primer BLAST在线设计引物扩增未知菌种
基于NCBI-primer blast的引物设计及验证

基于NCBI-primer blast的引物
设计及验证
流程
01 02引物的设计(已知模板设计引物)
引物的验证(已知引物查找模板/验证参数)
1-primer blast中引物的设计——模板
可以放序列:
如
也可以放NM号:
如NM_001289746.2
预期引物Tm 值:最适60,无特殊要求无需更改
预期产物大小:如80-150bp
数据库来源:根据模板设置,模板是
cDNA 就选mRNA
跨外显子设置:
RT-qPCR 定量引物为避免gDNA 干扰需设置跨外显子
物种种属设置:根据模板的种属设置
1-primer blast 中引物的设计——生成引物
生成引物:点击生成10对引物,可进行选择
最大扩增子设置:可不做更改,引物设计时前面已设置过产物大小
2-primer blast中已知引物的验证——只需放入引物序列
引物序列:
待验证引物序列放
于此处,注意引物
方向
2-primer blast中已知引物的验证——设置来源和种属
待验证引物来源数据库:
即是基于cDNA(mRNA反转)
的扩增引物,还是基于
gDNA的扩增引物;如定量
则肯定选mRNA
待验证引物来源种属:
即该引物扩增的模板的种属
注:如这两项未知,可先不做选择进行验证;如得不到完全匹配的结果,再进
行更改。
2-primer blast中已知引物的验证——进行验证
验证引物:
点击即可对上述输
入的引物进行验证,
可看到包括Tm值,
扩增子大小等引物
参数。
如何设计PCR扩增引物

如何设计PCR扩增引物1.找出这种细胞物种的PTN全长核苷酸序列2.采⽤primer premier 5.0软件设计引物设计应注意如下要点:● 1. 引物的长度⼀般为15-30 bp,常⽤的是18-27 bp,但不应⼤于38,因为过长会导致其延伸温度⼤于74℃,不适于Taq DNA聚合酶进⾏反应[2]。
● 2. 引物序列在模板内应当没有相似性较⾼,尤其是3’端相似性较⾼的序列,否则容易导致错配。
引物3’端出现3个以上的连续碱基,如GGG或CCC,也会使错误引发机率增加[2]。
● 3. 引物3’端的末位碱基对Taq酶的DNA合成效率有较⼤的影响。
不同的末位碱基在错配位置导致不同的扩增效率,末位碱基为A的错配效率明显⾼于其他3个碱基,因此应当避免在引物的3’端使⽤碱基A[3][4]。
另外,引物⼆聚体或发夹结构也可能导致PCR 反应失败。
5’端序列对PCR影响不太⼤,因此常⽤来引进修饰位点或标记物[2]。
● 4. 引物序列的GC含量⼀般为40-60%,过⾼或过低都不利于引发反应。
上下游引物的GC含量不能相差太⼤[2][5]。
● 5. 引物所对应模板位置序列的Tm值在72℃左右可使复性条件最佳。
Tm值的计算有多种⽅法,如按公式Tm=4(G+C)+2(A+T),在Oligo软件中使⽤的是最邻近法(the nearest neighbor method) [6][7]。
● 6. ΔG值是指DNA双链形成所需的⾃由能,该值反映了双链结构内部碱基对的相对稳定性。
应当选⽤3’端ΔG值较低(绝对值不超过9),⽽5’端和中间ΔG值相对较⾼的引物。
引物的3’端的ΔG值过⾼,容易在错配位点形成双链结构并引发DNA聚合反应[6]。
●7. 引物⼆聚体及发夹结构的能值过⾼(超过4.5kcal/mol)易导致产⽣引物⼆聚体带,并且降低引物有效浓度⽽使PCR反应不能正常进⾏[8]。
●8. 对引物的修饰⼀般是在5’端增加酶切位点,应根据下⼀步实验中要插⼊PCR产物的载体的相应序列⽽确定。
primer BLAST在线设计引物扩增未知菌种

进修生日志:primer BLAST在线设计引物扩增未知菌种兰会华1整理屈平华2审校1广西壮族自治区人民医院检验科,2广东省中医院检验科PCR的第一步就是引物设计。
引物设计的好坏,直接影响PCR的结果。
引物设计的软件很多,但是,多数时候是引物设计出来了,PCR也能扩增到目的条带,但非特异性的条带好几条,甚至比目的条带还要亮的,郁闷的有木有?再有,分离到一个新菌种,要扩增管家基因或蛋白基因,可引用参考文献的引物,却怎么也扩增不出来,泪奔的有木有?小编在这里给大家介绍利用细菌的全基因组序列,以primer BLAST在线设计引物和PCR扩增新菌种的方法,让您找到高大上的感觉。
一、实验目的:设计属特异性引物PCR扩增一未知菌弗朗西斯菌的sdhA基因二、引物设计过程(一)获得目的基因序列1、进入NCBI,点击gemone,搜索待扩增菌的属名Francisella2、点击search,结果得了相关菌种的全基因组序列。
2、点击其中的一个全基因组,如“广州弗朗西斯菌”4. 全基因组1658482 bp,这么大。
人海茫茫,该如何找到sdhA?这时,可使用网页搜索功能,按热键Ctrl+F,再输入要查找基因名称“sdhA”,如上图。
然后,就找到sdhA基因了。
请看,sdhA基因的位置在全基因组的第174351..176144之间哦(见下图)。
5. 再点击左侧的gene(上图),就得到了广州弗朗西斯菌1741bp的sdhA基因全序列,text文本保存。
(二)序列拼接,获得合并序列如果要扩增的是一个已知的菌种,如广州弗朗西斯菌,那么,只要得到上面的引物,你就可以直接去设计引物了。
但如果要扩增的是一个未知菌种呢?1个序列可不够;总之,你的序列得尽可能多,而且是完全不同的序列尽可能多;最好是弗朗西斯菌属内所有的已知的sdhA基因,甚至是与弗朗西斯菌亲缘关系相对较近的军团菌的sdhA基因当然,军团菌没有sdhA基因,这是后话。
实时定量PCR引物和探针设计操作步骤PrimerExpress软件

实时定量PCR引物和探针设计操作步骤Primer Express软件Primer Express 是实时定量PCR引物和探针设计的专用软件。
遵守以下三个原则有助于快速建立定量PCR反应体系:1.所有扩增按照同样的原则设计 (Primer Express);2.所有PCR反应在ABI PRISM ?7000/7900上使用同样的热循环条件;3.所有反应使用相同的PCR试剂。
引物和探针的设计原则下述原则的重要程度由上往下越来越低,请尽量满足编号靠前的条件。
它们中有的已经在Primer Expre软件中设置成缺省值,有的则需要在选择引物和探针时由设计者加以运用。
如果是设计SYBRGreen 引物,也要选择TaqMan Primer and Probe design并遵守这些规则,但是只需要合成引物就可以了。
TaqMan 探针:1. 保持G-C含量在30-80%之间。
2. 避免同一碱基重复过多。
特别是G,不可超过4个及以上。
3. 5' end不能是G。
4. 尽量使探针中的Cs多于Gs。
如果不能满足,则使用互补链上的探针。
5. 对于单探针反应,用Primer Express?软件计算出来的Tm值应当在68-70 °C 之间。
引物:1. 在探针确定以后再选择引物。
2. 引物要尽可能地接近探针,但是不要重叠。
3. 保持G-C含量在30-80%之间。
4. 避免同一碱基重复过多。
特别是G,不可超过4个及以上。
5. 用Primer Express?软件计算出来的Tm值应当在58-60 °C之间。
6. 3' end 的5个碱基中G and/or C碱基的总数不能超过2个。
实时TaqMan 引物和探针设计Begin by opening Primer Express and selecting "File", "New", and "TaqMan? Primer & Probe Design". The following screen will appear. You can close the TaqMan? Primer & Probe Data box as shown.输入或插入序列Import or paste a sequence into the window (Import shown). To paste a sequence from a Word or text file, first copy it to the clipboard. Be sure to only select the sequence (including numbers or annotations is OK); do not include extraneous information such as accession numbers etc. Next, select "Edit" and "Paste". The sequence will appear in the Sequence screen of Primer Express. Or, to Import a Sequence, click the "Import DNA File" button as shown. The software will then ask you to locate the sequence file. Select it from a folder, hard drive, disk, or desktop. Again, no annotations should be present in this sequence.A file is then imported after selecting the file location.保存输入的序列Select "File" and "Save" to give the sequence a name. This will be displayed in the File Name Box and will save the sequence in the Archive Folder.引物和探针设计参数Click the "Parameters" tab. This displays the Universal default parameters used to search for suitable TaqMan? primer & probe sets for real-time assays. It is strongly recommended that you do not adjust any of the parameters.引物和探针的排序及选择Primer Express is now ready to find Primers and Probes. Click the "Primers" tab, select "Options" and "Find Primers/Probes Now". The software will display the progress in the small window below the sequence.** Please disregard the "Optimal Primer Pairs Only" checkbox and the "Penalty" heading. By checking the Optimal Primer Pairs Only box, you will be severely limiting the range of your search, since the parameters it employs are not based on TaqMan? design guidelines. The Penalty score assigned to your Primer & Probe set is based on factors such as amplicon length. Since the default TaqMan? design parameters keep amplicons under 150 bp, this can be disregarded as well.Primer/probe sets will be listed when the search is complete. Scroll to the right to view the Probes. Click on the "Start" heading under probes to sort probes by sequence. This will group similar probes, simplifying the search.探针的选择Select a probe that is less than 30 bp in length and contains more C's than G's. The probes displayed are on the sense strand only. If the probes displayed do not have more C's than G's, then you will need to use the complement probe (as illustrated in this example). If you need to use the complement, make sure that the probe selected here does not have a C at the 3' end of the probe (otherwise, the complement will have a G at the 5' end ? whichis not allowed).The probe selected meets the first criteria above, but not the second (9 G's, 5 C's). Highlight this probe.Return to the sequence by clicking the "Sequence" tab.Lock in the probe sequence by clicking the Probe Button on the Tool Bar and highlight the probe sequence. The probe will turn green and be displayed in lower case when it is locked.引物选择Find compatible primers by returning to the "Primers" tab, selecting "Options" and "Find Primers & Probes Now". This will find new primer sets that will work with the probe you have selected. You can click on "Start" under Forward Primer to sort the displayed sequences.Search for a primer from the list displayed the meets the following criteria:1.No more than 2 G's and/or C's within the last 5 bases on the 3' end of the primer; and2.No runs of identical nucleotides, especially 4 or more G's.From the list of forward primers displayed, select a primer that has no more than 2 G's and/or C's within the last 5 bases on the 3' end of the primer. Highlight one of the primers that matches this criteria. If no forward primer matches this criteria then select a primer with 3 G's and/or C's. The example shown below matches the criteria and will serve as a suitable forward primer. Once you have selected the appropriate primer click on the "Sequence" tab to return to the Sequence window.Lock the forward primer by clicking the "Forward Primer" button on the toolbar, then highlighting the forward primer sequence. A blue arrow will be displayed under the forward primer showing that it is locked.Click on the "Primers" tab and perform a new search. Scroll to the Reverse Primers displayed and select a reverse primer following the same criteria for forward primer selection (G/C rule on the 3' end of primer).Return to the Sequence page and lock in on the Reverse Primer using the Reverse Primer Tool.This now displays the primers and probe you have selected. Return to the Primers tab and perform one final search to display your results.保存搜索结果Click on "Save List" at the bottom of the screen to save your selection in a tab delimitedformat. Click "Order" to generate an editable/printable text file of your sequences:互补探针的选择In the example above, you must use the complementary probe so as to insure that the probe has more C's than G's. Remember, the probe you use cannot have a G at the 5' end, thus the sense probe used for this search cannot have a C at the 3' end.In order to generate the probe complement, return to the Sequence screen. Highlight the probe sequence, select "Edit", and "Copy Complement". You will not see the complementary sequence at this point; it is copied to the clipboard:Return to the Order window and "Paste" the complement in this window, overwriting the probe displayed. You have the option of editing the primer/probe names, and adding the reporter/quencher dyes to the probe sequence.This document can now be saved and put into a Word document or attached to an e-mail message.在Results Archive中保存搜索结果Your search can also be saved in the Results Archive Folder. Click on the "Results" tab. The forward and reverse primers are displayed in their respective boxes, and the probe sequence is displayed in the "Cycle Params" box The probe sequence displayed is the original strand. To view/save the complementary strand, highlight the probe from the Sequence and select "Copy Complement". "Paste" the complement probe into the "Cycle Params". The complementary probe strand is now displayed. It is important to note that if you leave the Results page, the probe sequence will default back to the original. Each time you returnto the Results page you will need to re-paste the complementary probe strand. Note: The information displayed below the selected primer and probe sequences should be ignored when performing TaqMan Assays. The Universal TaqMan? Guidelines do not require you to perform optimizations, thus, the cycling/concentration, etc. information displayed here can be ignored. Save the Results by selecting "Save Results". A message will display showing the results were saved.打印结果 To print the Results, select "Open Results" from the "File" menu. The last (newest) results file will be the last one in the list (at the bottom of the list): Highlight and click "Open".This is the relevant information needed to order your primer/probe set. To print, click and drag, highlighting the information you want and selecting "Copy" from the "Edit" menu, placing it on the clipboard. This should be everything from the Sequence name through the TaqMan? probe annealing information.This is the relevant information needed to order your primer/probe set. To print, click and drag, highlighting the information you want and selecting "Copy" from the "Edit" menu, placing it on the clipboard. This should be everything from the Sequence name through the TaqMan? probe annealing information.You can then paste your sequence information in to a Word document; from here you can print a copy for your records.订购信息Be sure to include information on your needed synthesis scale and the corresponding part number, your reporter dye(s), your quencher (TAMRA), and your personal information (name, institution, address, phone fax etc.).。
Primer-BLAST-NCBI 的引物设计和特异性检验工具

Primer-BLAST:NCBI的引物设计和特异性检验工具一、Primer-BLAST介绍Primer-BLAST,在线设计用于聚合酶链反应(PCR)的特异性寡核苷酸引物。
Primer-Blast可以直接从Blast主页(/)找到,或是直接用下面的链接进入:/tools/primer-blast/这个工具整合了目前流行的Primer3软件,再加上NCBI的Blast进行引物特异性的验证。
Primer-BLAST免除了用另一个站点或工具设计引物的步骤,设计好的引物程序直接用Blast进行引物特异性验证。
并且,Primer-BLAST能设计出只扩增某一特定剪接变异体基因的引物。
Primer-BLAST有许多改进的功能,这样在选择引物方面比单个的用Primer3和NCBI BLAST更加准确。
二、Primer-BLAST的输入Primer-BLAST界面包括了Primer3和BLAST的功能。
提交的界面主要包括三个部分:target template(模板区), the primers(引物区), 和specificity check(特异性验证区)。
跟其它的BLAST 一样,点击底部的“Advanced parameters”有更多的参数设置。
(1)模板(Template)在“PCR Template”下面的文本框,输入目标模板的序列,FASTA格式或直接用Accession Number。
如果你在这里输入了序列,是用于引物的设计。
Primer-BLAST就会根据你输入的序列设计特异性引物,并且在目标数据库(在specificity check区选择)是唯一的。
(2)引物(Primers)如果你已经设计好了引物,要拿来验证引物的好坏。
可以在Primer Parameters区填入你的一条或一对引物。
并且选择好验证的目标数据库(在specificity check区选择)。
根据需要可设置产物的大小,Tm值等。
Primer-Blast在线引物设计工具

Primer-Blast:在线引物设计工具生物信息学引物设计用于PCR 聚合酶链式反应⏹PCR:Polymerase Chain Reaction⏹PCR 是一种用于放大扩增特定的DNA片段的分子生物学技术,它可看作是生物体外的特殊DNA复制,PCR的最大特点,是能将微量的DNA大幅增加。
Why PCR?⏹在某个特定物种内1.qPCR用于测定mRNA表达量:mRNA/cDNA水平2.基因克隆:基因水平——引物设计的要求:引物在该基因组内或cDNA库中具备特异性⏹在一个已知基因序列的基因组A内设计引物,然后在另一个还未基因注释的(近缘)基因组B内扩增引物——引物设计的要求:(1)引物在该A基因组内或cDNA 库中具备特异性,并且(2)引物在A和B内是保守的,所以设计的引物在A基因组内往往处于基因的编码区域(编码区域在不同物种间比非编码区更保守)How to do PCR?变性退火延伸How: 引物设计原则⏹退火温度(Tm):两个引物的Tm值相差不能大于5℃,扩增产物与引物的Tm值相差不能大于10℃决定引物退火温度Tm值的最主要因素是引物长度Tm = 4*(G+C)+ 2*(A+T)⏹碱基组成(G+C含量):40%~60%,4种碱基要分布均匀⏹引物长度⏹不能有大于3bp的方向重复序列或自身互补序列存在⏹一个引物的3’末端序列不能结合到另一个引物的任何位点上⏹不要有局部的GC rich或AT rich(特别是3’端),避开T/C或A/G的连续结构较基础全面的引物设计解说Primer Design/GenWeb/Molecular/seq_anal/primer_design/primer_design.htm常用引物设计软件⏹Primer Premier 6 (P6)⏹Beacon Designer 8NCBI Primer-Blasthttps:///tools/primer-blast/index.cgi?LINK_LOC=BlastHome⏹NCBI Primer-Blast: Finding primers specific to your PCR template (using Primer3 and BLAST)⏹能在线设计引物,并验证设计好的引物。
qPCR引物设计原则及具体操作步骤
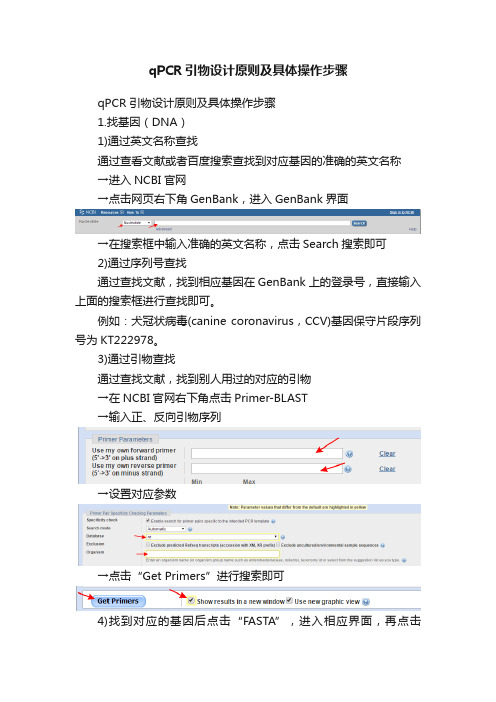
qPCR引物设计原则及具体操作步骤qPCR引物设计原则及具体操作步骤1.找基因(DNA)1)通过英文名称查找通过查看文献或者百度搜索查找到对应基因的准确的英文名称→进入NCBI官网→点击网页右下角GenBank,进入GenBank界面→在搜索框中输入准确的英文名称,点击Search搜索即可2)通过序列号查找通过查找文献,找到相应基因在GenBank上的登录号,直接输入上面的搜索框进行查找即可。
例如:犬冠状病毒(canine coronavirus,CCV)基因保守片段序列号为KT222978。
3)通过引物查找通过查找文献,找到别人用过的对应的引物→在NCBI官网右下角点击Primer-BLAST→输入正、反向引物序列→设置对应参数→点击“Get Primers”进行搜索即可4)找到对应的基因后点击“FASTA”,进入相应界面,再点击“Send to”选择相应格式,保存序列。
2.qPCR引物和TaqMan探针的设计1)引物设计注意事项a)引物长度17bp-25bp为佳。
太短的引物容易导致扩增效率降低;太长的引物会导致出现引物高级结构的几率增加。
两者都会干扰定量结果的准确性b)扩增片段长度为:90-150 bp(最低不能超过70,最高不能超过180)c)引物的Tm值为:最小57℃,最大63℃,最适为60℃,两条引物之间退火温度得差距不超过1℃,推荐使用Primer Premier 5进行Tm值计算;d)引物A、G、C、T整体分布尽量要均匀,避免使用GC或者TA 含量高的区域,尤其是3’端,必须避开GC含量不均匀的区域。
e)引物设计时请尽量避开TC或者AG的连续结构。
f)3’端不能超过3个以上碱基互补,自互补碱基数不超过3;3’端最后一个碱基绝对不能搭上g)特异性要有保证,与非特异模板3’端互搭碱基数不超过3,不连续出现4个及以上的GC互搭h)引物3’端最后五个碱基不能包含超过2个以上的G或者Ci)引物的GC含量控制在40%-60%之间为好,最佳为45%-55%之间j)正向或者反向引物应尽量接近探针序列但是不能和探针序列有重合区域k)在Primer-BLAST设计时,在Organism 处选择相应物种l)需跨外显子设计,避免基因组污染2)TaqMan探针设计指南a)探针序列应尽量接近正向或者反向引物,但是不能与之有重合区域;一般相隔1~5个碱基(一般10个以内,最好是1个碱基)。
NCBI在线Blast的图文说明

NCBI在线Blast的图文说明Blast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST 结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP 是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN 是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线blast:/Blast.cgi1、进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
NCBI在线blast页面2、粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
NCBI在线blast页面3、blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
blast参数设置4、注意一下你输入的序列长度。
图解blast验证引物教程
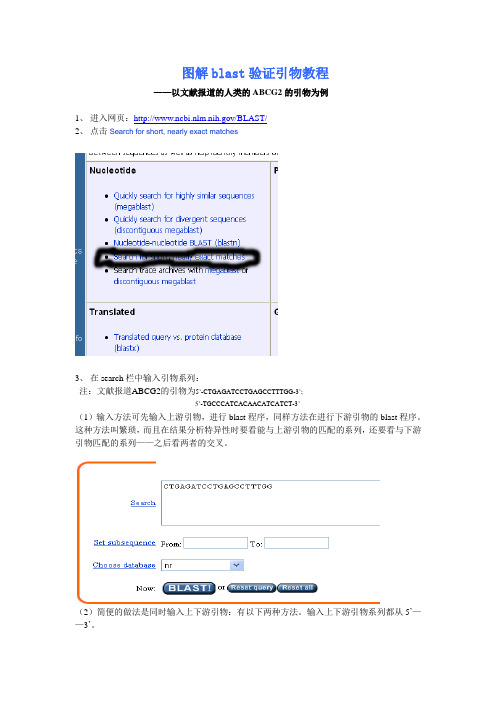
图解blast验证引物教程——以文献报道的人类的ABCG2的引物为例1、进入网页:/BLAST/2、点击Search for short, nearly exact matches3、在search栏中输入引物系列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物B、输入上游引物回车输入下游引物4、在options for advanced blasting中:select from 栏通过菜单选择Homo sapiens【ORGN】Expect后面的数字改为105、在format中:select from 栏通过菜单选择Homo sapiens【ORGN】Expect后面的数字填上0 106、点击网页中最下面的“BLAST!”7、出现新的网页,点击Format!8、等待若干秒之后,出现results of BLAST的网页。
该网页用三种形式来显示blast的结果。
(1)图形格式:图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:从左到右分别为:A、数据库系列的身份证:点击之后可以获得该序列的信息B、系列的简单描述C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
blast引物设计流程

blast引物设计流程Blast引物设计是基因组、转录组和蛋白质组研究中不可或缺的一环。
BLAST(基本局部序列比对工具)是用于在数据库中相似序列的一种工具。
它能够在数据库中找到一条或多条与查询序列相似的序列,并计算相似度分数。
利用BLAST工具进行引物设计可以帮助研究人员快速鉴定和选择目标基因、转录本或蛋白质,并进行后续实验。
下面将详细介绍BLAST引物设计的流程。
1.确定目标序列:确定要设计引物的目标序列,可以是基因组、转录组或蛋白质组中的一个特定基因、转录本或蛋白质。
2. 数据库选择:选择适当的数据库进行BLAST。
根据实际需要,可以选择不同的数据库,包括NCBI的GenBank、RefSeq、EST、非冗余蛋白质数据库等。
3.引物设计参数:根据实验需求和目标序列的特点,设置合适的引物设计参数。
参数包括引物长度、引物Tm值、引物GC含量、引物之间的距离等。
4. 引物设计工具选择:选择合适的引物设计工具进行设计。
目前有许多在线工具和软件可以进行BLAST引物设计,例如Primer-BLAST、Primer3、Geneious等。
5.引物设计:根据设置的参数和目标序列,使用选择的引物设计工具进行引物设计。
工具通常会生成多个潜在的引物序列,根据设计要求和实验条件进行筛选。
6.引物特性分析:对设计的引物进行分析,包括引物的Tm值、互补性、二聚体形成、酶切位点等。
高Tm值可增加引物与目标序列的稳定性,互补性较低可以避免二聚体的形成,酶切位点可能会影响后续实验的结果。
7.引物合成:选择合适的引物合成厂商进行引物合成。
根据实验需求,可以选择合成标记有荧光染料或其他分子的引物。
8.引物验证:将合成的引物进行验证,可以通过聚合酶链反应(PCR)或其他测序技术进行验证。
验证引物的特异性和效率,并根据需要进行优化。
9.实验应用:将验证通过的引物应用于实验中,如PCR扩增、基因表达分析、基因组重组等。
BLAST引物设计的流程如上所述,通过合理设置参数、使用适当的工具和对引物进行验证,在基因组、转录组和蛋白质组研究中可以选择出最合适的引物。
生物软件应用primer引物设计方法

引物设计软件Premier Primer 5学校专业姓名学号“Premier Primer 5”软件由加拿大的Premier公司开发的专业用于PCR或测序引物以及杂交探针的设计,评估的软件,其主要界面是分为序列编辑窗口(Genetank),引物设计窗口(Primer Design),酶切分析窗口(Restriction Sites)和纹基分析窗口(Motif)。
引物自动搜索可采用“Premier Primer 5”软件,引物设计的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。
Primer Premier 5.0软件可以用来做1、引物设计2、限制性内切酶位点分析3、DNA 基元(motif)查找4、同源性分析,这篇论文我们主要做引物设计,因为后三项有其专业化软件,而且它们不是“Premier Primer 5”软件的强项。
目的:根据松鼠的核酸序列设计PCR引物设计,扩增模板DNA序列。
首先在百度中搜索松鼠的拉丁名,放入NCBI的搜索框中,选择Nucleotide。
中文学名:松鼠拉丁学名:Sciurus vulgaris将序列复制到记事本上程序运行打开软件Premier Primer 5,点击file→open→DNA Sequence接着出现加载序列Preimer Premier 启动界面加载file点击接着点击出现点击,进入引物设计窗口点击,对下图的各种参数可以进行设置将DNA的双螺旋结构翻译成蛋白质序列,或者蛋白质翻译成DNA序列名称Primer的有用信息正义链或反义链基序分析酶切分析序列比对接着点击[如果选择按钮则出现下图,可以改变下图参数,经过多次选择,选High 时,Rating 最高。
]引物类型搜索模式5’引物位置范围3’引物位置范围产物大小范围引物长度如果想要False Priming也有参数,我们可以通过点击这个图上面的那个图片中的,点击下图的False Priming,上图的False Priming会出现参数。
blast引物设计流程

blast引物设计流程BLAST (Basic Local Alignment Search Tool) 引物设计是分子生物学研究中一个非常重要的步骤。
它用于通过比对已知的DNA或RNA序列来选择特定区域的引物,以进行PCR(聚合酶链式反应)、RT-qPCR(逆转录定量聚合酶链式反应)和荧光原位杂交等实验技术。
在本文中,我们将详细介绍设计BLAST 引物的流程。
1.收集目标序列数据:首先,我们需要收集目标序列的数据。
目标序列可以是基因序列、mRNA序列或其他DNA/RNA序列。
这些数据可以从公共数据库(如GenBank)或实验室内部的数据库获得。
2.确定引物长度:下一步是确定引物的长度。
通常,引物的长度在18到25个碱基对之间,相对长度和GC含量对于PCR引物尤为重要。
3.构建BLAST数据库:在设计引物之前,我们需要构建一个BLAST数据库。
选择适当的引物长度和需要比对的目标序列,将这些序列导入数据库中。
BLAST数据库可以通过使用NCBI(National Center for Biotechnology Information)或其他Bioinformatics软件来构建。
4.查询引物序列:使用BLAST软件,将具有已收集的目标序列信息的引物序列输入到BLAST数据库中。
BLAST会在数据库中寻找与引物序列相似的序列。
基于比对结果,可以评估引物的特异性和亲合性。
5.分析BLAST结果:根据BLAST比对的结果,需要对引物进行评估和筛选。
主要考虑以下几个方面:-特异性:引物应该与目标序列非常特异性地结合,而不会与其他非靶DNA或RNA结合。
特异性可以通过比对结果中的E值(期望值)和匹配长度来评估。
-互补性:引物应与目标序列互补,以便正确结合并形成PCR产物。
通过比对结果来评估引物序列与目标序列的互补性。
- 引物结构: 引物应具有适当的物理参数,如长度、GC含量和熔解温度等。
这些特征可以使用Bioinformatics工具来评估和优化。
PrimerBLAST操作说明

引物验证与实验
将筛选出的引物序列导出,进行合成和实验 验证。
在实验过程中,注意引物的特异性、灵敏度 、扩增效率和产物纯度等方面的检测和评估
。
03 PrimerBLAST参数设置
引物长度
总结词
引物长度是影响PCR扩增效率和特异性的重 要因素。
详细描述
引物长度过短可能降低扩增效率和特异性, 而长度过长可能导致引物与模板的结合能力 下降,同样影响扩增效果。通常,引物长度 在18-30个核苷酸之间较为适宜,但具体长 度应根据实验需求和引物设计原则进行选择。
详细描述
非特异性扩增通常在较低的温度下发生。通过提高PCR反 应温度,可以降低非特异性扩增的可能性。然而,过高的 温度可能导致引物结合效率降低,因此需要根据实际情况 选择一个合适的温度。
总结词
控制引物浓度
总结词
优化PCR循环参数
详细描述
如前所述,PCR循环参数的优化有助于提高引物的扩增效 率,同时也可以减少非特异性扩增的可能性。通过梯度 PCR或逐轮PCR的方法,可以找到最佳的循环参数组合。
引物特异性分析
引物特异性分析
PrimerBLAST会通过比对引物序列和基因序列,评估引 物的特异性。如果引物在基因序列上有多个匹配位点, 则可能导致非特异性扩增。用户可以通过查看比对结果 ,了解引物的特异性情况。
引物特异性验证
为了进一步验证引物的特异性,PrimerBLAST提供了基 于PCR实验的引物特异性验证功能。用户可以将筛选得 到的引物送至指定的实验室进行实验验证,以确认引物 的特异性。
综合的在线工具
PrimerBLAST是一个综合的在线 工具,用于设计PCR引物并评估 其潜在的基因组结合位点。
结合多种功能
blast验证引物

blast验证引物
一、概述
BLAST是Basic Local Alignment Search Tool 的英文缩写,意即碱基局部对准检索工具,是一种序 列类似性检索工具。它采用统计学记分系统,能将真 正配对的序列同随机产生的干扰序列区别开来;同时 采用启发式算法系统,即采用的是局部对准算法 (Local Alignment Algorithm),而不是全序列对准算 法(Global Alignment Algorithm)。
特殊blast
先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻 译成蛋白质序列,然后再将两种翻译结果从蛋白质水平进行查询。
ATGTTGGGGAAATGCTTGACC
引物序 列输入 窗口 数据库的选择 在此,我们选择 第三个数据库 输入方法可先输入上游引物,进行blast程序,同样 方法在进行下游引物的blast程序。在结果分析特异 性时要看能与上游引物的匹配的系列,还要看与下 游引物匹配的系列——之后看两者的交叉。 显示结 果在新 的窗口
(3)结果详细信息
序列的信息
上游引物与该序列的正链【Plus/Plus】的 匹配情况: 共有21个碱基匹配,得分42.1分【21×2+ 0.1=42.1】,E值为0.014 上游引物与序列的1~21位点匹配
结果判断: ①验证文献报道的引物是否正确:如果你可以在所显示 的结果中找出你的目的基因,一般说明你的引物正确性 没问题。如果你blast后没有发现你的目的基因,或者分 值很低,该引物就可能不适合用 ②检测该对引物是否可与其它序列匹配,引起PCR的非 特异性扩增。如果找到了你的目的基因名称,而且找到 了一大批同物种的不同基因,(上下游引物分别搜索到 相同的基因),而且分数也较高。这时表明你的引物设 计的特异性不高,极有可能在你的扩增产物中出现非特 异性产物。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
进修生日志:primer BLAST在线设计引物扩增未知菌种
兰会华1整理屈平华2审校
1广西壮族自治区人民医院检验科,
2广东省中医院检验科
PCR的第一步就是引物设计。
引物设计的好坏,直接影响PCR的结果。
引物设计的软件很多,但是,多数时候是引物设计出来了,PCR也能扩增到目的条带,但非特异性的条带好几条,甚至比目的条带还要亮的,郁闷的有木有?再有,分离到一个新菌种,要扩增管家基因或蛋白基因,可引用参考文献的引物,却怎么也扩增不出来,泪奔的有木有?小编在这里给大家介绍利用细菌的全基因组序列,以primer BLAST在线设计引物和PCR扩增新菌种的方法,让您找到高大上的感觉。
一、实验目的:设计属特异性引物PCR扩增一未知菌弗朗西斯菌的sdhA基因
二、引物设计过程
(一)获得目的基因序列
1、进入NCBI,点击gemone,搜索待扩增菌的属名Francisella
2、点击search,结果得了相关菌种的全基因组序列。
2、点击其中的一个全基因组,如“广州弗朗西斯菌”
4. 全基因组1658482 bp,这么大。
人海茫茫,该如何找到sdhA?
这时,可使用网页搜索功能,按热键Ctrl+F,再输入要查找基因名称“sdhA”,如上图。
然后,就找到sdhA基因了。
请看,sdhA基因的位置在全基因组的第174351..176144之间哦(见下图)。
5. 再点击左侧的gene(上图),就得到了广州弗朗西斯菌1741bp的sdhA基因全序列,text文本保存。
(二)序列拼接,获得合并序列
如果要扩增的是一个已知的菌种,如广州弗朗西斯菌,那么,只要得到上面的引物,你就可以直接去设计引物了。
但如果要扩增的是一个未知菌种呢?1个序列可不够;总之,你的序列得尽可能多,而且是完全不同的序列尽可能多;最好是弗朗西斯菌属内所有的已知的sdhA基因,甚至是与弗朗西斯菌亲缘关系相对较近的军团菌的sdhA基因当然,军团菌没有sdhA基因,这是后话。
小编最终只找了弗朗西斯菌属内8个sdhA基因。
DNAMAN软件进行了序列拼接,Sequence,Sequence Assembly,加载序列文件。
当然,为了防止序列差异过大,应该适当修改参数,如Identity 90%,应该修改为80%,甚至70%。
拼接后的序列,有除A、G、C、T外,还有Y、R、W等兼并碱基什么的。
对了,要的就是这个结果。
(三)primer BLAST 设计引物
1、百度搜索Blast网站,或网页直接打开/Blast.cgi,点击Primer-BLAST进入引物设计。
2、在把目的序列粘贴到框内。
3、引物的特异性验证的参数设置:primer BLAST 设计引物的最大优势,就是选择物种名称进行引物的特异性验证。
引物的特异性验证的参数设置方法见上面,根据我们的目的,在Database一栏,选择了Genome(chromosomes from all organisms),在Organism一栏中,选择了Francisella(见下图)。
4、点击“Get Primers”按钮,将显示所有与含目的基因序列相近的全基因组和菌株。
鉴于我们的目的是扩增一未知菌弗朗西斯菌的sdhA基因,为PCR增加成功的可能性,自然是勾选“All”(见下图)。
5、然后,然后,引物就设计好了……给我们设计了10条呢!
看看引物的特异性吧,土拉弗朗西斯菌(Francisella tularensis)挺不错,引物序列完全匹配,蜃楼弗朗西斯菌(Francisella philomiragia)就不那么乐观了,当初真应该把全部48个已公布全基因组菌种的sdhA基因都找出来啊。
三、实验结果
第1次的引物设计不太完美,但意想不到的是,居然还是成功地扩增到所需的目的片段,经基因测序验证,也确实是sdhA基因。
只能说,我们太幸运了。
四、小结:
设计PCR引物扩增一个已知菌种且已知序列的靶基因,应该没什么难度。
但如果要设计一个属特异性的引物,用于该属未知菌种的PCR扩增,还是需要那么一点点技巧和运气。
因为引物设计受靶基因序列的长度、碱基排列顺序和不同菌种序列的差异程度等多种因素的影响,所以并不是每一个靶基因都能设计出能扩增整个菌属的属特异性的引物。
故有时候,我们需要退而求其次,要么换一个更保守一点的基因(如管家基因)作为靶基因;要么找一个或几个与待测菌种亲缘关系接近菌种的靶序列来进行设计引物;只求能扩增到靶基因,而不必追求能设计出适合属内所有菌种的属特异性引物。
在我们的这些引物设计中,虽然在线的特异性验证结果并不完美,但最终仍然成功地扩增到靶基因,主要原因在于,该属内的多个菌种已经测序了全基因组,并且,与待菌株亲缘关系密切的广州弗朗西斯菌也测序了全基因组。
五、参考文献:
Ye J, Coulouris G, Zaretskaya I, et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction.[J]. Bmc Bioinformatics, 2012, 13(6):: 134.。