成就之一.然而,近年来,滥用抗生素导致了各种耐药性病原菌的大量出现,极大地威胁了人类健康,也向医药领域提出了严峻挑战.越来越多的细菌出现耐药性,其耐药水平也越来越高,细菌对大环内酯类抗生素的耐药性也随之出现并日益加剧.在现有的条件下,通过限制抗生素的滥用、制备新型抗生素或制备疫苗来治疗耐药细菌感染性疾病还存在一定困难.相比较而言,通过对现有抗生素的结构改造来寻找高效、低毒、抗耐药菌的药物成为目前解决抗生素耐药性的一条重要途径[2].基于大环内酯类抗生素的作用机制,在药物分子中引入易于与碱基形成氢键的结构基团将有助于化合物与靶酶的相互作用,在阿奇霉素结构中的克拉定糖羟基、去克拉定糖羟基或去氧氨基糖氮原子上通过不同的连接基团引入不同的糖基,获得高效、低毒、抗菌谱广、能克服耐药性的大环内酯糖基衍生物具有十分重要的意义.去克拉定糖阿奇霉素是衍生物的中间体,该物质的合成和纯化对生产阿奇霉素衍生物具有重要的意义.目前,国产阿奇霉素的主要杂质为去克拉定糖阿奇霉素,该杂质化合物的制备和物化性质健全了阿奇霉素杂质的数据库,为提高阿奇霉素的质量标准和控制要求提供了保障.文献报道的合成条件是用盐酸在溶剂或者水体系中进行.文献[3]研究了去克拉定糖阿奇霉素来自甲基化过程中的阿奇霉素的酸性降解.在甲基化反应过程中控制不好反应液的酸用量,会造成15元环3-O上脱去克拉定糖而生成杂质.1.1 溶剂体系中生产去克拉定糖文献[2]在室温下将阿奇霉素溶解在无水甲醇中,用1 mol/L盐酸调节pH至1.0~1.5,搅拌24 h,用1 mol/L的氢氧化钠溶液中和至pH=10.5~11.0,在室温下继续搅拌24 h.加入5%NaHCO3至反应停止,用二氯甲烷萃取,有机层用无水硫酸钠干燥,减压浓缩得白色固体,收率为92.1%.见图1.文献[4]和文献[5]先将阿奇霉素溶解在甲醇和水的混合溶剂中,用2 mol/L的盐酸调节pH至1.79,搅拌24 h后升温至50 ℃,搅拌4.5 h,降至室温后继续搅拌24 h.蒸发甲醇溶剂,使用二氯甲烷萃取3次并调节pH至1.79,分层弃去有机相,水层加入二氯甲烷萃取3次并调节pH至9.02,分层,收集有机相并用饱和食盐水清洗6次.有机相用无水Na2SO4干燥,减压浓缩,得白色固体,收率为74.0%. 文献[6]研究了阿奇霉素或阿奇霉素的中间体脱去克拉定糖化合物的制备工艺,提出将阿奇霉素或阿奇霉素中间体溶解于低级醇、低级醚、卤代烃、低级酮或低级酯中,加入适量的水后,在-5~40 ℃,滴加无机酸或有机酸或者无机酸和有机酸的混合酸进行水解反应,反应液pH值控制在0<pH≤6.0,反应时间为0.5~8 h;反应结束,加入氢氧化钠,将pH值调至7≤pH≤10,浓缩去溶剂,水相中析出固体,离心抽滤,滤饼在低级醇与水或者低级酮与水的混合体系下进行精制结晶.该工艺流程简单、收率高.1.2 水体系中生产去克拉定糖文献[7-8]将阿奇霉素溶在6 mol/L的盐酸溶液中,0 ℃反应4 h,反应结束后加入冰和50%氢氧化钠溶液,使用乙酸乙酯萃取,分层后在水层加入50%的氢氧化钠溶液,再用二氯甲烷萃取,有机层用Na2SO4干燥,减压蒸馏,得无色固体,收率100%.文献[9]将氮杂红霉素溶在水和35%的甲醛溶液中,加入甲酸,回流反应30 h,反应结束后降至室温,用乙酸乙酯萃取,分层,水层用氢氧化钠溶液调pH至11,继续用乙酸乙酯萃取,合并有机层并用水洗,有机相干燥、浓缩得白色固体.用甲基叔丁基醚洗涤固体,抽滤,干燥得目标物,收率61%.见图2.文献[10]在研究阿奇霉素相关物时,提出将阿奇霉素加入1 mol/L盐酸,待完全溶解后,保持室温反应1 h,然后加入乙酸乙酯萃取,有机层分别用水和饱和NaCl溶液洗涤,然后有机层用无水硫酸钠干燥,减压蒸干得白色固体,收率30.0%.文献[11]在制备阿奇霉素相关物时,提出将阿奇霉素悬浮于水中,在磁力搅拌下滴加浓盐酸溶解,用稀盐酸调节pH至2.0左右,TLC(薄层色谱)监控反应,反应结束后,反应液用甲基叔丁基醚萃取2~3次去除克拉定糖,分层,去除有机相,水相用氨水中和至中性,析出去克拉定糖阿奇霉素,抽滤,粗品用丙酮-水精制3次以上至HPLC(高效液相色谱)纯度大于95%,得白色纯品,收率40.6%.见图3.文献[12]在研究大环内酯类抗菌化合物及其药学上可接受的盐中,以去克拉定糖阿奇霉素制备3-O-取代阿奇霉素糖基衍生物时提出,将阿奇霉素悬浮于水中,滴加浓盐酸调pH至1~2,搅拌3 h,反应结束后,冰浴条件下滴加氨水至pH=8~9,析出白色固体,抽滤,滤液加入氯化钠固体至饱和,继续析出白色固体,抽滤,固体经水洗,干燥,丙酮结晶,得白色固体,收率91%.见图4.从工业化角度综合考虑原料成本、产率高低、操作难易、步骤长短、污染是否严重以及是否易于实现工业化生产等因素,本文采用以水为反应溶剂来进行研究.这是因为它收率高,而且成本低,不污染环境,后处理简便,易于操作,适合工业化生产.2.1 仪器与试剂2.1.1 仪器本试验所用仪器见表1.2.1.2 试剂阿奇霉素(自制)、盐酸、硫酸、磷酸、甲酸、乙酸、氨水、丙酮、甲醇、乙醇、乙酸乙酯、二氯甲烷、正己烷,均为分析纯.2.2 实验方法和步骤将10 g阿奇霉素置于250 mL单口瓶中,加入60 mL水,室温条件下滴加浓盐酸至pH=1~2,磁力搅拌3 h,反应结束后,冰浴条件下滴加浓氨水至pH=8~11,析出白色固体,抽滤,固体经水洗,得粗品.粗品用丙酮和水(2∶1)重结晶,真空干燥至恒重,得产品7.17 g,收率91%.2.3 色谱条件色谱柱:Waters XbridgeTm shield RP 18(5μm)(L=0.25 m,φ=4.6 mm).流动相:流动相A用稀磷酸或稀氢氧化钠溶液调节1.8 g/L无水磷酸氢二钠溶液的pH值至8.9;流动相B为甲醇∶乙腈=250∶750.流速:1.0 mL/min.柱温:60 ℃.检测器:210 nm.进样体积:50 μL.3.1 催化剂种类的选择为了选择合适的催化剂,分别考察了盐酸、硫酸、磷酸、甲酸、乙酸作为催化剂对结果的影响,见表2.由表2可见,不同酸对产品收率和质量的影响不大.由于盐酸物美价廉,工业运输、储存和使用均比较成熟,为此本研究选盐酸作为催化剂.3.2 反应pH和温度的选择分别以不同浓度的盐酸和水作为反应溶剂,盐酸作为催化剂,调节反应溶液的pH值,均在室温下进行反应,结果见表3-4.由表3和表4可以看出,当反应溶液的H+浓度很低时,不利于反应进行,pH值为2.5时,24 h原料的转化率只有51.3%;pH值大于2.5时,48 h基本不反应.但当H+浓度过高,pH<1.5时,收率基本保持一致,只是缩短了反应时间;同时,盐酸浓度越高,中和所需的氨水消耗量越大.综合经济效果和反应时间,本研究认为pH值为1.0时最佳.在试验过程中,升高温度即可加快反应速度,但也会使副产物增加.因此,选择室温下进行反应.3.3 后处理的选择以水为反应溶剂,后处理可选择提取法和析出法.提取法:反应结束后溶液可采用有机溶剂,如乙酸乙酯、二氯甲烷等萃取去除克拉定糖,分层,水相用碱析出.析出法:反应溶液用碱直接调pH析出.提取法收率一般在95%左右,操作复杂,配套设备较多,投资大;而析出法操作简便,但收率稍低,一般为90%~95%.从工业化角度,采用析出法更加合适.3.4 析出法pH的选择后处理可选用氢氧化钠固体或溶液、氨水等调节pH值.由于氨水价格便宜,因此本研究的后处理选用氨水调节pH值,同时考察了不同pH值下的反应结果,见表5.由表5可见,后处理采用氨水调节pH值至10.0时,收率最高,继续滴加氨水,收率没有明显变化.为此,后处理pH值调至10.0时最佳.3.5 精制溶剂的选择为了保证产品的质量,分别考察了不同溶剂对产品精制的影响,结果见表6和图5. 由表6可见,使用丙酮与水作为精制溶剂,收率和质量最高.为此,本研究选用丙酮和水来精制产品.由图5和表7可见,选用丙酮和水精制产品,HPLC含量达99.9%.去克拉定糖阿奇霉素作为大环内酯类抗菌化合物3-O-取代阿奇霉素糖基衍生物的关键中间体,优化其合成工艺具有重要意义.以水为反应溶剂时,收率高,而且溶剂成本低,不污染环境,后处理方便,操作简单,适合工业化生产.【相关文献】[1] 孙立权,付艳杰,刘聪,等.阿奇霉素相关物的研究进展[J].中国抗生素杂志,2013,38(5):393-396.[2] ZHANG LING,SONG LINCHEN,LIU ZHAOPENG,et al.Synthesis and antibacterial activityof novel 3-O-carbamoyl derivatives of clarithromycin and 11,12-cyclic carbonate azithromycin[J].European Journal of Medicinal Chemistry,2010,45(3):915-918.[3] 付艳杰,孙立权,范新苑,等.浅谈阿奇霉素药典标准提高与药品生产工艺的关系[J].中国药品标准,2012,13(2):83-86.[4] ALIHODZIC,SULEJMAN,STARCEVIC KRISTINA,et al. 3-N-substituted 9-deoxo-9a-methyl-9a-aza-homoerythromycin Having Antimalarial Activity:WO,2010086351[P].2010-08-05.[5] KRISTINA STARCEVIC,DIJIANA PESIC,ANA TOPLAK,et al.Novel Hybrid Molecules Based on 15-membered as Potential Antimalarial Agens[J].European Journal of Medicinal Chemistry,2012,49(2):365-369.[6] 覃鹏,吴忠,张俊山,等.阿奇霉素或阿奇霉素的中间体脱去克拉定糖化合物的制备工艺:中国,201310125166[P].2013-08-07.[7] BURNET MICHAEL,GUSE JAN-HINRICH,GUTKE HANS JURGEN,et al.Conjugates Of Biologically Active Compounds,Methods For Their Preparation And Use,Formulation And Pharmaceutical Applications Thereof:WO,2003070174[P].2003-08-28.[8] GUSE JAN-HINRICH,BAEUERLEIN CHRISTIANE,EGGERS MARY,et al.Anti-inflammatory Macrolides:WO,2012051126[P].2012-04-19.[9] DAUVERGNE,JEROME,WANG SHOUMING,et al.3-Descladinosyl-O-carbamoyl Functionalized Derivatives of Azithromycin:WO,2012131396[P].2012-10-04.[10] 刘啸,陈宇瑛,王瑛瑛,等.阿奇霉素中相关物的研究[J].国外医药抗生素分册,2013,34(2):77-80.[11] 刘聪,付艳杰,聂乙娇,等.阿奇霉素相关物的制备[J].北京理工大学学报,2014,34(1):106-110.[12] 柴晓云,吴秋业,俞世冲,等.一种大环内酯类抗菌化合物及其制备方法和应用:中国,201310056140[P].2013-06-05.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
最近, 孟彩虹[ 17] 比较了 3 种不同合成红霉素 A 肟的方法, 发现预先从 盐酸羟胺、硫氰酸红霉 素 A 中游离出羟胺和红霉素 A, 然后在 pH = 6 7 条件下
30
化工技术与开发
第 38 卷
羟胺和红霉素 A 于 58~ 60 ! 反应 28 h, 红霉素 A 肟的收率为 81% , 红霉素 A( E) 肟含量为 91 1% 。 1 3 红霉素 A 肟的纯化
2001 年, 石岩等人[ 10] 采用盐酸羟胺 - 三乙胺 - 乙酸体系将红霉素 A 转化为红霉素 A 肟, 主要考 察了酸度对反应的影响。在最佳反应条件下, 红霉 素 A 肟的收率为 97% , 纯度达 87 9% 。2002 年, 廖 国礼等人[ 11] 报道了在合成克拉霉素过程中以红霉 素碱为原料在甲醇 乙酸钠体系中回流反应 24 h, 红 霉素 A 肟的收率 90% , 含量为 95 1% 。
由于红霉素在酸性和碱性介质中均不稳定易发
生降解, 为了避免传统红霉素 A 肟制备方法中红霉 素 A 易降解问题, 1997 年, 美国雅培( Abbot) 公司[ 9] 采用羟胺水溶液代替盐酸羟胺, 甲酸或乙酸代替盐 酸, 中等极性的异丙醇代替甲醇制备红 霉素 A 肟, 使红霉素 A 肟收率大幅度提高, 达到 98 9% 。并首 次提及了红霉素 A 肟合成过程中 E 式和 Z 式的比 例问题, 讨论了不同的重结晶条件对于产品收率、纯 度、晶型特别是 E/ Z 比例的影响, 认为用异丙醇水体系重结晶可提高产品 E/ Z 比例。
阿奇霉素与红霉素( eryt hromycin) 在化学结构 上和作用机制上具有共同性, 均通过与细菌细胞中 核糖体 50S 亚基结合, 阻碍细菌转肽过程和( 或) m - RNA 转位, 抑制蛋白质的合成而达到抗菌作用。 但它们的生物学特性却截然不同。阿奇霉素对某些 难对付的细菌具有杀菌作用, 特别是对革兰氏阴性 菌作用增强。它被广泛应用于呼吸系统、泌尿系统
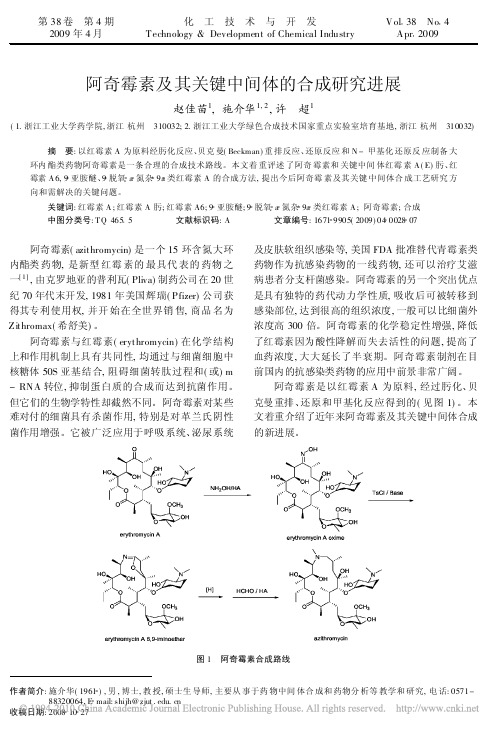
阿奇霉素是以红霉素 A 为原料, 经过肟化、贝 克曼重排、还原和甲基化反应得到的( 见图 1) 。本 文着重介绍了近年来阿奇霉素及其关键中间体合成 的新进展。
图 1 阿奇霉素合成路线
作者简介: 施介华( 1961 ) , 男 , 博士, 教 授, 硕士生 导师, 主要从 事于药 物中间 体合 成和 药物分 析等 教学和 研究, 电 话: 057188320064, E mail: shijh@ zjut . edu. cn
关键词: 红霉素 A ; 红霉素 A 肟; 红霉素 A6; 9 亚胺醚; 9 脱氧 a 氮杂 9a 类红霉素 A ; 阿奇霉素; 合成
中图分类号: T Q 465 5
文献标识码: A
文章编号: 1671 9905( 2009) 04 0028 07
阿奇霉素( azit hromycin) 是一个 15 环含氮大环 内酯类 药物, 是新型 红霉 素的 最具代 表的 药物 之 一[ 1] , 由克罗地亚的普利瓦( Pliva) 制药公司在 20 世 纪 70 年代末开发, 1981 年美国辉瑞( P fizer) 公司获 得其专利使用权, 并开 始在全世界销 售, 商品 名为 Z it hromax( 希舒美) 。
为了避免使用价格较贵的盐酸羟胺, 以及在使 用盐酸羟胺过程中产生大量无机盐所带来的问题, 1989 年, T akehiro 等人[ 7] 采用羟胺的水溶液作为肟 化试剂合成红霉素 A 肟, 收率达 82% 。1991 年, 法 国罗素( Roussel) 公司 Gasc 等人[ 8] 在合成罗红霉素 过程中, 提出了用三乙胺作为游离盐酸羟胺的碱制 备红霉素 A 肟。
收稿日期: 2008 10 27
第 4期
赵佳苗等: 阿奇霉素及其关键中间体的合成研究进展ቤተ መጻሕፍቲ ባይዱ
29
1 红霉素 A 的肟化反应
由于红霉素 A 是一个大环内酯类化合物, 具有 多羟基和 9 位的羰基, 且在其 3 位和 5 位有分别连 接的糖类化合 物。在氢离子作用下, 红霉素 A6 位 的羟基进攻 9 位的羰基形成红霉素 A 6, 9 半缩酮 ( Eryt hromycin A 6, 9 hemiacetal) , 接着 9 位的羰基 与 8 位的氢在酸催化下脱水形成红霉素 A 6, 9 烯醇 醚( Erythromycin A 6, 9 enolether) , 然后 12 位的羟 基进攻 C8 C9 双键, 生成 6, 9: 9, 12 螺缩酮( Ery t hromycin A 6, 9: 9, 12 spiroket al) 。在 更强的酸 性 条件下( pH 3) , 红霉素 A 会进一步水解生成红霉 素胺和克拉定糖。在碱性体系下, 这个内酯基团容 易发生开裂, 或 11 位的羟基与 13 位发生酯交换反 应从而 生成十二 元内酯环 的假红霉 素( pseudoery t hromycin) 结构。因此, 以红霉素 A 或红霉素 A 盐 为原料通过与盐酸羟胺反应制备红霉素肟需要在极 其苛刻的反应条件下反应, 才能保证不发生红霉素 A 降解, 从而得到好的转化率。关于红霉素 A 肟的 合成工艺改进, 长期以来化学工作者主要围绕反应 溶剂和介质 pH 值对肟化反应的影响开展了大量研 究。 1 1 以红霉素 A 为原料
图 2 红霉素 A ( E) 肟贝克曼重排反应 过程
1979 年, 德国赫尔布兰德大药厂的 Gabrijela 等 素 A( E) 肟重排中 的异构化 现象进行 了系统 的研
人[ 21~ 22] 首次提供了一种在丙酮 水介质中, 碳酸氢 究, 发现以乙醚作溶剂, 在吡啶存在下对甲基苯磺酰
钠作为碱, 在 5 ! 下以对甲基苯磺酰氯作离去基团 氯与红霉素 A( E) 肟反应时, 低温有利于红霉素 A9,
1966 年, Djokic 等人[ 2~ 3] 首次报道在无水甲醇 介质中, 以碳酸钡游离盐酸羟胺与红霉素 A 反应合 成了红霉素 A 肟盐酸盐。1984 年, 辉瑞( Pfizer ) 公 司的研究人员在研究红霉素的衍生物时, 用吡啶作 溶剂制备 红霉素 A 肟[ 4] 。1987 年, A dachi 等 人[ 5] 用咪唑游离盐酸羟胺, 于室温下在甲醇溶液中与红 霉素 A 反应 4 d, 制得红霉素 A 肟, 收率达到了 87 6% 。但由于反应时间过长以及咪唑价格比较昂 贵且不易处理, 所以没有投入工业化生产。接着, 他 们进一步进行了改进, 用醋酸钠代替咪唑游离盐酸 羟胺, 得 到令 人 满意 结 果, 红 霉素 A 肟 的收 率 达 84 9 % [ 6] 。此法对实现工业化比较有利。
2004 年, 楼科侠[ 20] 研究了以碳酸氢铵为 游离 碱, 甲醇为溶剂, 以红霉素 A 硫氰酸盐为原料, 盐酸
羟胺为肟化试剂, 在 70 ! , pH = 6 1~ 6 2 反应制备 红霉素 A ( E) 肟。并 比较了 不同体 系对红 霉素 A ( E) 肟纯化的影响, 发现在二氯甲烷体系中进行纯 化处理, 纯化收率达 92% , 红霉素 A( E) 肟含量大于 90% ( HPL C) 。
摘 要: 以红霉素 A 为原料经肟化反应、贝克 曼( Beckman) 重 排反应、还原反应 和 N - 甲基化 还原反 应制备 大 环内 酯类药物阿奇霉素是一条合理的合成技术路线。本文着 重评述 了阿奇 霉素和 关键中间 体红霉 素 A ( E) 肟、红 霉素 A 6, 9 亚胺醚、9 脱氧 a 氮杂 9a 类红霉素 A 的合成方法, 提出今后阿奇霉 素及其关键 中间体合 成工艺研究 方 向和需解决的关键问题。
1999 年, 西班牙的 Immaculada 等人[ 18~ 19] 对各 种红霉素 A 肟盐的中和反应进行了详细研究, 认为 红霉素 A 肟盐碱化时适宜在芳香类溶剂 水的混合 溶液中进行, 碱化试剂为氨水、氢氧化钠等, 反应温 度 50 ! 左右, 终点 pH = 9 0~ 10 0。在此条件下, 红霉素 A 肟盐转化为红霉素 A 肟的收率为 93% ( 纯 度为 98 5% ) 。
2003 年, 邓志华等人[ 12] 经过系统 地考察反应 体系的 pH 值、反应温度以及盐酸羟胺用量等影响 因素, 提出了在缓冲体系中合成红霉素 A 肟, 收率 可达 95% 以上, 纯度 92% , 产品中红霉素 E 肟 红霉 素 Z 肟= 7 82 1( 82 9 10 6) 。
最近, 汪松美等人[ 13] 报道了以红霉素 A 为原 料, 在甲醇 三乙胺( pH = 6 4) 体系中与盐酸羟胺回 流反应 24 h, 红霉素 A 肟盐酸盐的收率为 90% 。 1 2 以红霉素 A 硫氰酸盐为原料
第 38卷 第 4 期 2009 年 4 月
化工技术与开发 T echnology & Development of Chemical Industry
Vol 38 No 4 Apr 2009
阿奇霉素及其关键中间体的合成研究进展
赵佳苗1, 施介华1, 2 , 许 超1
( 1. 浙江工业大学药学院, 浙江 杭州 310032; 2. 浙江工业大学绿色合成技术国家重点实验室培育基地, 浙江 杭州 310032)