黄曲霉毒素检验方法
黄曲霉毒素国标检测方法

黄曲霉毒素国标检测方法
黄曲霉毒素(AF)是一种由黄曲霉属真菌产生的有毒代谢产物。
它们被广泛分布在食品、饲料和环境中,可能对人类和动物健康造成危害。
因此,对黄曲霉毒素的快速、准确检测成为了保障公众健康的关键。
国家标准委员会制定了黄曲霉毒素国标检测方法,以保证各个检验机构的检测质量和结果一致性。
1. 检测样品的制备
样品制备过程是检测过程的关键步骤。
样品必须被精确称量和混合,以确保最终检测结果的准确性和可重复性。
样品预处理的方法通常包括研磨、切碎、混合和抽样等步骤,具体方法取决于样品的特性,如水分含量、粘度和粒度分布等。
2. 样品提取
样品提取过程中,黄曲霉毒素从样品中被提取到适当的溶剂中。
提取的选择取决于样品的特性和黄曲霉毒素的特性。
一些常见的提取方法包括回收萃取、固相萃取和羧甲基纤维素柱萃取等。
3. 分离和净化
为了获得最终准确的结果,必须分离并净化提取的样品。
对于黄曲霉毒素的分离和纯化方法主要有和阳离子交换树脂和固相萃取等方法。
检测黄曲霉毒素的方法主要包括色谱检测法、酶联免疫检测法、毛细管电泳等方法。
其中酶联免疫检测法已成为广泛使用的检测方法之一。
该方法以特异性抗体为基础,利用抗原抗体相互作用,快速、敏感地检测黄曲霉毒素。
总的来说,黄曲霉毒素国标检测方法是一系列高效准确的方法,可以帮助检验机构检测食品或饲料中的黄曲霉毒素,保证公众健康。
黄曲霉毒素实验设计

ELISA试验精密度评价
2.精密度评价的内容:批内、批间、日内、 日间和总不精密度(日内+日间) 批内:在相同条件下对同一标本在尽可 能短的时间内进行多次重复测定 3.精密度评价实验 对稳定样品(低、中、高三个水平)重 复20次测定A,计算A的均值、s、S2和CV
ELISA试验临界值确定
1.临界值 实验结果处于(阴、阳性)分界点时的 样品中分析物浓度值 1.1当样品中分析物浓度处于临界值时,用 定性试验对样品进行多次重复检测,将产 生50%的阳性结果和50%的阴性结果
ELISA试验精密度评价
1.精密度
在相同条件下,对同一样品多次测量, 每次测量结果之间的接近程度 1.1不能用数字表示 1.2可数字表示的是其反义概念:不精密度 用标准差(s)或变异系数(CV)表示
ELISA试验精密度评价
1.精密度 1.3分析过程重复性的指标 1.4每个标本只做一次检验就发出结果 报告的重要误差(随机误差)来源 1.5临床上希望同一标本的多次检测结 果的精密度好,以减小随机误差
ELISA试验临界值确定
2.确立实验方法的临界值 2.1对已知浓度阳性样品进行系列稀释 2.2对各稀释度样品进行多次重复检测
2.3阴性和阳性结果各占50%的样品中 分析物浓度即为实验方法的临界值
实ห้องสมุดไป่ตู้设计的内容
1.实验原理 2.实验材料:仪器、试剂、耗材、标本等 3.实验方法:文条或表格(详细、具体) 4.实验结果判断 5.实验结果计算及统计学处理 6.室内质量控制 7.注意事项 8.影响因素
酶联免疫法(ELISA)检测饲料中黄曲霉毒素B1

15 mL分 液漏 斗 中 , 入 2 L三氯 甲烷 , 塞 2 加 0m 加
轻摇 3ri, 置 分层 。放 出下 层 三氯 甲烷 层 , n静 a 经 盛 有约 5 g 先用 三氯 甲烷湿 润 的无水 硫酸钠 过 预
滤 器 于 10 m 0 L蒸 发 皿 中 。再 加 5 mL三 氯 甲烷
恒 温培 养箱 、 量移 液器 、u : e型 酶标 微 S ns R e
仪( 天美)涡漩 式振荡 器 。 上海 、
一
1— 8
维普资讯
江西 饲料
2 o 第 6期 o 6年
于分 液 漏 斗 中 。 复振 摇 提 取 , 重 三氯 甲烷 层 一 并 滤 于 蒸 发 皿 中 。最 后 用 少 量 三 氯 甲烷 洗 涤 过 滤 器, 洗液 并 于蒸发 皿 中 ,5 6 ℃水 浴通 风挥 干 。挥 干 冷 却后 , 准确 加入 1. mL甲醇 水(: , 00 11 将蒸 发 皿 ) 中的凝 结物 充分 溶解 。
1 方法原 理
架 。每瓶酶标抗 原 中准确 加入 1 L酶标抗 原稀 .m 5 释液 。 充分溶解 , 配成 实验用酶标抗 原溶 液 ,— 28℃ 保存 。浓缩洗涤液用 30mL 0 蒸馏水配制成洗涤液。
3 样 品的制 备
31 一般 饲料 .
称 取 50 g样 品 于 10 . 0 mL具 塞 三 角 瓶 中 , 准 确 加 入 2. m 5 L甲醇 水(:) 液 , 荡 1 n 0 11溶 振 5mi,
甲醇 、 蒸馏 水 、 氯 甲烷 、 三 无水 硫 酸钠 、 曲 黄 霉毒 素 B 测试 盒 。 l
黄曲霉毒素 B 测试盒的组成 :包被抗体 的 l
反应 板 、 品稀释 液 、 F 样 A B标 准溶液 ( .,., , , 0105 15
2010版药典黄曲霉毒素检验法

实用标准文案2010版药典黄曲霉毒素检验法(文字版)附录Ⅸ V 黄曲霉毒素测定法本法系用高效液相色谱法(附录ⅥD)侧定药材饮片剂制剂中的黄曲霉毒素(以黄曲霉毒B1、黄曲霉毒索B2、黄曲霉毒素G1和黄曲霉毒素G2总量计),除另有规定外,按下列方法测定。
色谱条件与系统适用性试验以十八烷基硅硅烷键合硅胶为填充剂;以甲醇-乙腈-水(10:18:42)为流动相,流速每分钟0.8ml;采用柱后衍生法检测。
衍生溶液为0.05%的碘溶液(取碘0.5g,加人甲醇100ml使溶解,用谁稀释至1000ml制成),衍生化泵流速每挣钟0.3ml 衍生化温度70℃;以荧光检测器检测,激发波长λex=360nm(或365nm),发射波长λem=450nm。
两个相邻色谱峰的分离度应大于1.5。
混合对照品溶液的制备精密量取黄曲霉毒素混和标准品(黄曲霉毒B1、黄曲霉毒索B2、黄曲霉毒素G1、黄曲霉毒素G2标示浓度分别为1.0ug/ml、0.3ug/ml、1.0ug/ml、0.3ug/ml)0.5ml,置10ml量瓶中,用甲醇稀释至刻度,作为储备液。
精密量取储备液1ml,置25ml量瓶中,用甲醇稀释至刻度,即得。
供试品溶液的制备取供试品粉末约15g(过二号筛),精密称定,加氯化钠3g,置于均质瓶中,精密加入70%甲醇溶液75ml,高速搅拌2分钟(搅拌速度大于11000转/分钟),离心5分钟(离心速度2500转/分钟),精密量取上清液15ml,置50ml量瓶中,用水稀释至刻度,摇匀,用微孔滤膜(0.45um)滤过,量取续滤液20.0ml,通过免疫亲和柱(AflaT-est@P),流速每分钟3ml,用水20ml洗脱,洗脱液弃去,使空气进入柱子,将水挤出柱子,再用适量甲醇洗脱,收集洗脱液,置2ml量瓶中,并用甲醇稀释至刻度,摇匀。
即得。
测定法分别精密吸取上述混和对照品溶液5ul、10ul、15ul、20ul、25ul,诸如液相色谱仪,测定峰面积,以峰面积为纵坐标,进样量为横坐标,绘制标准曲线,另精密吸取上述供试品溶液20-25ul,注入液相色谱仪,测定峰面积,从标准曲线上读出供试品中相当于黄曲霉毒素B1、黄曲霉毒索B2、黄曲霉毒素G1、黄曲霉毒素G2的量,计算,即得。
糕点-黄曲霉毒素B1 的测定-薄层层析法

准确称取 1~1.2mg 黄曲霉毒素 B1 标准品,先加入 2mL 乙腈溶解,再用苯稀释定容至 100mL, 其浓度约为 10¦Ìg/mL,此标准溶液应通过紫外分光光度法测定其纯度,并以溶剂调整其浓度为 10¦Ìg/mL,避光,置于 4℃冰箱中保存。 3.9 黄曲霉毒素标准使用液,0.04¦Ìg/mL
3
将薄层板边缘附着的吸附剂刮净,在距薄层板下端 3cm 的基线上用微量注射器或血色素吸 管滴加样液。一块板可滴加 4 个点,点距边缘和间距约为 1cm,点直径约为 3mm。在同一板上滴 加点的大小应一致,滴加时可用吹风机用冷风边吹边加。滴加样式如下:
第一点:10μL 黄曲霉毒素 B1 标准使用液(0.04¦Ìg/mL).。 第二点:20μL 样液。 第三点:20μL 样液+10μL 0.04¦Ìg/mL 黄曲霉毒素 B1 标准使用液。 第四点:20μL 样液+10μL 0.2¦Ìg/mL 黄曲霉毒素 B1 标准使用液。 6.2.2 展开与观察 在展开槽内加 10mL 无水乙醚,预展 12cm,取出挥干。再于另一展开槽内加 10mL 丙酮- 三氯甲烷混合液(8+92),展开 10~12cm,取出。在紫外光下观察结果,方法如下。 由于样液点上加滴黄曲霉毒素 B1 标准使用液,可使黄曲霉毒素 B1 标准点与样液中的黄曲霉 毒素 B1 荧光点重叠。如样液为阴性,薄层板上的第三点中黄曲霉毒素 B1 为 0.0004¦Ìg,可用作检查 在样液内黄曲霉毒素 B1 最低检出量是否正常出现;如为阳性,则起定性作用。薄层板上的第四 点中黄曲霉毒素 B1 为 0.002¦Ìg,主要起定位作用。
饲料中黄曲霉毒素B1的检测

中国果菜质量控制黄曲霉毒素B1(AFB1)是二氢呋喃氧杂萘邻酮衍生物,含有一个双呋喃环和一个氧杂萘邻酮,是已知的化学物质中致癌性最强的一种。
AFB1污染的物质包括食品中的花生、玉米、稻谷、小麦、花生油等农产品及其加工产品;且以南方高温、高湿地区受污染最为严重。
黄曲霉毒素无臭、无味、无色,在饲料中及其稳定,对家禽、家畜的健康威胁极大,作为饲料强制性卫生监控指标,目前有半定量薄层色谱法[1]、免疫亲和柱净化———高效液相色谱法[2]、免疫亲和荧光光度法[3]、酶联免疫吸附法[4]。
高效液相色谱法虽然灵敏度高,但是样品前处理繁琐,操作复杂;薄层色谱法虽然简便,但是灵敏度差,因此研究能够准确、快速检测饲料中AFB1的检测方法是十分必要且具有现实意义的。
1实验部分1.1材料与试剂甲酸,优级纯;乙腈,色谱纯;超纯水;饲料中黄曲霉毒素B1的检测刘超景赞(乐山市食品药品检验检测中心,四川乐山614000)摘要:采用高效液相色谱串联质谱法对饲料中黄曲霉毒素B1进行检测。
建立了以Inertsil ODS-3为液相色谱柱,以0.1%甲酸水、乙腈(0.1%甲酸)为流动相,质谱使用电喷雾离子源,正离子扫描,以313.0/285.0为定量离子对的LC-MS/MS 检测方法。
结果表明:该方法所测得的黄曲霉毒素B1在0~100.0ng/mL 内具有良好线性,检出限为2.8μg/kg ,加标回收率在93.3%~98.3%。
该方法适合饲料中黄曲霉毒素B1的分析检测。
与传统的薄层色谱、高效液相色谱法相比较,该方法具有分析速度快、操作方便、重现性好、灵敏度高等特点。
关键词:饲料;黄曲霉毒素B1;液质联用中图分类号:TS207文献标志码:A文章编号:1008-1038(2015)12-0025-03Detection of AFB1in FeedLIU ChaoJING Zan(Leshan Food and Drug Inspection and Testing Center,LeShan 614000,China)Abstract:We tested aflatoxin B1in feed with LC -MS/MS method.A method was developed with novel HPLC columnInertsil ODS -3,Mobile phase was 0.1%methanoic acid water,0.1%methanoic acid acetonitrile,combined with MS,which used positive ion scanning,and quantification ion pair was 313.0/285.0.A good linear in 0~100.0ng/mL.detection limit was 2.8μg/kg.Adding standard recovery was 93.3%~98.3%.This method has advantages of being fast,easy,reproducible and relatively sensitive compared with TLC and HPLC.Key words:Feed;AFB1;LC-MS/MS收稿日期:2015-06-26作者简介:刘超(1984—),男,助理工程师,研究方向为食品安全25. All Rights Reserved.中国果菜质量控制AFB1标准品(2μg/mL,农业部环境保护科研所)。
(KJ201709)液体乳中黄曲霉毒素M1的快速检测胶体金免疫层析法

附件3液体乳中黄曲霉毒素M1的快速检测胶体金免疫层析法(KJ201709)1范围本方法规定了牛奶、羊奶、牦牛奶等液体乳中黄曲霉毒素M1的胶体金免疫层析快速检测方法。
本方法适用于生鲜乳、巴氏杀菌乳、灭菌乳中黄曲霉毒素M1的快速测定。
2原理本方法采用竞争抑制免疫层析原理。
样品中的黄曲霉毒素M1与胶体金标记的特异性抗体结合,抑制抗体和试纸条或检测卡中检测线(T线)上抗原的结合,从而导致检测线颜色深浅的变化。
通过检测线与控制线(C线)颜色深浅比较,对样品中黄曲霉毒素M1进行定性判定。
3试剂和材料除另有规定外,本方法所用试剂均为分析纯,水为GB/T 6682规定的二级水。
3.1试剂甲醇。
3.2参考物质黄曲霉毒素M1参考物质的中文名称、英文名称、CAS登录号、分子式、相对分子质量见表1,纯度≥90%。
表1 黄曲霉毒素M1参考物质中文名称、英文名称、CAS登录号、分子式、相对分子质量注:或等同可溯源物质。
3.3标准溶液的配制3.3.1黄曲霉毒素M1标准储备液(100 μg/mL):精密称取适量黄曲霉毒素M1标准品(3.2),置于10 mL容量瓶中,用甲醇(3.1)溶解并稀释至刻度,摇匀,制成浓度为100 μg/mL的黄曲霉毒素M1标准储备液;或可直接购买黄曲霉毒素M1标准储备液。
-20℃避光保存备用,有效期3个月。
—1—3.3.2黄曲霉毒素M1标准中间液(100 ng/mL):精密量取黄曲霉毒素M1标准储备液(100 μg/mL)(3.3.1)0.1 mL,置于100 mL容量瓶中,用甲醇(3.1)稀释至刻度,摇匀,制成浓度为100 ng/mL 的黄曲霉毒素M1标准中间液。
临用新制。
3.4材料黄曲霉毒素M1胶体金免疫层析试剂盒,适用基质为液体乳。
3.4.1金标微孔(含胶体金标记的特异性抗体)。
3.4.2试纸条或检测卡。
4仪器和设备4.1移液器:100 µL、200 µL和500 µL。
高效液相色谱法检测农产品中黄曲霉毒素B1的含量

分析检测高效液相色谱法检测农产品中黄曲霉毒素B1的含量韩 宇(四川省甘孜藏族自治州食品药品检验所,四川康定 626000)摘 要:目的:建立高效液相色谱法联合免疫磁固相萃取法测定农产品食品中黄曲霉毒素B1(Aflatoxin B1,AFB1)含量的分析方法。
方法:采用70%乙腈溶液提取花生、玉米、大豆、小麦、豌豆和绿豆样品中的AFB1,然后利用免疫磁珠净化、萃取和富集提取液中的AFB1,采用高效液相色谱法进行定量分析。
结果:方法的检出限为0.03~0.92 μg·kg-1,回收率为79.52%~97.53%。
利用该方法对市场上购买的20份花生、玉米、大豆、小麦、豌豆、绿豆样品中的AFB1进行检测,除了玉米和绿豆中未检测出AFB1外,其余实验样品中均检测出一定含量的AFB1。
结论:该方法简便、快速、准确,适用于复杂样品中黄曲霉毒素B1的测定。
关键词:高效液相色谱法;免疫磁固相萃取;黄曲霉毒素B1Determination of Aspergillus Flavus B1 in Agricultural Products by High-Performance Liquid ChromatographyHAN Yu(Ganzi Tibetan Autonomous Prefecture Institute for Food and Drug Control, Kangding 626000, China)Abstract: Objective: To establish a method for the determination of Aflatoxin B1 (AFB1) in agricultural products by high performance liquid chromatography combined with immunomagnetic solid phase extraction. Method: AFB1 was extracted from samples of peanuts, corn, soybeans, wheat, peas, and mung beans using a 70% acetonitrile solution, and then AFB1 was purified, extracted and enriched by immunomagnetic beads, and quantitative analysis was performed by HPLC. Result: The limits of detection were 0.03~0.92 μg·kg-1, and the recoveries were 79.52%~97.53%. AFB1 in 20 samples of peanut, corn, soybean, wheat, pea and mung bean purchased from the market was detected by this method. AFB1 content was detected in all the other samples except corn and mung bean. Conclusion: The method is simple, rapid, accurate and suitable for the determination of aflatoxin B1 in complex samples.Keywords: high-performance liquid chromatography; immunomagnetic solid-phase extraction; aspergillus flavus B1黄曲霉毒素是一种由黄曲霉菌产生的有毒代谢产物,在自然界中分布广泛,在粮食和饲料等农产品中广泛存在。
2020版《中国药典》解读一:真菌毒素分析方法验证

2020版药典真菌毒素分析方法验证*方法依据:《中国药典》2020版 第四部 9101 药品质量标准分析方法验证指导原则一 检测限、定量限信噪比法 1. 检测限:取黄曲霉毒素标准品,不断将其稀释(逐级稀释法),按照供试品进样体积进液相色谱检测,直至信噪比为3或略大于3,此时的标准品浓度(单位为μg/mL ),乘以5除以3得到的就是检测限,单位为μg/kg 。
2. 定量限:取黄曲霉毒素标准品,不断将其稀释(逐级稀释法),按照供试品进样体积进液相色谱检测,直至信噪比为10或略大于10,此时的标准品浓度(单位为μg/mL ),乘以5除以得到的就是定量限,单位为μg/kg 。
上述计算方法获得的数据须用含量相近的样品进行验证。
应附测试图谱,说明测试过程和结果。
二 准确度回收率验证: (以黄曲霉毒素为例)称取未检出黄曲霉毒素的样品6份,每份15g ,分别加入75μL 浓度为2.6ppm 的黄曲霉毒素混合标准品,按照2020版药典《通则2351 黄曲霉毒素测定法》进行操作。
回收率计算如下:1.黄曲霉毒素B 1、G 1回收率计算公式:%1005)(-)()(111111⨯=含量样品含量加标样品回收率黄曲霉毒素G B G B G B2.黄曲霉毒素B 2、G 2回收率计算公式:%1005.1)(-)()(222222⨯=含量样品含量加标样品回收率黄曲霉毒素G B G B G B表1 样品中待测定成分含量和回收率限度三 精密度1. 重复性在规定范围内,取同一浓度的供试品,用至少6份的测定结果进行评价。
或设计至少3种不同浓度,每种浓度分别制备至少3份供试品溶液进行测定,用至少9份样品的测定结果进行评价。
2. 中间精密度考查随机变动因素,如不同日期、不同分析人员、不同仪器对精密度的影响,应进行中间精密度试验。
3. 重现性国家药品质量标准采用的分析方法,应进行重现性试验,如通过不同实验室协同检验获得重现性结果。
表2 样品中待测定成分的含量与精密度可接受范围关系。
黄曲霉毒素检测标准方法汇总
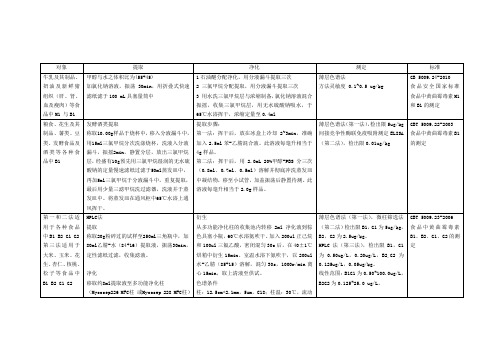
流动相的梯度变化表(GBT 5009.23-2006 食品中黄曲霉毒素B1、B2、G1、G2的测定)
标准方法检出限(GBT 23212-2008)
次氯酸钠溶液(消毒用):
取100g漂白粉,加入500ml水,搅拌均匀。
另将80g工业用碳酸钠(Na2CO3·10H2O)溶于500ml温水中,再将两液混合、搅拌、澄清后过滤。
此滤液含次氯酸浓度约为25g/L。
若用漂粉精制备,则碳酸钠的量可以加倍。
所得溶液的浓度约为50g/L。
污染的玻璃仪器用10g/L次氯酸钠溶液浸泡半天或用50g/L次氯酸钠溶液浸泡片刻后,即可达到去毒效果。
(GBT 5009.22-2003 食品中黄曲霉毒素B1的测定)
AFT B1标准溶液配制
1 仪器校正,测定重铬酸钾溶液的摩尔消光系数,以求出使用仪器的校正因素f
2 标准溶液配制,配制AFT B1标准溶液,用紫外分光光度计测此溶液最大吸收峰的波长及该波长的吸光度值,计算AFT B1标准液浓度,后用苯-乙腈混合液调到标准液为10.0ug/ml,并用分光光度计核对浓度。
(GBT 5009.22-2003 食品中黄曲霉毒素B1的测定)。
薄层层析法检验黄曲霉毒素

样品处理:一、国标1.玉米、大米、麦类、面粉、薯干、豆类、花生、花生酱【称取20g粉碎过筛试样(面粉或花生酱不需粉碎)置于250ml具塞锥形瓶中加30ml正已烷或石油醚和100mL50%甲醇水溶液(50ML甲醇+50mlddwater)在瓶塞上涂上一层水,盖严防漏。
振荡30min,静置片刻。
以叠成折叠式的快速定性滤纸过滤于分漏斗中,待下层甲醇溶液分清后,放出甲醇水溶液于另一具塞锥形瓶内】。
取20ml甲醇水溶液(相当于4g 试样)置于另一125mL分液漏斗中,加20ml三氯甲烷振摇2min。
静置分层,如出现乳化现象可滴加甲醇促使分层。
放出三氯甲烷层,经盛有约10g预先用三氯甲烷湿润的无水硫酸钠的定量慢速滤纸过滤于50ml蒸发皿中,再加5ml三氯甲烷于分液漏斗中。
重复振摇提取,三氯甲烷层一并滤于蒸发皿中。
最后用少量三氯甲烷洗过滤器,洗液并于蒸发皿中。
将蒸发皿放在通风柜于65℃水浴上通风挥干,然后放在冰盒上冷却2至3min后准确加入1ml苯-乙腈的混合液或将三氯甲烷用浓缩蒸馏器减压吹气蒸干后,准确加入1ml苯-乙腈混合液。
用带橡皮头的滴管的管尖将残渣充分混合,若有苯的结晶析出,将蒸发皿从冰盒上取出,继续溶解,混合,晶体即消失。
再用此滴管吸取上清液转移于2ml具塞试管中2.薄层板的活化新制薄层板100℃活化2h或制备好的薄层板105-110℃活化1h3.点样:在距薄层板下端3cm的基线上用微量注射器或血色素吸管滴加样液。
一块板可滴加4个点,点距边缘和点间距约为1cm,点直径约为3mm。
在同一块板上滴加点的大小应一致,滴加时可用吹风机的冷风边吹边加。
4.展开:展开槽(内长25cm、宽6cm、高4cm)在展开槽中加入展开剂静置平衡30min以上,然后在展开槽内加10ml无水乙醚,预展12cm。
取出挥干,再于另一展开槽内加10cm 丙酮-三氯甲烷(8:92)展开10-12cm取出。
食品中黄曲霉毒素B族和G族的测定

食品安全国家标准食品中黄曲霉毒素B族和G族的测定1范围本标准规定了食品中黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2(以下简称A F TB1㊁A F TB2㊁A F T G1和A F T G2)的测定方法㊂本标准第一法为同位素稀释液相色谱-串联质谱法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F TB1㊁A F TB2㊁A F TG1和A F TG2的测定㊂本标准第二法为高效液相色谱-柱前衍生法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F T B1㊁A F T B2㊁A F T G1和A F T G2的测定㊂本标准第三法为高效液相色谱-柱后衍生法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F T B1㊁A F T B2㊁A F T G1和A F T G2的测定㊂本标准第四法为酶联免疫吸附筛查法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F TB1的测定㊂本标准第五法为薄层色谱法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品中A F TB1的测定㊂第一法同位素稀释液相色谱-串联质谱法2原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液提取,提取液用含1%T r i t o nX-100(或吐温-20)的磷酸盐缓冲溶液稀释后(必要时经黄曲霉毒素固相净化柱初步净化),通过免疫亲和柱净化和富集,净化液浓缩㊁定容和过滤后经液相色谱分离,串联质谱检测,同位素内标法定量㊂3试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂3.1试剂3.1.1乙腈(C H3C N):色谱纯㊂3.1.2甲醇(C H3O H):色谱纯㊂3.1.3乙酸铵(C H3C O O N H4):色谱纯㊂3.1.4氯化钠(N a C l)㊂3.1.5磷酸氢二钠(N a2H P O4)㊂3.1.6磷酸二氢钾(K H2P O4)㊂3.1.7氯化钾(K C l)㊂3.1.8盐酸(H C l)㊂3.1.9 T r i t o nX-100[C14H22O(C2H4O)n](或吐温-20,C58H114O26)㊂3.2试剂配制3.2.1乙酸铵溶液(5mm o l/L):称取0.39g乙酸铵,用水溶解后稀释至1000m L,混匀㊂3.2.2乙腈-水溶液(84+16):取840m L乙腈加入160m L水,混匀㊂3.2.3甲醇-水溶液(70+30):取700m L甲醇加入300m L水,混匀㊂3.2.4乙腈-水溶液(50+50):取50m L乙腈加入50m L水,混匀㊂3.2.5乙腈-甲醇溶液(50+50):取50m L乙腈加入50m L甲醇,混匀㊂3.2.610%盐酸溶液:取1m L盐酸,用纯水稀释至10m L,混匀㊂3.2.7磷酸盐缓冲溶液(以下简称P B S):称取8.00g氯化钠㊁1.20g磷酸氢二钠(或2.92g十二水磷酸氢二钠)㊁0.20g磷酸二氢钾㊁0.20g氯化钾,用900m L水溶解,用盐酸调节p H至7.4ʃ0.1,加水稀释至1000m L㊂3.2.81%T r i t o nX-100(或吐温-20)的P B S:取10m LT r i t o nX-100(或吐温-20),用P B S稀释至1000m L㊂3.3标准品3.3.1 A F TB1标准品(C17H12O6,C A S:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.2 A F TB2标准品(C17H14O6,C A S:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.3 A F T G1标准品(C17H12O7,C A S:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.4 A F T G2标准品(C17H14O7,C A S:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.5同位素内标13C17-A F TB1(C17H12O6,C A S:157449-45-0):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.6同位素内标13C17-A F TB2(C17H14O6,C A S:157470-98-8):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.7同位素内标13C17-A F T G1(C17H12O7,C A S:157444-07-9):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.8同位素内标13C17-A F T G2(C17H14O7,C A S:157462-49-7):纯度ȡ98%,浓度为0.5μg/m L㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂3.4标准溶液配制3.4.1标准储备溶液(10μg/m L):分别称取A F TB1㊁A F TB2㊁A F TG1和A F TG21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂3.4.2混合标准工作液(100n g/m L):准确移取混合标准储备溶液(1.0μg/m L)1.00m L至100m L容量瓶中,乙腈定容㊂此溶液密封后避光-20ħ下保存,三个月有效㊂3.4.3混合同位素内标工作液(100n g/m L):准确移取0.5μg/m L13C17-A F TB1㊁13C17-A F TB2㊁13C17-A F T G1和13C17-A F T G2各2.00m L,用乙腈定容至10m L㊂在-20ħ下避光保存,备用㊂3.4.4标准系列工作溶液:准确移取混合标准工作液(100n g/m L)10μL㊁50μL㊁100μL㊁200μL㊁500μL㊁800μL㊁1000μL至10m L容量瓶中,加入200μL100n g/m L的同位素内标工作液,用初始流动相定容至刻度,配制浓度点为0.1n g/m L㊁0.5n g/m L㊁1.0n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁8.0n g/m L㊁10.0n g/m L的系列标准溶液㊂4仪器和设备4.1匀浆机㊂4.2高速粉碎机㊂4.3组织捣碎机㊂4.4超声波/涡旋振荡器或摇床㊂4.5天平:感量0.01g和0.00001g㊂4.6涡旋混合器㊂4.7高速均质器:转速6500r/m i n~24000r/m i n㊂4.8离心机:转速ȡ6000r/m i n㊂4.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂4.10固相萃取装置(带真空泵)㊂4.11氮吹仪㊂4.12液相色谱-串联质谱仪:带电喷雾离子源㊂4.13液相色谱柱㊂4.14免疫亲和柱:A F TB1柱容量ȡ200n g,A F TB1柱回收率ȡ80%,A F TG2的交叉反应率ȡ80%(验证方法参见附录B)㊂注:对于不同批次的亲和柱在使用前需进行质量验证㊂4.15黄曲霉毒素专用型固相萃取净化柱或功能相当的固相萃取柱(以下简称净化柱):对复杂基质样品测定时使用㊂4.16微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂4.17筛网:1mm~2mm试验筛孔径㊂4.18p H计㊂5分析步骤使用不同厂商的免疫亲和柱,在样品上样㊁淋洗和洗脱的操作方面可能会略有不同,应该按照供应商所提供的操作说明书要求进行操作㊂警示:整个分析操作过程应在指定区域内进行㊂该区域应避光(直射阳光)㊁具备相对独立的操作台和废弃物存放装置㊂在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施㊂5.1样品制备5.1.1液体样品(植物油㊁酱油㊁醋等)采样量需大于1L,对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),将所有液体样品在一个容器中用匀浆机混匀后,其中任意的100g(m L)样品进行检测㊂5.1.2固体样品(谷物及其制品㊁坚果及籽类㊁婴幼儿谷类辅助食品等)采样量需大于1k g,用高速粉碎机将其粉碎,过筛,使其粒径小于2mm孔径试验筛,混合均匀后缩分至100g,储存于样品瓶中,密封保存,供检测用㊂5.1.3半流体(腐乳㊁豆豉等)采样量需大于1k g(L),对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),用组织捣碎机捣碎混匀后,储存于样品瓶中,密封保存,供检测用㊂5.2样品提取5.2.1液体样品5.2.1.1植物油脂称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液(3.4.3)振荡混合后静置30m i n㊂加入20m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n,取上清液备用㊂5.2.1.2酱油㊁醋称取5g试样(精确至0.01g)于50m L离心管中,加入125μL同位素内标工作液振荡混合后静置30m i n㊂用乙腈或甲醇定容至25m L(精确至0.1m L),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.2固体样品5.2.2.1一般固体样品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.2.2婴幼儿配方食品和婴幼儿辅助食品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(50+50)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.3半流体样品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.3样品净化5.3.1免疫亲和柱净化5.3.1.1上样液的准备准确移取4m L上清液,加入46m L1%T r i t i o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂5.3.1.2免疫亲和柱的准备将低温下保存的免疫亲和柱恢复至室温㊂5.3.1.3试样的净化待免疫亲和柱内原有液体流尽后,将上述样液移至50m L注射器筒中,调节下滴速度,控制样液以1m L/m i n~3m L/m i n的速度稳定下滴㊂待样液滴完后,往注射器筒内加入2ˑ10m L水,以稳定流速淋洗免疫亲和柱㊂待水滴完后,用真空泵抽干亲和柱㊂脱离真空系统,在亲和柱下部放置10m L刻度试管,取下50m L的注射器筒,加入2ˑ1m L甲醇洗脱亲和柱,控制1m L/m i n~3m L/m i n的速度下滴,再用真空泵抽干亲和柱,收集全部洗脱液至试管中㊂在50ħ下用氮气缓缓地将洗脱液吹至近干,加入1.0m L初始流动相,涡旋30s溶解残留物,0.22μm滤膜过滤,收集滤液于进样瓶中以备进样㊂5.3.2黄曲霉毒素固相净化柱和免疫亲和柱同时使用(对花椒㊁胡椒和辣椒等复杂基质)5.3.2.1净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂5.3.2.2免疫亲和柱净化用刻度移液管准确吸取上述净化液4m L,加入46m L1%T r i t i o n X-100(或吐温-20)的P B S[使用甲醇-水溶液提取时,加入23m L1%T r i t i o n X-100(或吐温-20)的P B S],混匀㊂按5.3.1.2和5.3.1.3处理㊂注:全自动(在线)或半自动(离线)的固相萃取仪器可优化操作参数后使用㊂5.4液相色谱参考条件液相色谱参考条件列出如下:a)流动相:A相:5mm o l/L乙酸铵溶液;B相:乙腈-甲醇溶液(50+50);b)梯度洗脱:32%B(0m i n~0.5m i n),45%B(3m i n~4m i n),100%B(4.2m i n~4.8m i n),32%B(5.0m i n~7.0m i n);c)色谱柱:C18柱(柱长100mm,柱内径2.1mm;填料粒径1.7μm),或相当者;d)流速:0.3m L/m i n;e)柱温:40ħ;f)进样体积:10μL㊂5.5质谱参考条件质谱参考条件列出如下:a)检测方式:多离子反应监测(MR M);b)离子源控制条件:参见表1;c)离子选择参数:参见表2;d)子离子扫描图:参见图C.1~图C.8;e)液相色谱-质谱图:见图C.9㊂表1离子源控制条件电离方式E S I+毛细管电压/k V3.5锥孔电压/V30射频透镜1电压/V14.9射频透镜2电压/V15.1表1(续)离子源温度/ħ150锥孔反吹气流量/(L/h)50脱溶剂气温度/ħ500脱溶剂气流量/(L/h)800电子倍增电压/V650表2离子选择参数表化合物名称母离子(m/z)定量离子(m/z)碰撞能量e V定性离子(m/z)碰撞能量e V离子化方式A F TB13132852224138E S I+13C17-A F TB13302552330135E S I+A F TB23152872525928E S I+13C17-A F TB23323032527328E S I+A F T G13292432528325E S I+13C17-A F T G13462572529925E S I+A F T G23312453028527E S I+13C17-A F T G23482593030127E S I+5.6定性测定试样中目标化合物色谱峰的保留时间与相应标准色谱峰的保留时间相比较,变化范围应在ʃ2.5%之内㊂每种化合物的质谱定性离子必须出现,至少应包括一个母离子和两个子离子,而且同一检测批次,对同一化合物,样品中目标化合物的两个子离子的相对丰度比与浓度相当的标准溶液相比,其允许偏差不超过表3规定的范围㊂表3定性时相对离子丰度的最大允许偏差相对离子丰度/%>5020~5010~20ɤ10允许相对偏差/%ʃ20ʃ25ʃ30ʃ505.7标准曲线的制作在5.4㊁5.5的液相色谱串联质谱仪分析条件下,将标准系列溶液由低到高浓度进样检测,以A F TB1㊁A F TB2㊁A F TG1和A F TG2色谱峰与各对应内标色谱峰的峰面积比值-浓度作图,得到标准曲线回归方程,其线性相关系数应大于0.99㊂5.8试样溶液的测定取5.3处理得到的待测溶液进样,内标法计算待测液中目标物质的质量浓度,按第6章计算样品中待测物的含量㊂待测样液中的响应值应在标准曲线线性范围内,超过线性范围则应适当减少取样量重新测定㊂5.9空白试验不称取试样,按5.2和5.3的步骤做空白实验㊂应确认不含有干扰待测组分的物质㊂6分析结果的表述试样中A F TB1㊁A F TB2㊁A F T G1和A F T G2的残留量按式(1)计算:X=ρˑV1ˑV3ˑ1000V2ˑmˑ1000 (1)式中:X 试样中A F TB1㊁A F TB2㊁A F T G1或A F T G2的含量,单位为微克每千克(μg/k g);ρ 进样溶液中A F TB1㊁A F TB2㊁A F T G1或A F T G2按照内标法在标准曲线中对应的浓度,单位为纳克每毫升(n g/m L);V1 试样提取液体积(植物油脂㊁固体㊁半固体按加入的提取液体积;酱油㊁醋按定容总体积),单位为毫升(m L);V3 样品经净化洗脱后的最终定容体积,单位为毫升(m L);1000 换算系数;V2 用于净化分取的样品体积,单位为毫升(m L);m 试样的称样量,单位为克(g)㊂计算结果保留三位有效数字㊂7精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%㊂8其他当称取样品5g时,A F TB1的检出限为:0.03μg/k g,A F TB2的检出限为0.03μg/k g,A F TG1的检出限为0.03μg/k g,A F T G2的检出限为0.03μg/k g;A F TB1的定量限为0.1μg/k g,A F TB2的定量限为0.1μg/k g,A F T G1的定量限为0.1μg/k g,A F T G2的定量限为0.1μg/k g㊂第二法高效液相色谱-柱前衍生法9原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液的混合溶液提取,提取液经黄曲霉毒素固相净化柱净化去除脂肪㊁蛋白质㊁色素及碳水化合物等干扰物质,净化液用三氟乙酸柱前衍生,液相色谱分离,荧光检测器检测,外标法定量㊂10试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂10.1试剂10.1.1甲醇(C H3O H):色谱纯㊂10.1.2乙腈(C H3C N):色谱纯㊂10.1.3正己烷(C6H14):色谱纯㊂10.1.4三氟乙酸(C F3C O O H)㊂10.2试剂配制10.2.1乙腈-水溶液(84+16):取840m L乙腈加入160m L水㊂10.2.2甲醇-水溶液(70+30):取700m L甲醇加入300m L水㊂10.2.3乙腈-水溶液(50+50):取500m L乙腈加入500m L水㊂10.2.4乙腈-甲醇溶液(50+50):取500m L乙腈加入500m L甲醇㊂10.3标准品10.3.1 A F TB1标准品(C17H12O6,C A S号:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.2 A F TB2标准品(C17H14O6,C A S号:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.3 A F T G1标准品(C17H12O7,C A S号:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.4 A F T G2标准品(C17H14O7,C A S号:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂10.4标准溶液配制10.4.1标准储备溶液(10μg/m L):分别称取A F T B1㊁A F T B2㊁A F T G1和A F T G21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂10.4.2混合标准工作液(A F TB1和A F T G1:100n g/m L,A F TB2和A F T G2:30n g/m L):准确移取A F TB1和A F T G1标准储备溶液各1m L,A F TB2和A F T G2标准储备溶液各300μL至100m L容量瓶中,乙腈定容㊂密封后避光-20ħ下保存,三个月内有效㊂10.4.3标准系列工作溶液:分别准确移取混合标准工作液10μL㊁50μL㊁200μL㊁500μL㊁1000μL㊁2000μL㊁4000μL至10m L容量瓶中,用初始流动相定容至刻度(含A F T B1和A F T G1浓度为0.1n g/m L㊁0.5n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁10.0n g/m L㊁20.0n g/m L㊁40.0n g/m L,A F T B2和A F T G2浓度为0.03n g/m L㊁0.15n g/m L㊁0.6n g/m L㊁1.5n g/m L㊁3.0n g/m L㊁6.0n g/m L㊁12n g/m L 的系列标准溶液)㊂11仪器和设备11.1匀浆机㊂11.2高速粉碎机㊂11.3组织捣碎机㊂11.4超声波/涡旋振荡器或摇床㊂11.5天平:感量0.01g和0.00001g㊂11.6涡旋混合器㊂11.7高速均质器:转速6500r/m i n~24000r/m i n㊂11.8离心机:转速ȡ6000r/m i n㊂11.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂11.10氮吹仪㊂11.11液相色谱仪:配荧光检测器㊂11.12色谱分离柱㊂11.13黄曲霉毒素专用型固相萃取净化柱(以下简称净化柱),或相当者㊂11.14一次性微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂11.15筛网:1mm~2mm试验筛孔径㊂11.16恒温箱㊂11.17p H计㊂12分析步骤12.1样品制备12.1.1液体样品(植物油㊁酱油㊁醋等)采样量需大于1L,对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),将所有液体样品在一个容器中用匀浆机混匀后,其中任意的100g(m L)样品进行检测㊂12.1.2固体样品(谷物及其制品㊁坚果及籽类㊁婴幼儿谷类辅助食品等)采样量需大于1k g,用高速粉碎机将其粉碎,过筛,使其粒径小于2mm孔径试验筛,混合均匀后缩分至100g,储存于样品瓶中,密封保存,供检测用㊂12.1.3半流体(腐乳㊁豆豉等)采样量需大于1k g(L),对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),用组织捣碎机捣碎混匀后,储存于样品瓶中,密封保存,供检测用㊂12.2样品提取12.2.1液体样品12.2.1.1植物油脂称取5g试样(精确至0.01g)于50m L离心管中,加入20m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n,取上清液备用㊂12.2.1.2酱油㊁醋称取5g试样(精确至0.01g)于50m L离心管中,用乙腈或甲醇定容至25m L(精确至0.1m L),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.2固体样品12.2.2.1一般固体样品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.2.2婴幼儿配方食品和婴幼儿辅助食品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(50+50)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.3半流体样品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.3样品黄曲霉毒素固相净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂12.4衍生用移液管准确吸取4.0m L净化液于10m L离心管后在50ħ下用氮气缓缓地吹至近干,分别加入200μL正己烷和100μL三氟乙酸,涡旋30s,在40ħʃ1ħ的恒温箱中衍生15m i n,衍生结束后,在50ħ下用氮气缓缓地将衍生液吹至近干,用初始流动相定容至1.0m L,涡旋30s溶解残留物,过0.22μm滤膜,收集滤液于进样瓶中以备进样㊂12.5色谱参考条件色谱参考条件列出如下:a)流动相:A相:水,B相:乙腈-甲醇溶液(50+50);b)梯度洗脱:24%B(0m i n~6m i n),35%B(8.0m i n~10.0m i n),100%B(10.2m i n~11.2m i n),24%B(11.5m i n~13.0m i n);c)色谱柱:C18柱(柱长150mm或250mm,柱内径4.6mm,填料粒径5.0μm),或相当者;d)流速:1.0m L/m i n;e)柱温:40ħ;f)进样体积:50μL;g)检测波长:激发波长360n m;发射波长440n m;h)液相色谱图:参见图D.1㊂12.6样品测定12.6.1标准曲线的制作系列标准工作溶液由低到高浓度依次进样检测,以峰面积为纵坐标-浓度为横坐标作图,得到标准曲线回归方程㊂12.6.2试样溶液的测定待测样液中待测化合物的响应值应在标准曲线线性范围内,浓度超过线性范围的样品则应稀释后重新进样分析㊂12.6.3空白试验不称取试样,按12.2㊁12.3和12.4的步骤做空白实验㊂应确认不含有干扰待测组分的物质㊂13分析结果的表述试样中A F TB1㊁A F TB2㊁A F T G1和A F T G2的残留量按式(2)计算:X=ρˑV1ˑV3ˑ1000V2ˑmˑ1000 (2)式中:X 试样中A F TB1㊁A F TB2㊁A F T G1或A F T G2的含量,单位为微克每千克(μg/k g);ρ 进样溶液中A F TB1㊁A F TB2㊁A F T G1或A F T G2按照外标法在标准曲线中对应的浓度,单位为纳克每毫升(n g/m L);V1 试样提取液体积(植物油脂㊁固体㊁半固体按加入的提取液体积;酱油㊁醋按定容总体积),单位为毫升(m L);V3 净化液的最终定容体积,单位为毫升(m L);1000 换算系数;V2 净化柱净化后的取样液体积,单位为毫升(m L);m 试样的称样量,单位为克(g)㊂计算结果保留三位有效数字㊂14精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%㊂15其他当称取样品5g时,柱前衍生法的A F TB1的检出限为0.03μg/k g,A F TB2的检出限为0.03μg/k g, A F TG1的检出限为0.03μg/k g,A F TG2的检出限为0.03μg/k g;柱前衍生法的A F TB1的定量限为0.1μg/ k g,A F TB2的定量限为0.1μg/k g,A F TG1的定量限为0.1μg/k g,A F TG2的定量限为0.1μg/k g㊂第三法高效液相色谱-柱后衍生法导语:下述方法的仪器检测部分,包括碘或溴试剂衍生㊁光化学衍生㊁电化学衍生等柱后衍生方法,可根据实际情况,选择其中一种方法即可㊂16原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液的混合溶液提取,提取液经免疫亲和柱净化和富集,净化液浓缩㊁定容和过滤后经液相色谱分离,柱后衍生(碘或溴试剂衍生㊁光化学衍生㊁电化学衍生等),经荧光检测器检测,外标法定量㊂17试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂17.1试剂17.1.1甲醇(C H3O H):色谱纯㊂17.1.2乙腈(C H3C N):色谱纯㊂17.1.3氯化钠(N a C l)㊂17.1.4磷酸氢二钠(N a2H P O4)㊂17.1.5磷酸二氢钾(K H2P O4)㊂17.1.6氯化钾(K C l)㊂17.1.7盐酸(H C l)㊂17.1.8 T r i t o nX-100[C14H22O(C2H4O)n](或吐温-20,C58H114O26)㊂17.1.9碘衍生使用试剂:碘(I2)㊂17.1.10溴衍生使用试剂:三溴化吡啶(C5H6B r3N2)㊂17.1.11电化学衍生使用试剂:溴化钾(K B r)㊁浓硝酸(HN O3)㊂17.2试剂配制17.2.1乙腈-水溶液(84+16):取840m L乙腈加入160m L水㊂17.2.2甲醇-水溶液(70+30):取700m L甲醇加入300m L水㊂17.2.3乙腈-水溶液(50+50):取500m L乙腈加入500m L水㊂17.2.4乙腈-水溶液(10+90):取100m L乙腈加入900m L水㊂17.2.5乙腈-甲醇溶液(50+50):取500m L乙腈加入500m L甲醇㊂17.2.6磷酸盐缓冲溶液(以下简称P B S):称取8.00g氯化钠㊁1.20g磷酸氢二钠(或2.92g十二水磷酸氢二钠)㊁0.20g磷酸二氢钾㊁0.20g氯化钾,用900m L水溶解,用盐酸调节p H至7.4,用水定容至1000m L㊂17.2.71%T r i t o nX-100(或吐温-20)的P B S:取10m LT r i t o nX-100,用P B S定容至1000m L㊂17.2.80.05%碘溶液:称取0.1g碘,用20m L甲醇溶解,加水定容至200m L,用0.45μm的滤膜过滤,现配现用(仅碘柱后衍生法使用)㊂17.2.95m g/L三溴化吡啶水溶液:称取5m g三溴化吡啶溶于1L水中,用0.45μm的滤膜过滤,现配现用(仅溴柱后衍生法使用)㊂17.3标准品17.3.1 A F TB1标准品(C17H12O6,C A S号:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.2 A F TB2标准品(C17H14O6,C A S号:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.3 A F T G1标准品(C17H12O7,C A S号:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.4 A F T G2标准品C17H14O7,C A S号:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂17.4标准溶液配制17.4.1标准储备溶液(10μg/m L):分别称取A F T B1㊁A F T B2㊁A F T G1和A F T G21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂17.4.2混合标准工作液(A F TB1和A F T G1:100n g/m L,A F TB2和A F T G2:30n g/m L):准确移取A F TB1和A F T G1标准储备溶液各1m L,A F TB2和A F T G2标准储备溶液各300μL至100m L容量瓶中,乙腈定容㊂密封后避光-20ħ下保存,三个月内有效㊂17.4.3标准系列工作溶液:分别准确移取混合标准工作液10μL㊁50μL㊁200μL㊁500μL㊁1000μL㊁2000μL㊁4000μL至10m L容量瓶中,用初始流动相定容至刻度(含A F T B1和A F T G1浓度为0.1n g/m L㊁0.5n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁10.0n g/m L㊁20.0n g/m L㊁40.0n g/m L,A F T B2和A F T G2浓度为0.03n g/m L㊁0.15n g/m L㊁0.6n g/m L㊁1.5n g/m L㊁3.0n g/m L㊁6.0n g/m L㊁12n g/m L 的系列标准溶液)㊂18仪器和设备18.1匀浆机㊂18.2高速粉碎机㊂18.3组织捣碎机㊂18.4超声波/涡旋振荡器或摇床㊂18.5天平:感量0.01g和0.00001g㊂18.6涡旋混合器㊂18.7高速均质器:转速6500r/m i n~24000r/m i n㊂18.8离心机:转速ȡ6000r/m i n㊂18.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂18.10固相萃取装置(带真空泵)㊂18.11氮吹仪㊂18.12液相色谱仪:配荧光检测器(带一般体积流动池或者大体积流通池)㊂注:当带大体积流通池时不需要再使用任何型号或任何方式的柱后衍生器㊂18.13液相色谱柱㊂18.14光化学柱后衍生器(适用于光化学柱后衍生法)㊂18.15溶剂柱后衍生装置(适用于碘或溴试剂衍生法)㊂18.16电化学柱后衍生器(适用于电化学柱后衍生法)㊂18.17免疫亲和柱:A F TB1柱容量ȡ200n g,A F TB1柱回收率ȡ80%,A F T G2的交叉反应率ȡ80% (验证方法参见附录B)㊂注:对于每个批次的亲和柱使用前需质量验证㊂18.18黄曲霉毒素固相净化柱或功能相当的固相萃取柱(以下简称净化柱):对复杂基质样品测定时使用㊂18.19一次性微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂18.20筛网:1mm~2mm试验筛孔径㊂19分析步骤使用不同厂商的免疫亲和柱,在样品的上样㊁淋洗和洗脱的操作方面可能略有不同,应该按照供应商所提供的操作说明书要求进行操作㊂警示:整个分析操作过程应在指定区域内进行㊂该区域应避光(直射阳光)㊁具备相对独立的操作台和废弃物存放装置㊂在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施㊂19.1样品制备同12.1㊂19.2样品提取同12.2㊂19.3样品净化19.3.1免疫亲和柱净化19.3.1.1上样液的准备准确移取4m L上述上清液,加入46m L1%T r i t o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂19.3.1.2免疫亲和柱的准备将低温下保存的免疫亲和柱恢复至室温㊂19.3.1.3试样的净化免疫亲和柱内的液体放弃后,将上述样液移至50m L注射器筒中,调节下滴速度,控制样液以1m L/m i n~3m L/m i n的速度稳定下滴㊂待样液滴完后,往注射器筒内加入2ˑ10m L水,以稳定流速淋洗免疫亲和柱㊂待水滴完后,用真空泵抽干亲和柱㊂脱离真空系统,在亲和柱下部放置10m L刻度试管,取下50m L的注射器筒,2ˑ1m L甲醇洗脱亲和柱,控制1m L/m i n~3m L/m i n的速度下滴,再用真空泵抽干亲和柱,收集全部洗脱液至试管中㊂在50ħ下用氮气缓缓地将洗脱液吹至近干,用初始流动相定容至1.0m L,涡旋30s溶解残留物,0.22μm滤膜过滤,收集滤液于进样瓶中以备进样㊂19.3.2黄曲霉毒素固相净化柱和免疫亲和柱同时使用(对花椒㊁胡椒和辣椒等复杂基质)19.3.2.1净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂19.3.2.2免疫亲和柱净化用刻度移液管准确吸取上部净化液4m L,加入46m L1%T r i t o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂按19.4.1.3处理㊂注:全自动(在线)或半自动(离线)的固相萃取仪器可优化操作参数后使用㊂19.4液相色谱参考条件19.4.1无衍生器法(大流通池直接检测)液相色谱参考条件列出如下:a)流动相:A相,水;B相,乙腈-甲醇(50+50);b)等梯度洗脱条件:A,65%;B,35%;。
黄曲霉毒素检测方法介绍

黄曲霉毒素检测方法介绍黄曲霉毒素(AFT)是一类化学结构类似的化合物,均为二氢呋喃香豆素的衍生物。
黄曲霉毒素是主要由黄曲霉(aspergillus flavus) 寄生曲霉(a.parasiticus)产生的次生代谢产物,在湿热地区食品和饲料中出现黄曲霉毒素的机率最高。
B1是最危险的致癌物,经常在玉米,花生,棉花种子,一些干果中常能检测到。
它们在紫外线照射下能产生荧光,根据荧光颜色不同,将其分为B族和G族两大类及其衍生物。
AFT目前已发现20余种。
AFT主要污染粮油食品、动植物食品等;如花生、玉米,大米、小麦、豆类、坚果类、肉类、乳及乳制品、水产品等均有黄曲霉毒素污染。
其中以花生和玉米污染最严重。
家庭自制发酵食品也能检出黄曲霉毒素,尤其是高温高湿地区的粮油及制品种捡出率更高。
主要的几种检验检疫方法:1,薄层层析法薄层层析(Thin-Layer Chromatography,TLC)是在黄曲霉毒素研究方面应用最广的分离技术.自1990年,它被列为AOAC(Association of Official Agricultural Chemists)标准方法,该方法同时具有定性和定量分析黄曲霉毒素的功能.2,液相色谱法液相色谱(LiquidChromatography,LC)与薄层层析在许多方面具有相似性,二者互相补充.通常用TLC进行前期的条件设定,选择适宜的分离条件后,再用LC进行黄曲霉毒素的定量测定.3,免疫化学分析方法利用具有高度专一性的单克隆抗体或多克隆抗体设计的黄曲霉毒素的免疫分析方法,也是最常用的黄曲霉毒素检测方法.这类方法通常包括放射免疫分析方法(Radioimmunoassay,RIA),酶联免疫法(Enzyme-linked of Immunosorbent Assay,ELISA)和免疫层析法(Immunoaflinity Column Assay,ICA).它们均可以对黄曲霉毒素进行定量测定.(1) 免疫亲和柱-荧光分光光度法和免疫亲和术-HPLC法免疫亲和柱法和酶联免疫吸附法虽然都可达到速简便效果,但酶联免疫吸附法仅能检测单一毒素(如黄曲霉毒素B1)含量,而且易出现假阳性结果,难以控制.免疫亲和柱法(包括荧光光度法和HPLC法)却能达到既定量准确又快速简便的要求.免疫亲和柱的使用可以避免传统TLC和HPLC的缺点,同时免疫亲和柱与TLC和HPLC法结合可以大大提高工作效率,提高灵敏度和准确度.黄曲霉毒素免疫亲和柱-荧光光度计法是以单克隆免疫亲和柱为分离手段,用荧光计,紫外灯作为检测工具的快速分析方法.它克服了TLC和HPLC法在操作过程中使用剧毒的真菌毒素作为标定标准物和在样品预处理过程中使用多种有毒,异味的有机溶剂,毒害操作人员和污染环境的缺点.同时黄曲霉毒素免疫亲和柱-荧光光度计法分析速度快,一个样品只需10-15min,比传统方法快几个小时甚至几天时间;仪器设备轻便容易携带,自动化程度高,操作简单,直接读出测试结果,可以在小型实验或现场使用.可以进行黄曲霉毒素总量(B1B2G1G2)的测定,检测限可达到1ug/kg,达到黄曲霉毒素标准限量值以下测定范围为1-300ug/kg.黄曲霉毒素免疫亲和柱-高效液相色谱法比传统的HPLC法更加安全,可靠,灵敏度和准确度高.它采用单克隆抗体免疫技术,可以特效性地将黄曲霉毒素或其他真菌毒素分离出来,分离效率和回收率高.分析原理试样中的黄曲霉毒素用一定比例的甲醇/水提取液经过过滤,稀释后,用免疫亲和柱净化,以甲醇将亲和柱上的黄曲霉毒素淋洗下来,在淋洗液中加入溴溶液衍生,以提高测定灵敏度,然后用荧光分光光度计进行定量.也可以将甲醇-黄曲霉毒素淋洗液的一部分注入HPLC中,对黄曲霉毒素B1,B2,G1,B2分别进行定量分析.免疫亲和柱是用大剂量的黄曲霉毒素单克隆抗体固化在水不溶性的载体上,然后装柱而成.该方法的测定范围0-300ug/kg.(2) 酶联免疫吸附法:1996年,Nakane建立了辣根过氧化物酶标记抗体的测定技术.由于该方法简便,敏感,特异,可作为多种抗原或抗体的测定,20世纪70年代后期,该方法引入真菌毒素的检测中,下面介绍的是竞争性酶联免疫吸附间接法检测黄曲霉毒素B1.原理:将已知抗原吸附在固态载体表面,洗除末吸附抗原,加入一定量抗体与待测样品(含有抗原)提取液的混合液,竞争培养后,在固相载体表面形成抗原抗体复合物.洗除多余抗体成分,然后加入酶标记的抗球蛋白的第二抗体结合物,与吸附在固体表面的抗原抗体结合物相结合,再加入酶底物.在酶的催化作用下,底物发生降解反应,产生有色物质,通过酶标检测仪测出酶底物的降解量,从而推知被测样品中的抗原量.(3) 微柱筛选法可以用来半定量测定各种食品中黄曲霉毒素B1,B2,G1,G2的总量.原理:样品提取液中的黄曲霉毒素被微柱管风硅镁型吸附层吸附后,在波长365nm紫外光灯下显示蓝紫色荧光环,其荧光强度与黄曲霉毒素在一定的光密度范围内成正比例关系.若硅镁型吸附剂层未出现蓝紫色荧光,则样品为阴性(方法灵敏度为5-10ug/kg).由于在微柱上不能分离黄曲霉毒素B1,B2,G1,G2,所以测得结果为总的黄曲霉毒素含量.(4) 一步式黄曲霉毒素检测金标试纸法一步式黄曲霉毒素检测金标试纸法是利用单克隆抗体而设计的固相免疫分析法.由此产生的一步式黄曲霉毒素快速检测试纸可在5-10分钟完成对样品中黄曲霉毒素的定性测定.借助黄曲霉毒素标准样品,这种方法能估算黄曲霉毒素的含量,非常适用于现场测试和进行大量样品的初选.一步式黄曲霉毒素检测金标试纸法是利用单克隆抗体而设计的固相免疫分析法.由此产生的一步式黄曲霉毒素快速检测试纸可在5-10分钟完成对样品中黄曲霉毒素的定性测定.借助黄曲霉毒素标准样品,这种方法能估算黄曲霉毒素的含量,非常适用于现场测试和进行大量样品的初选.检测方法分析薄膜层析法和液相色谱法是目前国内绝大多数检测机构都在使用的方法,由于其检测周期长,程序复杂,所需试剂繁多等缺点已远远不能满足现代检测要求.随着现代科学技术的不断发展,特别是免疫学,生物化学,分子生物学的不断发展,人们已创建了不少快速,简便,特异,敏感,低耗且适用的黄曲霉毒素检测方法.而且以金标试纸为代表的这些方法已经被先进国家所广泛使用,引进和消化这些先进的方法是我们检测领域的当务之急.免疫亲和柱法优点很多,但由于检测费用过高,而无法普及.而一步式黄曲霉毒素检测金标试纸法似乎更适用于我国,值得推广.。
黄曲霉毒素方法标准

黄曲霉毒素方法标准嘿,咱今儿个就来唠唠黄曲霉毒素方法标准这档子事儿。
黄曲霉毒素啊,那可不是啥好惹的家伙!就像隐藏在我们生活里的小恶魔,稍不注意,它就可能蹦出来捣乱。
你想想,要是吃的东西里有了它,那对咱身体的伤害可不得了哇!那怎么才能把这个小恶魔给揪出来呢?这就得靠那些专门的方法标准啦!就好比我们去抓坏人,得有一套厉害的抓捕手段和规则,不然怎么能把坏人绳之以法呢?这些黄曲霉毒素方法标准呢,就像是一道道关卡,严格地检验着各种物品。
比如说食品吧,从原材料的挑选到加工制作,再到最后的成品,每一个环节都得按照标准来。
不然的话,那些黄曲霉毒素就可能偷偷混进去,到时候遭殃的不还是咱老百姓嘛!咱可以打个比方,标准就像是一个超级严格的老师,食品就是学生。
老师得时刻盯着学生,看看他们有没有犯错,有没有违反规定。
只有这样,才能保证学生们个个都优秀,都能让人放心。
再说说检测方法,那也是相当重要啊!这就好比医生看病,得有厉害的诊断手段才能准确知道病人得了啥病。
检测黄曲霉毒素的方法也得够精准、够灵敏,不能放过一丝一毫的蛛丝马迹。
不然,那些隐藏得很深的黄曲霉毒素不就逍遥法外了吗?而且啊,这些方法标准还得不断更新完善呢!为啥?因为黄曲霉毒素也会变着法儿地躲避检测呀!就跟那狡猾的狐狸似的。
所以我们的方法标准也得与时俱进,不断升级自己的“武器装备”,才能跟得上黄曲霉毒素的变化。
你说,要是没有这些严格的黄曲霉毒素方法标准,我们的生活得变成啥样?那还不得整天提心吊胆的,不知道吃进去的东西安不安全。
所以啊,咱可得重视这些方法标准。
政府部门得严格监管,企业得认真执行,咱老百姓也得有这个意识,多了解了解这些知识。
这样一来,我们才能把黄曲霉毒素这个小恶魔给彻底打败,让我们的生活更加健康、安全。
你想想,要是没有这些标准,那些不良商家不就可以随便乱来啦?那我们还能放心地吃东西吗?那肯定不行啊!所以说,黄曲霉毒素方法标准真的是太重要啦!它们就是我们健康的守护神,保护着我们不受黄曲霉毒素的侵害。
黄曲霉素

黄曲霉的致癌性
• 由于黄曲霉毒素具有基因毒性,可使基 因发生突变,由此导致动物细胞发生畸 变、诱变及癌变,AF是目前已知的最强 的致癌物。大量研究表明,长期低剂量 或短期摄入较大剂量的AF均可诱发大鼠、 小鼠、豚鼠、雪貂、鸭雏、狗、猫、兔、 猴等动物原发性肝癌,其中以大鼠和鳟 鱼最敏感。
黄曲霉的致癌性
黄曲霉毒素
• 结构类似的一组化合物,均为二呋喃香豆素 (difuranocoumarin)的衍生物。虽然AF的 衍生物有20多种,目前已分离鉴定出的AF包 括AFB1、AFB2、AFG1、AFG2、AFM1、 AFM2、AFP1、AFQ、AFH1、AFGM、 AFB2a和毒醇等12种,但天然污染粮油食品 的AF是B组和G组两大类,即AFB1、AFB2 、 AFG1和AFG2。
黄曲霉毒素
• AFB1对热非常稳定, 268-269℃方可 破坏, 故一般烹调温度不破坏其毒性。 某些化学试剂如5%次氯酸钠和丙酮可使 AFB1完全分解,因此在实验室中用此方 法对接触过AFB1的器皿作解毒处理。但 在紫外线照射可使之分解。
黄曲霉毒素
• AFB1和AFB2在紫外光下可产生蓝紫色 荧光, AFG1和AFG2则产生黄绿色荧光, 该特性是检测粮油食品中黄曲霉毒素的 重要依据。
黄曲霉毒素
• 迄今为止,该病的病因尚未明了, 其发病与 多种因素有关,其中膳食暴露AFB1作为一重 要的危险因子已引起世界的广泛注意。来自中 国、肯尼亚、莫桑比克、瑞典、泰国及菲律宾 的研究结果表明,膳食低剂量长期暴露AFB1 与人类原发性肝细胞癌呈正的剂量反应关系。 与城区相比,该病的发生在以谷物为膳食主要 来源的农村更为常见,且有年轻化的趋势,严 重威胁居民的身体健康。
黄曲霉的生长
• 在实验的1660株黄曲霉中,来自广西的145 中有86株(59.3%)产毒,其中23(15.9%) 株为强毒株,产毒量最高者达400 mg/kg,从 产毒菌株数和产毒量上均为全国之最。该研究 结果与17省区粮油食品中AFB1的污染水平和 原发性肝癌的发病率呈正相关关系。Gabal等 报道,产毒的黄曲霉中有40%的菌株可同时 产生AFB1、、AFB2、AFG1和AFG2。
黄曲霉毒素的危害及检测方法研究进展

黄曲霉毒素的危害及检测方法研究进展赵晓野,王 儒,王 婷*,张 乐,林达峰(海南省食品药品检验所海口分所,海南海口 570311)摘 要:黄曲霉毒素(Aflatoxins,AFT)主要是由真菌寄生曲霉和黄曲霉产生的次生代谢产物,在自然界中普遍存在,尤其在湿热的环境中极易产生,由于其具有极强的致癌性,研究其检测方法极其重要。
本文主要介绍黄曲霉毒素的危害、检测技术及黄曲霉的残留限量标准,并对应用较广的高效液相色谱-质谱联用法、液相色谱法、薄层色谱和酶联免疫吸附等测定黄曲霉毒素的方法、原理及应用进行了综述,以期为黄曲霉毒素检测技术的研究提供一定的参考。
关键词:黄曲霉毒素;危害;限量标准;检测技术Research Progress on the Harm and Detection Methods ofAflatoxinsZHAO Xiaoye, WANG Ru, WANG Ting*, ZHANG Le, LIN Dafeng(Hainan Provincial Institute for Food and Drug Control Haikou Branch, Haikou 570311, China)Abstract: Aflatoxins (AFT) are mainly secondary metabolites mainly produced by Aspergillus flavus and Aspergillus parasiticus. They are widely distributed in nature, especially in humid and hot environment. Because of their strong carcinogenicity, it is very important to study their detection methods. This paper mainly introduces the harm and detection technology of aflatoxins and the residue limit standard of aflatoxins; and summarizes widely used methods, principles and applications of high performance liquid chromatography-mass spectrometry, liquid chromatography, thin layer chromatography and enzyme-linked immunosorbent assay for the determination of aflatoxins, in order to provide some reference for the research of aflatoxins detection technology.Keywords: aflatoxins; hazard; limit standard; detection technology黄曲霉是一种广泛分布的环境习居菌,经常在种植、贮藏、加工和运输过程污染玉米、花生等富含脂肪酸的粮食及相关食品和饲料,并会产生多种有毒次生代谢产物,统称为黄曲霉毒素。
高效液相色谱法定量分析食用油中的黄曲霉毒素B1

分析检测高效液相色谱法定量分析食用油中的黄曲霉毒素B1乔佳璐,王 丽(宝鸡市食品药品检验检测中心,陕西宝鸡 721000)摘 要:目的:建立高效液相色谱测定食用油中黄曲霉毒素B1的方法。
方法:样品用甲醇水提取,经黄曲霉毒素B1免疫亲和柱净化,用甲醇洗脱,洗脱液用柱后碘衍生法上机测定。
结果:该方法线性回归方程为Y=27 217.3X,相关系数R2=0.999 6,回收率在96.8%~101.2%,方法重复性的RSD为0.6%,质控样的Z 值为0.5,检出限为0.029 µg·kg-1,定量限为0.098 µg·kg-1。
结论:该方法线性关系良好,回收率优良,质控样、检出限、定量限均符合要求,具有简单、快速、高效、节约试剂耗材的特点。
关键词:食用油;高效液相色谱法;黄曲霉毒素B1Quantitative Determination of Aflatoxins B1 in Edible Oil by High Performance Liquid ChromatographyQIAO Jialu, WANG Li(Baoji Food and Drug Testing Center, Baoji 721000, China)Abstract: Objective: In order to establish the determination method of aflatoxins B1 in edible oil by high performance liquid chromatography. Method: The samples were extracted with methanol water, purified by aflatoxins B1 immunoaffinity column, eluted with methanol, and determined by post-column iodine derivatization. Result: The linear regression equation was Y=27 217.3X, the correlation coefficient was R2=0.999 6. The recovery rate of method was from 96.8% to 101.2%, the RSD of reproducibility was 0.6%, the Z value of quality control samples was 0.5, and the detection limit was 0.029 µg·kg-1, the limit of quantification was 0.098 µg·kg-1. Conclusion: The method has good linearity, good recovery, quality control sample, detection limit, quantitative limit are in line with the requirements, with simple, fast, efficient and save reagent consumables characteristics.Keywords: edible oil; high performance liquid chromatograph; aflatoxins B1黄曲霉毒素是一种由黄曲霉菌产生的真菌毒素,主要包括黄曲霉毒素B1、B2、G1、G2等羟基化代谢产物[1]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
一、目的
规范实验室检验方法。
二、内容
(一)试验准备
1、标定荧光计。
(1)、将仪器后的电源开关打开。
(2)、按SELECTTEST键,直至显示出AflaTest,按ENTER。
(3)、荧光计显示:START CALBRATION…OPEN THE LID
INSERT RED VIAL
打开仪器盖,将红色的真菌毒素标定管放入。
一定要确认标定管是
否已放到了仪器的底部。
(4)、仪器显示:HIGHCAL 22PPB
如果这个值正好是所选分析方法规定的设定值,请按ENTER键。
否则,从键盘上所规定的设定值,确认准确无误后,按ENTER键。
(5)、仪器显示:READING HIGH CAL…SA VING HIGH INENSTTY
OPEN THE LID
INSERT GREEN VIAL
打开仪器盖取出红色的标定管,插入绿色的真菌毒素标定管。
一定
要确认标定管是否已充分插入仪器的底部。
(6)、仪器显示:LOW CAL 1.0PPB
如果这个值正好是所选分析方法规定的设定值,请按ENTER键.否则,从
键盘上所规定的设定值,确认准确无误后,按ENTER键。
(7)、仪器显示:READING LIW CAL…SA VING HIGH INTENSITJY 接着显示:OPEN THE LID
INSERT GREEN VIAL
打开仪器盖取出绿色的标定管。
(8)、仪器显示:VICAM V1.3 READY
按SELECT TEST键,仪器显示: AFLATEST, 按ENTER键。
(9)、仪器显示:START RUN TEST OPEN THE LID (10)、插入黄色的真菌毒素标定管,其读数应该在分析方法的测定范围内。
(11)、荧光屏计标定工作完毕,可经进行样品分析了。
荧光计每
周需要标定一次。
如果需要重新标定,请按STOP键,再按OPIONS 键,直至仪器显示:CALIBRATE TEST
然后按ENTER键,仪器显示:AFLATEST
如果不是这样,按SELECT TEST键,直至仪器显示:AFLATEST,
按
ENTER键。
然后,按照上面所述操作步骤进行。
如果进行样品分析,按SELECT TEST键,直至仪器显示:AFLATEST键。
2、配制AflaTest显色液(一天配制一次)。
3、配制甲醇/水(60:40体积比)溶液(每周配制一次,或根据需要
配制)。
4、试剂空白试验(1ml甲醇+1ml显色液置于测试管中),荧光计读数
应为0。
5、纯水空白试验(2ml纯水置于测试管中),荧光计读数应为0。
(二)样品提取
1、称取25克磨细的样品,5克氯化钠,置于搅拌杯中。
2、加入125ml甲醇/水(60:40)溶液。
3、盖上搅拌杯的盖子,高速成搅拌1分钟。
4、取下盖子,将提取物到入折叠滤纸上,滤液收集于干净的容器中。
(三)提取物的稀释
1、移取20ml上步的滤液,置于干净的容器中。
、
2、用20ml纯水将滤液稀释,混匀。
3、将上步稀释液通过玻璃纤维滤纸过滤,滤液收集于玻璃注射器筒中,量取10ml。
(四)分离柱色谱操作
1、将上步10 ml滤液(10ml=1.0g的样品),以1-2滴/秒的流速全部通过AflaTest-P亲和柱,直到空气进入到亲和柱中。
2、将10 ml纯水以2滴/秒的流速通过亲和柱中。
3、将7.2步再重新操作一次, 直到空气进入到亲和柱中。
4、用1ml色谱级甲醇以1-2滴/秒的流速淋洗亲和柱,将所有样品淋洗液收ml玻璃试管中。
5、将1ml AflaTest显色液加到测试管的淋洗液中,混匀后,将测试管置于已标定好了的荧光计中。
60秒后,读数。
(五)、如果要结束试验,按STOP键。